
Abstract
Antibody–drug conjugates (ADCs) are an emerging class of anticancer therapeutics, delivering highly cytotoxic molecules directly to cancer cells. ADCs are composed of an antibody, a small molecule drug, and a linker attaching one to another. Antibodies are directed to a large variety of antigens overexpressed on tumor cells, tumor vasculature, or tumor-supporting stroma. After internalization, the ADC is transferred to lysosomes where the cytotoxic component is released, finally killing the target cell. All ADCs are administered via intravenous injection. Once in the circulation, linker stability in plasma is of high importance. In vivo studies in animals address the release of payload over time and typically measure total antibody, conjugated ADC, and free drug. ADC development is driven by ICH (International Council for Harmonisation) guidelines S6(R1) and S9. Dose-limiting toxicities of current ADCs are mainly associated with the payload and correlate well between clinical trials and nonclinical studies in rodents and nonrodents. This mini review is intended to provide general information about ADCs in oncology and shall assist the toxicologic pathologist in correctly interpreting morphological findings acquired in toxicity studies with this entity.
Keywords
Introduction
Antibody–drug conjugates (ADCs) have emerged as an important class of anticancer therapeutics (Hedrich et al. 2018). They are designed to deliver highly cytotoxic small molecules directly to cancer cells via tumor-specific antibodies, hence providing a much wider therapeutic window than conventional chemotherapy. The basic concept is that (i) ADCs specifically bind to tumor cells or tumor-supporting cells, that (ii) they become internalized upon antigen binding, and that (iii) within the target cell the cytotoxic component of the ADC is released and destroys the target cell (Figure 1).
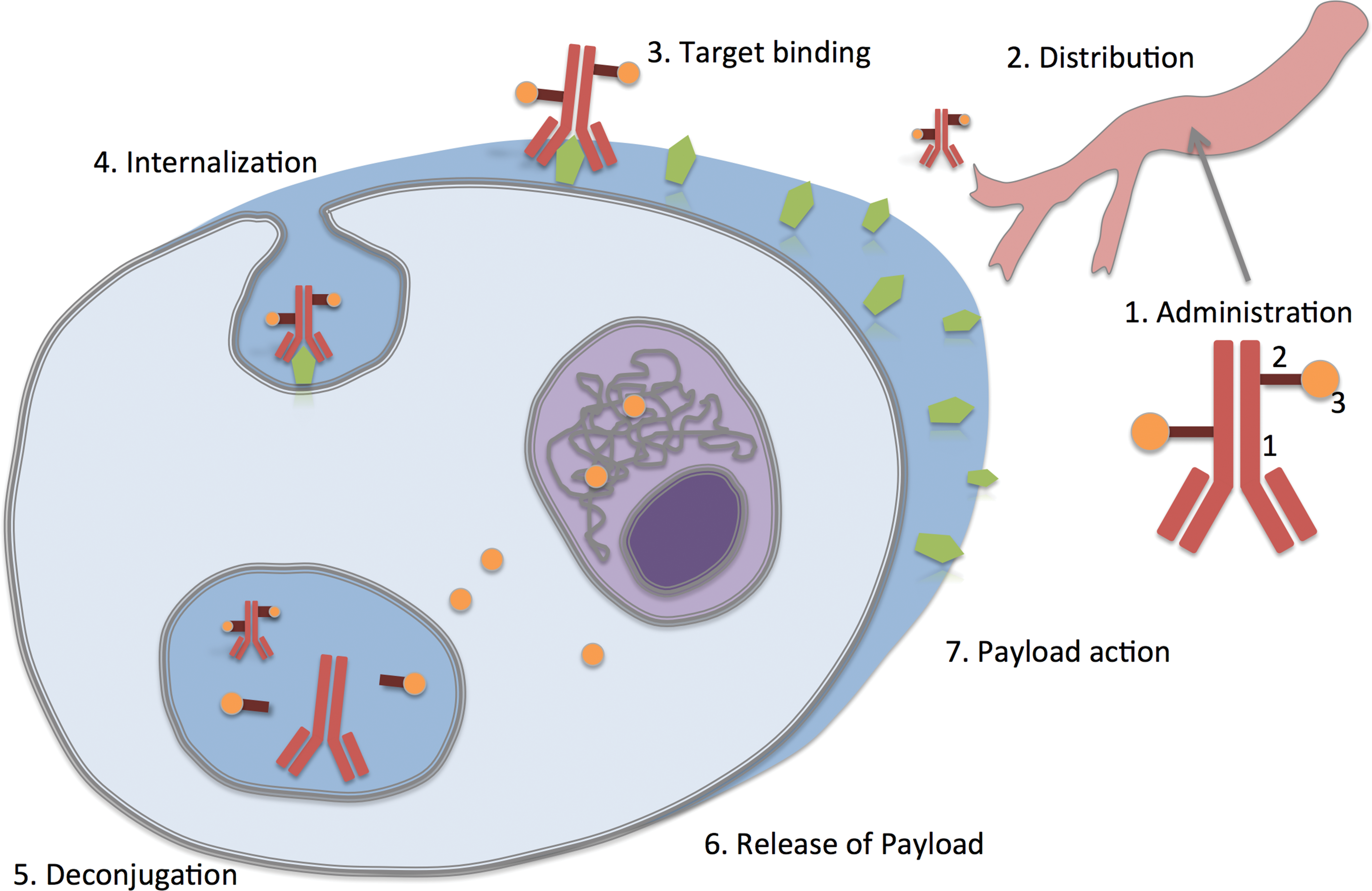
Concept of ADCs in oncology. An ADC is composed of antibody (1), linker (2), and drug (3). After intravenous administration (1), it is distributed throughout the body (2) and binds to an epitope-expressing target cell (3). Upon target binding, it becomes internalized (4) and is transferred to lysosomes. Therein the ADC is deconjugated (5), the payload is released (6), and diffuses into the cytoplasm and the cell nucleus, where it is active (in this example, interacting with DNA), causing a cytotoxic effect (7). ADC = antibody–drug conjugate.
Four different ADCs have so far received approval for the treatment of various types of hematological and solid cancer. Gemtuzumab ozogamicin (Mylotarg; Pfizer Inc., Philadelphia, PA, USA), which is an anti-CD33 antibody conjugated to calicheamicin, was the first ADC receiving approval by the U.S. Food and Drug Administration (U.S. FDA) in 2000. It was used in the treatment of acute myelogenous leukemia but was withdrawn from the market in 2011 due to an unfavorable risk–benefit evaluation; for the same reason, the European Medicines Agency (EMA) refused approval in 2008. The second marketed ADC, receiving U.S. FDA approval in 2011 and EMA approval in 2013, was brentuximab vedotin (Adcetris; Seattle Genetics Inc., Bothell, WA, USA). This is an anti-CD30 antibody that is conjugated to monomethyl auristatin E (MMAE) and that is used to treat non-Hodgkin lymphoma and anaplastic large cell lymphoma (EMA 2012). The third ADC receiving marketing approval by U.S. FDA and EMA in 2013 is ado-trastuzumab emtansine (Kadcyla; Genentech/Roche, Grenzach-Wyhlen, Germany). This is an anti-HER2 antibody conjugated to DM-1, which is used to treat HER2-positive metastatic breast cancer (EMA 2013). In 2017, inotuzumab ozogamicin (Besponsa; Pfizer, Sandwich, UK) was approved by U.S. FDA and EMA. This is an antibody against CD22 conjugated to calicheamicin and used in the treatment of CD22-positive B-cell precursor acute lymphoblastic leukemia (EMA 2017).
Many of the current ADCs still have relatively narrow therapeutic index and limited clinical success (Lin, Guo, and Wang 2018). Consequently, there are many more ADCs in the drug development pipeline and in clinical trials. As of August 2018, the database clinicaltrials.gov lists 35 actively recruiting studies which investigate ADCs in hematologic and solid tumors, and more than 200 ADCs under development are listed on https://adcreview.com/adc-university/adc-drugmap/. As the number of ADCs entering the clinical stage of development increases, the likelihood rises that toxicologic pathologists become exposed to this class of compounds. This mini review is intended to provide general information about this class and shall assist the toxicologic pathologist in correctly interpreting morphological findings acquired in toxicity studies with ADCs. In the scope of this review, only anticancer ADCs are discussed, while the general principles would also apply to ADCs developed for nononcology indications.
Structure
ADCs are composed of an antibody, a small molecule drug, and a linker attaching one to another. Antibodies may be directed to a large variety of antigens that are overexpressed on tumor cells, on tumor vasculature, or on the tumor-supporting stroma. The antibody is the primary component that directs the molecule to its site of action. Consequently, target antigens generally show high interindividual penetrance in patients, high density on the cell surface, and accurate discrimination between target cells and normal cells, which should not be affected by the therapy. Antigens are selected to allow for appropriate internalization of the ADC. While all currently marketed ADCs are composed of humanized antibodies, some ADCs are under development that contain modified antibody versions such as single-domain antibody fragments (known as nanobodies; Fang et al. 2016) or bispecific antibodies (Sellmann et al. 2016), which are supposed to optimize the pharmacokinetic and pharmacologic properties of the ADC.
After the ADC has been internalized and transferred to lysosomes, the cytotoxic component of the ADC is released. This is mainly driven by the chemical structure of the linker. There are basically two linker concepts: cleavable linkers and noncleavable linkers. Noncleavable linkers (such as thioether or maleimidocaproyl) require that the entire molecule is digested within lysosomes. They have the advantage of being very stable in plasma but may show limited efficacy at the target cell. Cleavable linkers may have the disadvantage of instability in plasma, which results in the uncontrolled release of the cytotoxic molecule and side effects, paired with the advantage that they generally show higher efficacy at the target cell. There are generally 3 different types of cleavable linkers used, that is, protease-sensitive linkers (e.g., dipeptides), acid-sensitive linkers (e.g., hydrazine), and glutathione-sensitive linkers (e.g., disulfide). The third and pharmacodynamically most important component of ADCs is the small molecule drug, also known as payload or warhead. Since only a few molecules will become internalized and released within the target cell, the payload needs to be highly cytotoxic with potency in the subnanomolar range. Therefore, conventional cytotoxic drugs cannot be used. Most cytotoxic payloads in ADCs target tubulin and thus impair cell division, or they bind to DNA and cause DNA damage, which blocks cell division. Payloads interacting with DNA are able to exert their mode of action at any phase in the cellular cycle, whereas tubulin binders will only attack tumor cells when they are in a mitotic state. It has been discussed that DNA damaging agents might be more efficacious in tumor cell killing than tubulin binders, particularly in case of solid tumors (www.synthon.com). With the scope to improve efficacy or the therapeutic index of ADCs, several new payloads are under development such as highly potent anthracyclines, tubulysin derivatives, aplyronines, epothilones, thailanstatin, exatecan derivatives, cryptophycins, hemiasterlins, and amanitins.
Pharmacology
The desired pharmacologic effect of ADCs in oncology is to destroy the target cell upon internalization. However, neighboring cells may be affected by extracellular cleavage of the ADC or by diffusion of the payload, which is known as the bystander effect (Staudacher and Brown 2017). All three components of an ADC molecule, that is, antibody, linker, and payload, determine its pharmacologic properties (Lucas et al. 2018). The antibody component, typically of the IgG isotype, drives efficacy by target binding and internalization. While it is generally believed that there is a correlation between available antigen expression and ADC efficacy, target antigens with low expression may still be effective, if they are efficiently internalized (Lucas et al. 2018). Since internalization by endocytosis is so important for ADCs, antibodies are typically selected that show limited or no potency to induce direct or antibody-mediated cytotoxicity. In vitro tests are typically used to characterize target binding and internalization, and in vivo studies in nude mice bearing xenogenic tumors are typically performed to characterize the effect on tumor growth or regression in reference to different dosing regimens (EMA 2013, 2017).
An important factor in pharmacological efficacy of ADCs is the number of cytotoxic molecules that is attached to a single antibody (Lucas et al. 2018). This number is known as the drug–antibody ratio (DAR). For most ADCs, the DAR is not a precise number but rather a range between 2 (two drug molecules attached to an antibody) and 14 (14 drug molecules attached to a single antibody). Under conditions of random chemical conjugation, a mixture of ADCs with different DARs arises. This heterogeneity in DAR may negatively impact efficacy and tolerability of the ADC. While it has been demonstrated in vitro that a higher DAR leads to increased potency, in vivo studies have shown decreased efficacy of agents with high DAR, due to faster distribution and clearance (Lucas et al. 2018). In addition, the site of conjugation seems to play an important role (Lucas et al. 2018). Therefore, strategies such as site-specific conjugation have been developed that deliver ADCs with a very narrow band of DARs, resulting in a wider therapeutic index.
Pharmacokinetics
All ADCs are administered via intravenous injection, either as a bolus or via infusion over an extended period of time. Tissue distribution is mainly driven by the expression profile of the epitope to which the ADC binds, and it is typically investigated using ADCs with radiolabeled drug.
Once in the circulation, stability of the linker is crucially important for tolerability. Hence, linker stability is typically investigated in vitro, using plasma from humans and various animal species. Briefly, the ADC is incubated in plasma from different species at 37°C. At different time points, samples are drawn in which the amount of total antibody and conjugated antibody as well as the amount of free drug (or drug linker) is quantified. The ideal linker has high stability in plasma but rapidly releases the payload once taken up by lysosomes.
In vivo studies in animals also need to address the release of payload over time. Consequently, in vivo studies require repeated blood sampling and analysis for total (conjugated and unconjugated) antibody, conjugated ADC, and free drug (Kraynov et al. 2016). A typical pharmacokinetic profile is shown in Figure 2. The concentration of total (conjugated and unconjugated) antibody declines over time, driven by uptake into antigen-expressing target cells and by antibody clearance. The pharmacokinetic profile of the currently marketed ADCs shows high interpatient variability, and the reasons behind this are still insufficiently understood (Lucas et al. 2018). Factors influencing antibody clearance are uptake by the mononuclear phagocyte system (which is mainly mediated via various forms of Fc-γ receptors), and neonatal Fc receptor (FcRn)-mediated recycling. The FcRn is widely expressed throughout the body, but it only binds in the acidic environment of an endocytic vacuole, where binding results in recycling of the ADC to the extracellular compartment. The occurrence of anti-ADC antibodies (i.e., immunogenicity) may further influence the pharmacokinetic profile of an ADC, similar to what is known for unconjugated therapeutic antibodies.
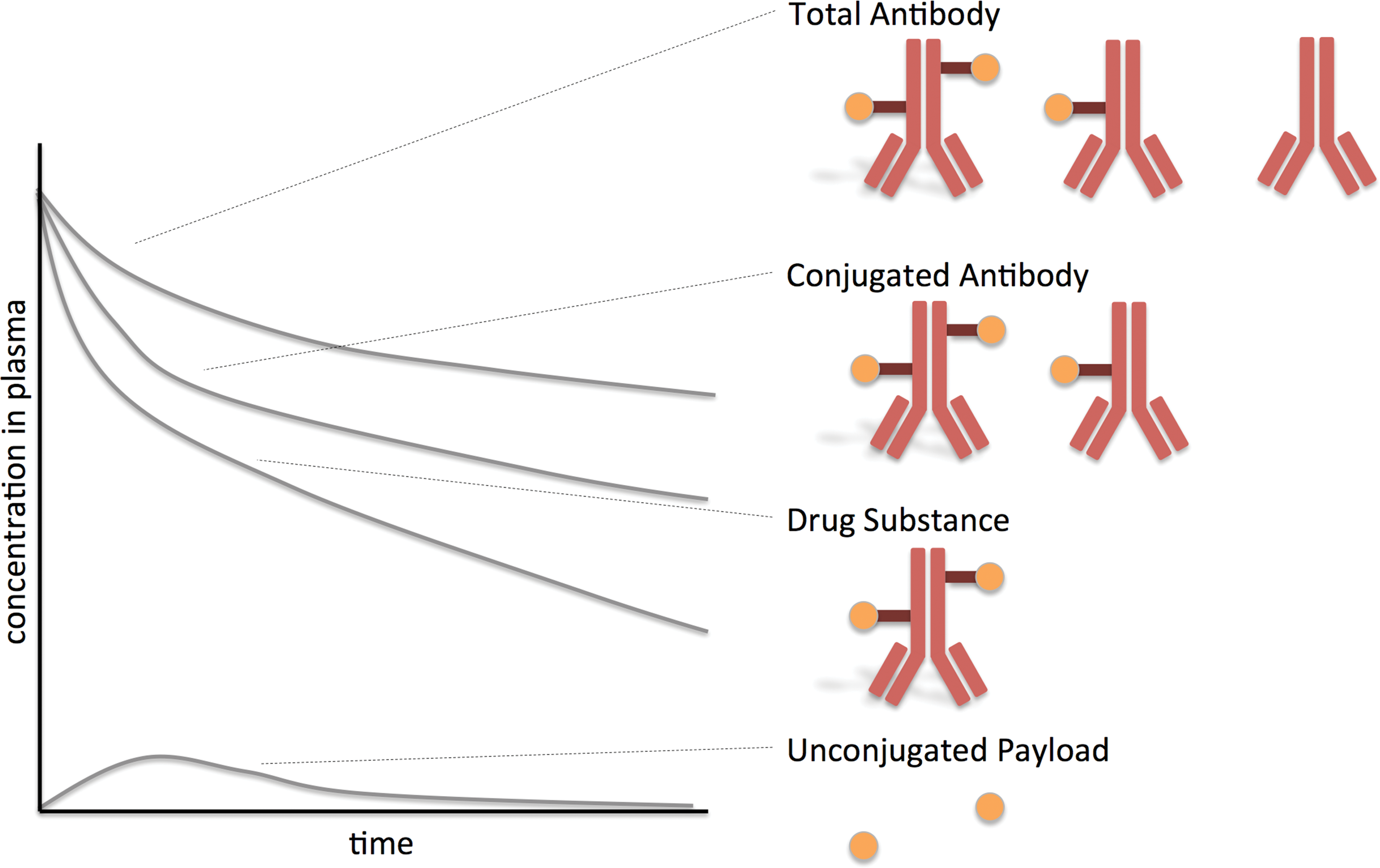
Prototypical pharmacokinetic profile of an ADC. The graph shows the plasma concentration over time. Plasma concentrations of the drug substance declines rapidly, due to target binding, elimination, and deconjugation. Deconjugation results in conjugated antibody forms with a reduced DAR and in the presence of unconjugated payload in the plasma. ADC = antibody–drug conjugate; DAR = drug–antibody ratio.
The concentration of conjugated antibody also declines due to some unspecific extracellular cleavage of the linker. This results in the occurrence of unconjugated antibody and of free drug within the circulation. The latter is then eliminated via metabolism and clearance. Metabolism and clearance of the free payload can be influenced by drug–drug interactions and impaired elimination pathways such as liver and kidney disease.
Toxicity
Since ADCs developed for targeted chemotherapy contain highly potent cytotoxic drugs, they are toxic molecules which require a thorough preclinical safety assessment (Hinrichs and Dixit, 2015). As a biotechnology-derived medicinal product, preclinical safety assessment of ADCs is guided by the ICH guideline S6(R1). One of the main aspects from this guidance is that toxicity should be evaluated in species that express the epitope to which the antibody is binding and that show binding kinetics to this epitope in the same range as humans. For most ADCs, this results in safety studies conducted in the nonhuman primate (mostly the cynomolgus macaque). Additional guidance is given by the guideline ICH S9 on anticancer drugs, which contains a section specifically referring to conjugation products. A question and answer document to ICH S9, issued in 2016, recommends that toxicity of an ADC is tested in at least one species, even if the epitope is not present in any animal species. At least two doses of the ADC should be administered in order to support initial clinical trials. Studies of the unconjugated antibody in binding species are generally not warranted, unless there are particular concerns regarding understanding of in vivo pharmacology (resulting from antibody binding to target) and/or a lack/poor understanding regarding antigen expression and the potential for direct antigen-dependent toxicity. If the toxicity of the payload/linker has not previously been characterized, evaluation of the free drug in the rat, to assess antigen-independent toxicity, is typically included in the toxicology program.
The nonclinical safety program for an ADC needs to be determined case by case and in consideration of the clinical program that is to be supported. According to ICH S9, the starting dose for a first-in-human clinical trial is typically 1/6 of the highest nonseverely toxic dose in the repeat-cycle GLP toxicity study in the nonrodent species (typically the nonhuman primate). Interestingly, ICH S9 recommends allometric scaling based on body surface area (as described in the U.S. FDA’s Guidance for Industry: Estimating the Maximum Safe Starting Dose in Initial Clinical Trials for Therapeutics in Adult Healthy Volunteers from 2005), since most of the toxicity is caused by the small molecule payload rather than the antibody (see below).
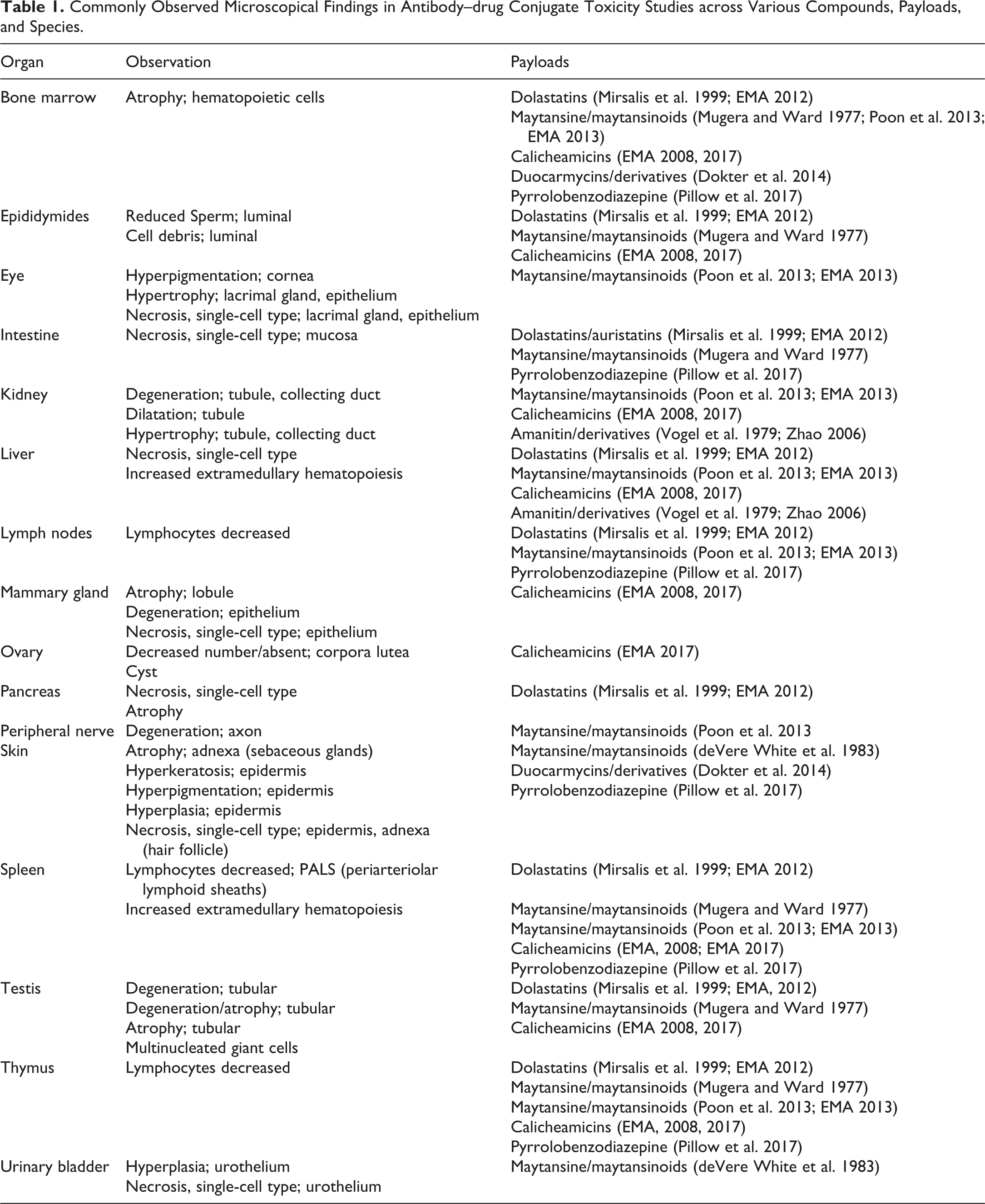
Hinrichs and Dixit (2015) compared clinical toxicities from ADCs with 5 different warheads (MMAE, MMAF [Monomethyl auristatin F], DM-1, DM-4, and calicheamicin) and summarized that myelotoxicity (resulting in peripheral neutropenia or thrombocytopenia) was most common, followed by peripheral neuropathy, hepatotoxicity, skin rash, ocular toxicity, and toxicity of the gastrointestinal tract. While all components of an ADC are important for determining its tolerability, dose-limiting toxicities are mostly associated with the cytotoxic payloads (Hinrichs and Dixit, 2015), which are briefly summarized below. Commonly observed morphological findings in ADC toxicity studies in rodents and cynomolgus monkeys are listed in Table 1. Those depend not only on the type of compound but also on the time between administration of the test article and preservation of organs. Most test article–related microscopic findings increase in severity with dose and are reversible.
Commonly Observed Microscopical Findings in Antibody–drug Conjugate Toxicity Studies across Various Compounds, Payloads, and Species.
Dolastatins and Auristatins Such as MMAE and MMAF
Dolastatins are oligopeptides found in the wedge sea hare, Dolabella auricularia. They block polymerization of tubulin. Dose-limiting toxicity of the prototypic dolastatin 10 is myelotoxicity. The maximum tolerated dose after a single intravenous dose is 450 μg/kg in mice, 75 μg/kg in rats, and <20 μg/kg in dogs (Mirsalis et al. 1999). MMAE is a synthetic derivative of dolastatin and was first used as payload in the drug brentuximab vedotin (Adcetris). Toxicity studies in rats and cynomolgus monkeys identified toxicity in bone marrow (hypocellularity), lymph nodes (lymphoid depletion), thymus (lymphoid necrosis), spleen (lymphoid depletion), liver (single-cell necrosis, biliary hyperplasia), intestine (single-cell necrosis), pancreas (single-cell necrosis), and testes (tubule degeneration; EMA 2012). Consistent with the preclinical findings, adverse clinical effects that are consistently reported for MMAE ADCs in clinical trials are anemia, neutropenia, and peripheral neuropathy (Masters et al. 2018). MMAF is another synthetic derivative of dolastatin and is clinically often associated with ocular toxicity.
Maytansine and Maytansinoids Such as DM-1 and DM-4
Maytansines are toxins that were first isolated from the plant Maytenus serrata. Maytansines bind to tubulin at the rhizoxin/vinblastine binding site and thus inhibit microtubule formation. Maytansines affect all dividing cell populations and cause toxicity in the gastrointestinal tract, thymus, spleen, bone marrow, and testis (Mugera and Ward 1977). A common histopathological finding in toxicity studies with maytansinoids is the presence of large atypical mitotic figures (cells arrested in metaphase) in a variety of organs (Mugera and Ward 1977; Poon et al. 2013; Figure 3). Further target organs of toxicity are urinary bladder (deVere White et al. 1983) and skin. Emtansine/mertansine (DM-1) and ravtansine (DM-4) are maytansinoids, that is, synthetic derivatives of maytansine. DM-1 was first used as payload in the drug ado-trastuzumab emtansine (Kadcyla). Primary target organs of toxicity of DM-1 in rats are liver (degeneration, necrosis), hematopoietic cells (mainly affecting platelets), and lymphoid organs (Poon et al. 2013; EMA 2013). Further organs of toxicity are kidney (tubular degeneration, necrosis), lacrimal glands (epithelial cell hypertrophy and decreased mucous cells), and peripheral nerves (axonal degeneration; EMA 2013). This correlates with common adverse effects in clinical trials reported as thrombocytopenia and hepatic toxicity (Masters et al. 2018).
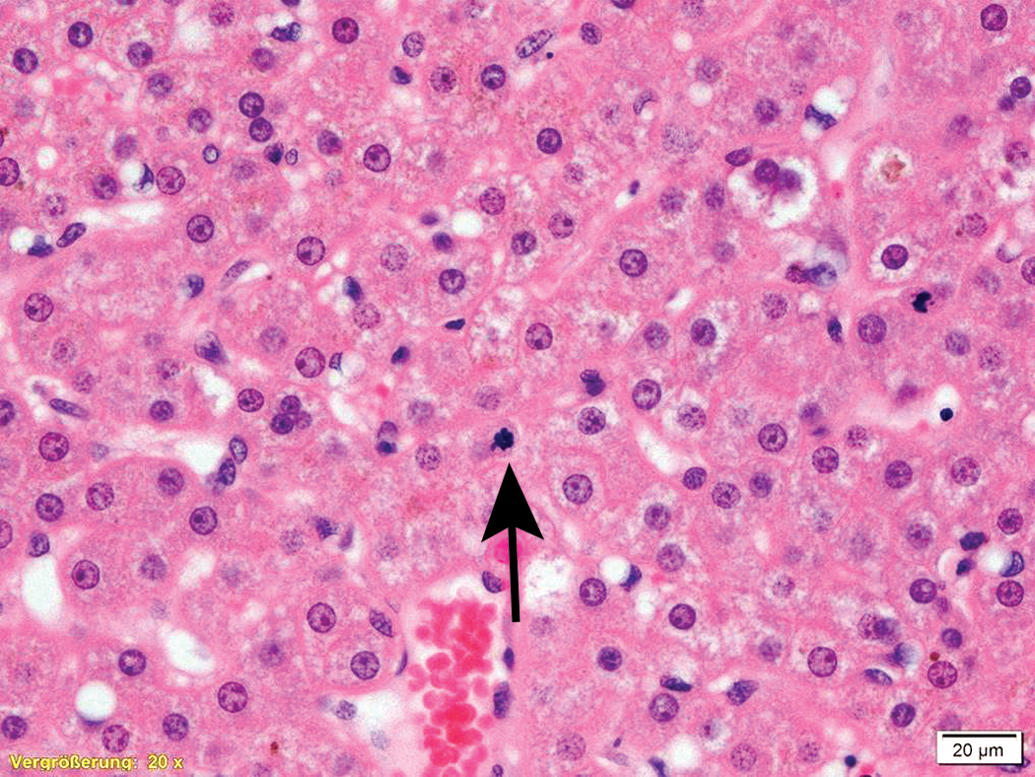
Atypical mitotic figure in the liver due to metaphase arrest caused by a dolastatin. Formalin-fixed, paraffin-embedded tissue from a cynomolgus monkey. Hematoxylin & eosin stain.
Calicheamicins and Their Derivatives
Calicheamicins derive from the bacterium Micromonospora echinospora. They bind to the minor groove of DNA and cause double-stranded DNA breaks at subpicomolar concentration. Ozogamicin, a synthetic calicheamicin derivative, is used in the ADC products Mylotarg (gemtuzumab ozogamicin) and Besponsa (inotuzumab ozogamicin). Toxicity of gemtuzumab ozogamicin was similar in rats and cynomolgus monkeys and primarily affected bone marrow, mammary glands, kidneys (tubules), liver, spleen, and testes/epididymides (EMA 2008). Toxicity of inotuzumab ozogamicin was investigated in rats and cynomolgus monkeys and affected liver (karyomegaly, single cell necrosis), lymphoid tissue (bone marrow atrophy, lymphoid atrophy in thymus, spleen, and gut), kidneys (proteinaceous intraluminal casts), testis (tubular degeneration), mammary gland (atrophy), ovaries (degenerating primary follicles), lung (alveolar macrophage accumulation), and peripheral nerves (axonal degeneration; EMA 2017).
Duocarmycins and Their Derivatives
Duocarmycins are toxins isolated from Streptomyces bacteria. They bind to the minor groove of DNA and irreversibly alkylate adenine, which causes DNA damage. The LD50 of duocarmycin B2 in mice is 180 μg/kg and the LD10 is 140 μg/kg (Kobayashi et al. 1994). Several analogs of duocarmycins (such as adozelesin, bizelesin, and carzelesin) have been synthesized and explored as potential antineoplastic agents, but published reports on their toxicity profile are sparse. An ADC composed of a HER2-targeting antibody coupled to duocarmycin by a cleavable linker has shown mild and transient myelotoxicity as well as skin hyperpigmentation in cynomolgus monkeys (Dokter et al. 2014).
Anthramycin, Pyrrolobenzodiazepines, and Indolino-benzodiazepines
Pyrrolobenzodiazepines are produced by actinomycetes bacteria. They bind to the minor groove of DNA and cross-link specific sites of the DNA, thus blocking cell division. The prototypic pyrrolobenzodiazepine is anthramycin, which was first isolated from the Streptomyces refuineus bacterium. Synthetic pyrrolobenzodiazepines such as tesirine and talirine are used in ADCs that are currently undergoing clinical investigation. Published information on the toxicity of pyrrolobenzodiazepine is sparse. Single-dose intravenous toxicity studies in rats have shown myelotoxicity, lymphoid system toxicity, intestinal toxicity, and cutaneous toxicity (Pillow et al. 2017). Indolino-benzodiazepines are modified pyrrolobenzodiazepines in which the pyrrolo group is substituted by a single imine group. In mice, indolino-benzodiazepines have primarily shown myelotoxicity (Miller et al. 2016).
Amanitin and Derivatives
α-amanitin is a bicyclic oligopeptide that naturally occurs in mushrooms of the genus Amanita. α-amanitin is a potent inhibitor of the enzyme RNA polymerase II, which plays a major role in the transcription of DNA into mRNA. α-amanitin causes hepatotoxicity and nephrotoxicity in various species. Hepatotoxicity is characterized by hepatocellular necrosis, and nephrotoxicity is characterized by cytoplasmic swelling and vacuolar degeneration in renal tubules, progressing to extensive necrosis (Vogel, Braatz, and Mengs 1979; Zhao et al. 2006).
Conclusion
ADCs are complex molecules, and in the scope of regulatory toxicity studies, they cause a variety of microscopical findings. Those findings overlap across different payloads, yet there are specific differences related to their mode of action. Dolastatins and maytansine/maytansinoids block polymerization of tubulin and hence cause metaphase arrest of dividing cells, resulting in the presence of large atypical mitotic figures. These are not found in payloads that directly cause DNA damage. Most currently used payloads are associated with myelotoxicity, which may result in cytopenias and impaired host resistance leading to secondary infections and moribund conditions. Associated with their specificity for replicating cells, many ADCs cause microscopical findings in the male and female reproductive organs and the intestine. Maytansines have been shown to be associated with peripheral neuropathy, and amanitins are well known to cause findings in liver and kidney with limited or no hematopoietic toxicity. Overall, the pattern of microscopic findings is quite variable across ADCs, demonstrating that it does not only depend on the payload used but also the distribution (which is primarily determined by the antibody component) and the time point by which the organs are investigated.
Footnotes
Author Contribution
The author (LM) contributed to conception or design; data acquisition, analysis, or interpretation; drafting the manuscript; and critically revising the manuscript. The author gave final approval and agreed to be accountable for all aspects of work in ensuring that questions relating to the accuracy or integrity of any part of the work are appropriately investigated and resolved.
Declaration of Conflicting Interests
The author(s) declared no potential, real, or perceived conflicts of interest with respect to the research, authorship, and/or publication of this article
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.