
Abstract
Nonclinical implantation studies are a common and often critical step for medical device safety assessment in the bench-to-market pathway. Nonclinical implanted medical devices or drug-device combination products require complex macroscopic and microscopic pathology evaluations due to the physical presence of the device itself and unique tissue responses to device materials. The Medical Device Implant Site Evaluation working group of the Society of Toxicologic Pathology’s (STP) Scientific and Regulatory Policy Committee (SRPC) was tasked with reviewing scientific, technical, and regulatory considerations for these studies. Implant site evaluations require highly specialized methods and analytical schemes that should be designed on a case-by-case basis to address specific study objectives. Existing STP best practice recommendations can serve as a framework when performing nonclinical studies under Good Laboratory Practices and help mitigate limitations in standards and guidances for implant evaluations (e.g., those from the International Organization for Standardization [ISO], ASTM International). This article integrates standards referenced by sponsors and regulatory bodies with practical pathology evaluation methods for implantable medical devices and combination products. The goal is to ensure the maximum accuracy and scientific relevance of pathology data acquired during a medical device or combination drug-device implantation study.
Keywords
This Points to Consider article is a product of a Society of Toxicologic Pathology (STP) Working Group commissioned by the Scientific and Regulatory Policy Committee (SRPC) of the STP. It has been reviewed and approved by the SRPC and Executive Committee of the STP but does not represent a formal best practice recommendation of the Society; rather, it is intended to provide key “points to consider” in designing studies or interpreting data from toxicity and safety studies intended to support regulatory submissions. The views expressed in this article are those of the authors and do not represent the policies, positions, or opinions of their respective agencies and organizations. Readers of Toxicologic Pathology are encouraged to send their thoughts on these articles or ideas for new topics to the Editor.
Introduction
Medical devices are defined in Section 201(h) of the United States Food, Drug, and Cosmetic Act to include products intended to affect the structure or any function of the body of man or other animals, and which does not achieve its primary intended purposes through chemical action within or on the body of man or other animals or which is not dependent upon being metabolized for the achievement of its primary intended purposes.
1
This broad definition includes multiple classes of medical devices from relatively low-risk and low exposure Class I products, to intermediate-level Class II products, to high-risk life-sustaining invasive Class III products produced by complex or emerging technologies (e.g., active implanted medical devices). Importantly, medical devices are tested for both safety (e.g., appropriate tissue and host response [i.e., not adverse] using the final finished form [FFF] introduced in a clinically relevant location) and efficacy (e.g., functionally appropriate response in the tissue and host). In addition, small molecules or biologics may be fully integrated into medical devices, resulting in a FFF as a combination product (drug or biologic and/or a medical device). 2 The pathways for testing safety and efficacy of new biomedical products generally include multiple in vitro and in vivo studies, with the in vivo studies ranging from acute to chronic tissue tolerability of a simple biomaterial (e.g., intramuscular implantation) to multi-year implantation of the FFF in an animal model of the intended clinical indication (e.g., cardiac assist device in an animal model of congestive heart failure).
The wide range of medical device products and target organs/tissues has triggered the development of an equally broad range of tissue/organ and device-specific standards and guidance documents. 3 Assessment is further complicated by the ever-evolving range of biomaterials, which may include novel formulations, physiochemical modifications that alter tissue and immunologic responses, self-forming gels, and electronics with integrated software. In addition, the regulatory landscape for medical devices can be complex, with differing worldwide approaches for the classification of, standards for, and approval pathway of medical devices and drug-device combination products.
Pathologists encountering these documents for the first time, particularly the International Organization for Standardization (www.ISO.org) testing standards, can find the number and breadth of published standards to be overwhelming and access to the standards restrictive due to their for-purchase nature. Pathologists may be further challenged by the expectation of rigid application of pre-defined scoring systems for identifying histopathological findings and assigning microscopic severity grades which are described in the currently published standards; these histology grading schemes/rubrics are widely but not judiciously applied within the industry. In most cases, these pre-defined tissue response scores cannot be accurately applied to many medical devices or implant locations, and thus are insufficient to fully communicate the microscopic pathology findings in a study. The purpose of the pathology evaluation is to identify macroscopic and microscopic pathology findings within a study and to interpret them in context of the study design, and it is not possible to define or predict ahead of time all of the possible microscopic findings that can occur at an implant site. Medical device pathologists (often toxicologic pathologists experienced in nonclinical studies) also must have a working knowledge of commonly used terms and definitions that apply to regulation, testing, and evaluation of medical devices. Some relevant terms and definitions can be found in regulatory documents, but there is limited consensus on the definition of most terms.
To provide clarity for its members who work in this field, the Society of Toxicologic Pathology (STP) tasked its Scientific and Regulatory Policy Committee (SRPC) to empanel a working group to review the scientific, technical, and regulatory considerations for nonclinical studies designed to evaluate the safety of medical devices and combination drug-device products. The objective of the Medical Device Implant Site Evaluation working group (MDISE) was to compile a list of major elements to consider when performing the pathology evaluation for medical devices or combination products. As the subject material in this field is broad, the MDISE focused this article on points to consider when conducting macroscopic (gross) and microscopic analyses of target tissues, as a means of evaluating local biological responses to implantable medical devices or implantable drug-device combination products. Particular emphasis was placed on demonstrating that well-established principles of microscopic evaluation as applied routinely in toxicologic pathology are relevant to medical devices and combination products. A second critical goal was to provide key references that detail specific evaluation parameters for individual organ systems and organ/tissue-specific bioabsorbable devices, which have been provided as Supplemental Material. This points-to-consider document highlights approaches for conducting the macroscopic and microscopic pathology portions needed to assess the local safety of implanted materials during a nonclinical safety study. The specific objectives undertaken by the MDISE were as follows:
To provide background information and practical considerations for medical device implantation studies to pathologists, engineers, sponsors, and regulatory professionals;
To provide a summary of regulatory documents and considerations for study planning;
To recognize deficiencies in the pathology methodology and interpretations presented in currently accepted standards; and
To suggest suitable methods for evaluating, documenting, and reporting the pathology of implantation studies, and considerations for applying these approaches to the ISO-10993-6:2016 4 (the most commonly used standard for implant studies).
Background
Safety assessment has been applied to medical device materials only recently, and thus has evolved along a different path than that of small molecules and biologics. Historically, biomaterials were considered inert and biological/toxicological safety concerns did not bring about regulatory standards until the 1980s. Testing standards/recommendations from different organizations were based on historical protocols for chemical and materials testing protocols, and safety testing procedures were often driven by the considerations and needs of engineers rather than chemists, biologists, or pathologists. Thus, scientific approaches to implant biocompatibility assessment evolved largely as a separate discipline with only recent safety contribution from toxicologic pathologists. As a result of the diverse worldwide regulatory groups responsible for assessments of medical devices and combination products, nonclinical testing procedures for medical devices are generally based on standard testing of physical materials rather than the biological testing guidelines developed globally by national regulatory agencies for drug and biological nonclinical safety assessment. These differences in safety assessment of biomedical materials are amplified when drug and/or biologics are combined with medical devices as combination products.
While the nonclinical testing program for medical devices has many similarities to those for small molecules and biologics, there are notable differences. 5 The goal of nonclinical testing for pharmacologically active products is to develop understanding of target organ toxicity and safety margins prior to clinical testing. Uniformity of nonclinical testing and evaluation of pharmacological products has been refined over many decades through publication of guidance documents offered by multinational efforts such as The International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use (ICH) and the Organisation for Economic Cooperation and Development (www.OECD.org). There are also efforts at standardizing nomenclature through the development of globally recognized microscopic terminology such as the Global Open Registry Nomenclature Information System (goRENI [www.goRENI.org]), and the multiple organ- and species-focused toxicologic pathology lexicons prepared under the International Harmonization of Nomenclature and Diagnostic Criteria (INHAND) initiative (https://www.toxpath.org/inhand.asp#pubg). However, accepted microscopic nomenclature has not been produced for medical device nonclinical implant site evaluations, largely due to the diversity and complexity of tissue changes identified across the range of ever-evolving implantable medical devices and drug-device combination products. Consequently, the lack of a standardized nomenclature specific to biomaterials and variable application of the INHAND terminology fosters ambiguity in the microscopic criteria used in pathology evaluations for medical device and combination product nonclinical studies. Ultimately, striving for establishment of standardized nomenclature for medical device studies will require flexibility to accommodate novel biomaterials, device innovation, and the inevitable novel microscopic findings.
Except for the term biocompatibility (the ability of a material to perform with an appropriate host response in a specific situation.),1,6 consensus definitions for basic terms that apply to medical devices are not readily available. The choice of preferred terms and their denotations and connotations are frequently based on material sciences, engineering, and country-specific laws rather than a pathobiological foundation of tissue responses. Meetings to define consensus terminology by the biomaterial societies, and inclusion of definitions in ISO standards, US Food and Drug Administration (FDA) documents (webpages and legal definitions), and dictionaries are the current available sources for lexical items (Table 1). Where there is no available preferred terminology or clear definition, the MDISE’s collective experience indicates that any vested parties on an individual development team (e.g., engineers, pathologists, toxicologists, regulators, industry sponsors, and contract research organizations [CROs]) should agree on usage and, as appropriate, include definitions for preferred terms within the pathology and/or study reports.
Reference sources for general and selected specific definitions for medical device–related terms.
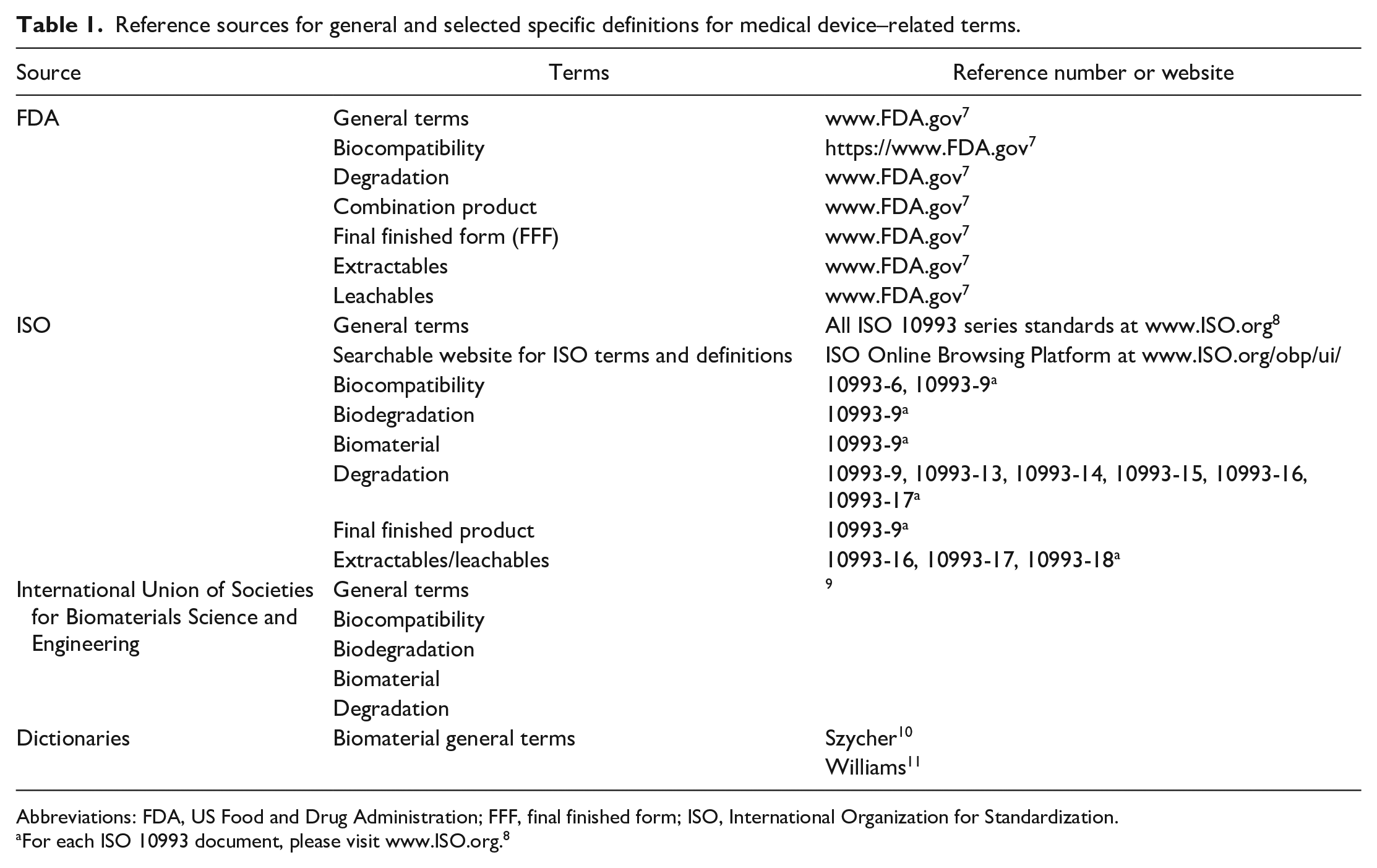
Abbreviations: FDA, US Food and Drug Administration; FFF, final finished form; ISO, International Organization for Standardization.
For each ISO 10993 document, please visit www.ISO.org. 8
Generally, the currently accepted algorithm for medical device evaluation begins with physical/chemical information (such as material composition and extractables and/or leachables) and often will include assays to assess cytotoxicity, sensitization, irritation, hemocompatibility, and pyrogenicity (bacterial and/or material-mediated endotoxins). 6 Dependent on duration and nature of contact, some assessment of genotoxicity, local tissue impacts, and acute through chronic systemic toxicity can be required, although some basic medical devices or materials may be de-risked by physical and chemical properties alone. In addition, considerations for manufacturing/packaging sterilization, particulates and contaminants, and degradation are also included in the risk assessment. When necessary, additional testing for safety pharmacology, reproductive and developmental toxicity, immunotoxicity, and carcinogenicity may be considered. These criteria are broadly addressed in the medical device nonclinical biocompatibility testing standards published by the ISO within the 10993 series. 8 The standards most relevant to toxicologic pathologists will be discussed in more detail below.
The timing of medical device safety studies may differ substantially compared with safety studies for most bio/pharmaceutical nonclinical programs. Nonclinical safety evaluations of medical devices are routinely conducted prior to any clinical evaluations, whereas pharmaceutically active products are filed as Investigational New Drug (IND) products that normally use a staged approach of nonclinical testing in a series of progressively longer studies which overlap with the early phases of clinical testing. Consequently, there may be many years before the completion of the required nonclinical testing for pharmaceuticals and biological products. Combination products also may encounter a staged approach whereby a certain amount of nonclinical device testing (in conjunction with testing for the drug or biologic) allows for clinical trials early in development. Another fundamental difference is that nonclinical efficacy studies are generally required for the final clinically packaged, and fully functioning form of a medical device (e.g., use of the FFF such as a wound dressing in a wounded animal model) prior to any initiation of clinical trials, whereas typically, safety is the primary focus for nonclinical assessment of small molecules and biologics. The relevance of identifying dose-dependent effects in device studies is highly study-contingent depending on the device material or presence of pharmaceutical activity. The FFF is frequently considered to be the maximum implantable device (i.e., the device is often not scalable to change the “dose” of the device, without affecting efficacy and/or function). For many functioning medical devices (e.g., pacemaker), a typical dose response is irrelevant, while in certain circumstances, a dose response may be relevant (e.g., implanted biomaterials where various volumes, weights, or numbers of implanted biomaterials may be evaluated).
Typical animal models utilized in safety studies for small molecules and biologics include rodents (mice and rats), beagle dogs, minipigs, and non-human primates (e.g., cynomolgus macaque or rhesus macaques).12,13 In addition to use of these species, large FFF devices that require efficacy testing in anatomically site-appropriate animal models may require more suitable test animals such as small ruminants (usually sheep), large hounds rather than beagle dogs, mini and full-size pigs, and other less common laboratory animal species. Compared with testing of small molecules and biologics, this wider range of test species and strains has greater genetic and environmental variability compared with conventional toxicology species. This increases the complexity of histopathologic evaluation and interpretation due to the decreased availability of standardized terminology, reduced documentation of normal variability (i.e., incidental background findings), reduced numbers of animals on study, and increased breadth of spontaneous and induced pathology that might be encountered in unusual species that are seldom used to test small molecules and biologics.
The primary objective of the pathology evaluation in nonclinical safety studies for pharmaceuticals/biologics is most often to identify and characterize the systemic dose-dependent morphological responses of both the intended target organ(s) and any off-target organ(s). In these cases, an appropriate assessment may result in a morphological diagnosis of “not remarkable” for many organs and tissues in such studies. Very detailed evaluations of the local impact (the site of primary administration or absorption) are often less critical for small molecules and biologics, unless it is relevant to the route of administration and/or mechanism of action. However, the local implantation site in most device implant studies, including the control groups implanted with reference materials, will merit characterization of microscopic findings using detailed histopathologic descriptions and potentially, selective photographic documentation. Possible systemic effects of implanted medical devices are evaluated, if warranted, by risk level, duration of contact, and site of implantation (such as devices with a higher degree of risk or longer duration of implantation). 6 In addition, a balance of local and/or systemic assessment may be more critical for combination drug-device products, as longer term systemic effects of a medical device may be de-risked by methods of extraction (methods which provide potential materials/chemicals from a device under more extreme conditions which do not degrade the device) and leachate (methods which provide potential materials/chemicals from a device by replicating normal physiologic conditions of use) profile analyses, which may replace longer in vivo studies. Virtually all implantable medical devices or implanted combination products result in local tissue changes related to alteration of regional topography due to the physical disruption imparted by the implant, the procedure required for the implant, and any response related to material composition. The cellular/tissue response to an implant reflects alteration of the local biological milieu; this pathophysiological response may vary based on the composition, location, intended purpose of the device. Observed changes do not always indicate that the tissue responses are abnormal or even undesirable. Some medical device–related tissue responses can be actively utilized to support and extend desirable tissue remodeling outcomes (e.g., osteointegration ensuring long-term stability of a bone implant). Just as the flexibility should exist for terminology and grading of systemic morphological findings in drug and biological studies, basic suggested descriptors limited to specific cell types and fixed numerical grades (as noted in ISO 10993-6:2016, Annex E) 4 are often not suited to the robust descriptions needed to fully characterize the variety of local tissue responses in medical device implant studies.
Considerations for Medical Device Implant Site Evaluation
Successful evaluation of medical device implant sites requires appropriate planning and standard operating procedures (SOPs). The following points to consider have been devised to make the generation, analysis, interpretation, and communication of accurate and useful pathology data a straightforward endeavor for toxicologic pathologists who participate in safety assessment for implantable medical devices or drug-device products. The points for medical devices are typically also relevant for drug-device combination products; thus, the references below are typical to “medical device.” Where applicable, variations are noted.
1. General Approaches for Implant Study Design
There is no one-size-fits-all study design for all implant studies. Implant study design must be tailored within the study scope to answer specific questions regarding pathology, safety (including biocompatibility), and sometimes functional performance (efficacy).14,15
Except for the most straightforward medical devices, implantation studies require a significant investment of time to ensure that the tissue trimming and sectioning approaches, methodology, and ancillary supporting imaging (such as scanning electron microscopy [SEM] or quantitative microCT) are appropriate for the study protocol.
For the majority of implantable devices, pathology evaluation is a major part of the risk assessment. Possible adverse cellular/tissue responses at the implant site are based on implant type, location, duration, and so on, as this ensures that the examination (gross, microscopic, ± ancillary imaging) is structured to capture all possible serious findings in advance of the final risk assessment.
The study design of a combination product in particular, may increase the need for participation of the pathologist in the study design to minimize duplication of in vivo studies and appropriately de-risk both the drug/biologic and medical device aspects. Often for complex combination products, involvement with regulatory personnel is needed prior to designing and finalizing an in vivo study design to ensure appropriate characterization of endpoints relevant to both the drug/biologic aspect and the medical device aspect.
The study pathologist should be involved in the implant study design prior to finalization of the protocol. A pathologist, preferably one experienced with medical device studies, should provide insight and advice on likely biological responses, appropriate control material(s), sample endpoints and timepoints, tissue sampling methods, histology preparations, advanced tissue techniques, and appropriate qualitative and semi-quantitative evaluations.14,16
Small pilot studies for medical device implant studies not performed in compliance with Good Laboratory Practice (GLP) guidance are strongly recommended as a sound investment ahead of any definitive or large-scale GLP study. 5 These investigative studies can confirm the implantation technique, determine whether specialized pathology approaches might provide meaningful macroscopic and microscopic data regarding local and systemic effects, confirm the appropriateness and adequacy of control groups and reference biomaterials, and can help identify implanted biomaterial effects from confounding surgical or insertional procedures. The FDA has a stated preference that definitive medical device animal studies be conducted in a GLP-compliant manner if intended for regulatory submission, aligning with the same requirements for pharmacologic studies. 7
2. Guidance Regarding Study Pathologist Qualifications
The study pathologist should possess appropriate training in anatomic pathology (gross and microscopic) and have advanced training and/or experience in the evaluation of medical devices.
While the initial necropsy and evaluation of gross lesions and photographic documentation of study findings may involve or be conducted by trained prosectors, having a pathologist experienced with medical device evaluation available on-site during the necropsy to review and adjudicate macroscopic findings may be helpful.
The study pathologist should familiarize themselves with the content of applicable guidance documents and standards.3,5 This potentially includes the entire ISO 10993 standard series, but particularly ISO 10993-6:2016 (for local toxicity and tolerability), 4 ISO 10993-11:2017 (for systemic toxicity) 17 (discussed in Alves, 2019), 18 ISO 10993-1:2018 (for developing the comprehensive risk assessment approach), 6 and FDA usage guidance for ISO 10993-1. 7 In addition, relevant (vertical) ISO standards other than series 10993 and other related (lateral) international regulatory documents, when relevant, may be consulted for evaluations of cardiovascular, eye, and dental implants (reviewed by Schuh and Funk 19 and Drevon-Gaillot 20 ). Unlike free government-issued guidance documents, the ISO, ASTM International (www.ASTM.org), and United States Pharmacopeia (USP; www.USP.org) standards documents and reference materials that apply to medical devices and implantation studies must be purchased.
3. Organization of the Sponsor and Implant Study Team
Ideally, the study pathologist should be part of the implant study team from testable prototype and development of surgical models through to regulatory submission, especially with regard to complex and more integrated medical devices and combination products.5,16,19 It may be valuable to have the study pathologist attend the initial implantation surgery to assess the surgical approach for complex or novel medical devices or combination products. In addition, the presence of the implanting surgeon at the necropsy could facilitate exchange of critical information regarding medical device orientation, changes attributable to the surgery itself, and findings associated with the medical device that will assist in the contextual histopathologic evaluation of the implant site.
The device development team should provide the study pathologist with as much relevant information as possible prior to the pathology evaluation. This may include (but is not limited to) the clinical indication, device diagrams detailing the engineering/physical features, the physiochemical characteristics and manufacturing processes, rate of degradation of the biomaterials and FFF, a summary of results from assays for leachables/extractables, results of other in vivo and in vitro biocompatibility assays, and data from proof-of-concept studies. See 3.f. for blinded or masked evaluations.
If known, the sponsor should also provide the study pathologist with information regarding the characteristics of the biomaterial in tissue sections and known biological responses to this or similar biomaterials as well as any predicate (cleared/approved marketed devices used as a comparator) or established medical devices within the same risk class and clinical indication.
The study pathologist should also be provided with known characteristics of the control materials. The physiochemical characteristics of the control material(s) are imperative to consider as the interpretation of study findings typically is based on differences between control and test article group(s).
The control group for many implantation studies uses a “reference material control” rather than a biologically relevant “negative control” (despite the usage of the term negative control within ISO 10993-6:2016). 4 Reference control materials frequently lack the physiochemical characteristics and degradation profile of the test article. The inflammatory and tissue responses to reference materials are known and previously characterized, but these biological responses may or may not be similar to tissue effects induced by the test device. These reference control materials are best thought of as a partial control for the study procedure, and if applicable, shape and volume controls. True biologically relevant negative controls (vehicle/base materials control) may be necessary with combination drug-device products (especially where a systemic as well as a local response may be anticipated) but are seldom possible with medical devices. Therefore, in addition to a reference material control group, consideration should be given to including one or more additional control groups such as implantation of predicate medical devices or inert base material control groups, sham surgical, saline, and/or an untreated group in the study designs containing a reference material control.
In general, pathology evaluations of an implanted medical device should be performed with knowledge of all information including the treatment (i.e., a “non-blinded” approach). Sponsors concerned that the study pathologist will not be able to provide an unbiased evaluation should discuss their concerns and the appropriateness of study masking (“blinding”) with the pathologist prior to finalizing the protocol. It is a common but erroneous notion that the pathologist must be blinded when performing a histopathologic evaluation to maximize the accuracy and sensitivity of the analysis. 21 Because information that can be gained through histopathologic analysis is context-driven, the diagnostic power of the pathology evaluation generally is improved when the study pathologist can compare findings between specimens of known provenance (i.e., treated versus concurrent control groups). In some cases, a blinded evaluation may not be possible (e.g., when test and control devices differ in color, shape, and/or size).
The use of pathology peer review is a well-established best practice across medicine. 22 Peer review is not a requirement per medical device standards and/or guidance documents, but, for GLP regulatory submissions, peer review is strongly encouraged as it is useful in confirming the accuracy, sensitivity, and consistency of pathology diagnoses and interpretations.
4. Considerations for the Surgeon/Interventionalist
The surgeon or interventionalist should inform the study pathologist about the general implantation procedure, and the pathologist should observe the implantation if not familiar with the medical device, implantation location, or implantation procedures.14,19
The surgeon should indicate to the study pathologist whether secondary surgical medical devices (sutures, staples, clips, and/or glue) will remain in or near the medical device implantation site.
The surgeon and study pathologist should discuss the need for or use of tissue markers (tattoo dye, sutures, and/or other labels) to delineate the original implantation site (e.g., to assist sampling of degraded biomaterials) or to permanently mark the spatial orientation of the medical device.
5. Necropsy Procedures for Nonclinical Implant Studies
The timing of sample collection, sampling procedures, and documentation requirements need to be carefully planned in advance according to the unique needs of the implant type and site of implantation.14,23
Imaging studies such as computed tomography (CT) or angiography used during the in vivo phase of a study may also be employed just prior to the necropsy to provide supplemental data to guide the conduct and interpretation of the gross evaluation.
For GLP studies where a comprehensive necropsy is required in the protocol, the necropsy report should document the implant site and all gross findings (designated as trackable gross lesions [TGL]). The TGL should be processed for microscopic evaluation even if they do not occur in proximity to the implant site.
The pathology data should contain a macroscopic-microscopic correlation for all TGL if a correlation can be made. Use of descriptive, not diagnostic, gross terminology for TGL documentation at necropsy is recommended to prevent confusion during time of macroscopic-microscopic correlation. As an example, an area of red discoloration at necropsy should be described as “Focus, red.” This finding should not be called “hemorrhage” during macroscopic description at necropsy, as macroscopic changes can look similar but have different microscopic diagnoses. For example, based on microscopic evaluation of tissue, “focus, red” could be correlated to the active process of “hemorrhage” with extravasation of erythrocytes, or passive intravascular blood accumulation as “congestion.”
The entire implant site and a wide margin around the tissue-device interface should generally be collected. For large or deep implants, the specimen may need to be carefully sectioned prior to fixation to allow penetration of the fixative into the deep tissues.
Depending on the study objectives and the pathology endpoints, the tissue-device interface should be stabilized and preserved as much as possible for histology processing. If the device needs to be explanted in the fresh state at necropsy for engineering or other evaluations, this manipulation will compromise the histopathologic evaluation,15,23 and if this occurs, it is generally noted in the pathology report.
Formalin (neutral buffered 10% formalin [NBF]) is commonly used for immersion fixation and is suitable for the majority of implantation sites. The ratio of NBF fixative volume to tissue volume should normally be at least 10:1. However, for large pieces of tissue or tissues containing medical devices, a ratio approaching 20:1 is often more appropriate. If warranted to ensure rapid exposure of deep tissues to fixative, perfusion fixation should be considered (with NBF or other aldehyde-based fixatives). 23 Other fixatives may be needed if the study protocol also requires immunohistochemistry (IHC) or transmission electron microscopy (TEM) to answer specific questions related to the biological response.23,24
Collection and gross examination of regional (“draining”) lymph nodes is a routine endpoint for implantation studies per ISO 10993-6:2016. The draining lymph nodes should be processed for histopathologic evaluation if required in the study protocol, if TGL are identified, or at the discretion of the study pathologist. 25 The most appropriate and most proximal draining lymph nodes to the implant may not be the typical lymph nodes collected for other GLP studies, and collection of lymph nodes outside the drainage region and/or contralateral lymph nodes may be warranted as control lymph nodes. Particularly for superficial implants (cutaneous, subcutaneous, and intramuscular), special sense organs, and mucosal tissues, species-specific lymphatic drainage patterns should be thoroughly investigated prior to finalizing the study protocol so that the most proximal draining lymph nodes or mucosa-associated lymphoid tissue will be examined.23-28 Collection and examination of distant superficial or internal lymph nodes or other lymphoid tissues (spleen, thymus, or bone marrow) may also be relevant as controls or to evaluate systemic distribution of implanted materials. 29
Macroscopic photographic documentation can be considered useful in implant site evaluation per ISO 10993-6:2016 and is often requested by regulatory agencies. When warranted, macroscopic photographs should be undertaken in a consistent manner after due consideration to document findings relevant to the study scope and to provide visual context when necessary to supplement, but not to supplant, complex gross descriptions.
If included in pathology reports, macroscopic photographs should be high-resolution (at least 300 DPI) color images annotated with appropriate identifying information; examples of relevant metadata include the date, study number, animal number, group (test article or control), site (tissue and any regional sub-location), and measurement scale. Ideally, images should be captured consistently for all animals using the same locations, angles, and magnification(s), and then presented with no or minimal digital post-processing or manipulation. 28 Macroscopic photographs may be a specific study endpoint for certain medical devices or models, but the extent of macroscopic photographic documentation needed for implant studies is highly study-dependent. Macroscopic photographs are considered raw data in GLP studies if used for diagnosis and/or morphometric purposes, and, thus would need to be archived in this instance. 28
However, photography is not required for all implant study necropsies. Macroscopic photographs will not add value to the pathology evaluation in the following scenarios: if the implantation site is not visible at necropsy (although the pathologist should be consulted to verify its absence), when the device is constructed of well-characterized material(s), when tissue response findings are adequately predicted by predicate devices, or when devices are applied/implanted in such a manner that the expected tissue responses are already well understood and can be accurately captured by descriptive macroscopic terminology.
Macroscopic photographs should generally be taken in a series so that findings can be evaluated by moving from a large field of view (low magnification) to a smaller field of view (if macroscopic images are relevant to the implant).
In addition to necropsy macroscopic photography, macroscopic photographs may also be taken during subsequent post-necropsy tissue review of fixed specimens (i.e., at the time of trimming) if additional gross observations are recognized that may be correlated to findings likely to be detected during the microscopic evaluation. Macroscopic photographic documentation of findings at tissue trimming may be necessary for some implant studies (e.g., where the implant site is removed and fixed while intact, or where fixation has stabilized an implant site). A potential opportunity to correlate macroscopic pathology with clinical observations and microscopic pathology may be missed if the gross observations on the day of necropsy are the only macroscopic pathology data collected. This additional round of macroscopic photography should be permitted but not required in the study protocol and should be performed at the discretion of the study pathologist.
The need to take macroscopic photographs of all, representative, or selected sites is dependent on many factors. Major considerations include the type of biomaterial or medical device, the tissue location, prior experience or knowledge with the tissue responses to the incorporated biomaterials or predicate devices, the uniqueness of the surgical procedure or model, and the presence of any unusual findings identified by the study pathologist. The pathologist should determine the macroscopic photographic approach (i.e., how many images are needed and from what perspectives to appropriately document the tissue responses within a study) by either observing the surgical implantation procedures or evaluating macroscopic photographs taken during surgery. 16
6. Trimming and Histology Processing Procedures for Nonclinical Implant Studies
GLP studies with implanted biomaterials and complex medical devices may benefit from proactively indicating in the study protocol that macroscopic observations to include macroscopic photography may be conducted at tissue trimming in addition to, or instead of, at necropsy. The study pathologist should decide when macroscopic photographs should be taken (i.e., at necropsy and/or at tissue trimming).
Documentation of implant site configuration after explantation/fixation at trimming may be necessary for some devices when changes are produced by fixation. The protein cross-linking of formalin fixatives can cause artificial contraction of muscular tissue and shrinkage of soft tissues that can alter the tissue-implant interface, fixatives may not prevent displacement of tissue compartments (e.g., artifactual detachment of retina), and may produce cellular swelling, vacuoles, fragmentation, and tinctorial changes. 26 ,30-32 Detailed documentation of implant site configuration is particularly helpful for studies in which in-life work was performed at one test facility but gross samples were sent to another test site for processing or when histopathologic evaluation is performed by a pathologist who did not observe the necropsy.
If the medical device or implantation area was not visible at necropsy, photographic documentation may be performed when fixed tissues are trimmed for histology processing. Delaying the detailed examination and macroscopic photography until trimming may also be warranted for delicate samples for which fragile tissues will be stabilized by fixation. 23 Photographs of trimmed tissues in cassettes may also be used to document chain of custody of specimens from macroscopic to microscopic evaluation.
No one-size-fits-all approach exists for optimal tissue trimming of tissues containing implantable medical devices. Instead, tissue sectioning planes and methods to be used for slide production will be determined on a case-by-case basis, determined by the study pathologist in consultation with the histotechnology team, the objectives of the study, any available literature on known biological responses incited by the biomaterials, and conformations of the test device and predicate medical devices.
As for all studies involving microscopic evaluation of tissue, improper collection, inadequate processing, poor microtomy technique, and/or poor slide production are likely to produce artifacts that render tissue uninterpretable.32,33 The selection of tissue sectioning planes in implantation studies which capture complete implant-tissue interface information is a pivotal step in histology processing. Errors in this step can reduce or prohibit complete microscopic tissue evaluation of the implant site. 23
Ideally, device-tissue sectioning planes are established by/with the study pathologist who will perform the microscopic evaluation. If this is not an option (e.g., if the pathologist is at another test site), then detailed information regarding device-tissue sectioning planes should be provided to the study pathologist ahead of the histopathologic analysis.
If the size of the device precludes proper fixation of the intact implantation site, the device-tissue interface should be selectively disturbed (e.g., by making one or several widely spaced channels or cuts into the unfixed tissue at necropsy) rather than completely separating the interface (e.g., by device removal from unfixed tissue at necropsy). Removal of an implanted medical device from tissue will almost always cause loss of information regarding changes at the tissue-device interface. Details of this manipulation ideally should be clearly documented in the study protocol (based on pilot study data), and/or it may be communicated in the pathology report.
Successful preservation of some implantation sites in bones may require removal of soft tissues prior to immersion fixation. If this cannot be accomplished, perfusion fixation should be used when necessary. 20
Where feasible, fixed tissue specimens trimmed after device removal may be embedded routinely in paraffin. This practice is suitable when the loss of interface information is interpreted as minimal (i.e., having negligible or no impact on the accuracy and sensitivity of the pathology evaluation). The relevance of this absent/lost information compared with the speed and efficiency of conventional paraffin embedding should generally be determined in consultation with, or by, the study pathologist. Sometimes, a pilot study will be necessary to determine whether this approach will be an acceptable procedure or previous interpretation of the tissue-device interface may inform the fixation for future studies.
Markers applied at necropsy (e.g., tattoo dye, sutures, or other materials) may be used to define the specific spatial orientation (front or back, up or down, left or right) of the tissue for consistent embedding. Any markers should not disrupt the tissue-device interface or impinge greatly on the implantation site.
Non-standard embedding methods (e.g., hard plastic resin) permit the generation of tissue sections while the device remains in situ (even for metal-based materials). This practice comes with many technical challenges. The pathologist and histotechnologist are the best guides for selecting an appropriate embedding method. Some studies may require a combination of embedding methods (e.g., hard plastic and paraffin embedding for parallel samples) to permit a complete evaluation.
Histology scoring rubrics suggested in existing ISO standards may be applicable for small uncomplicated studies and/or biomaterials, but should not be forced to fit devices with multiple biomaterials and functional components. These standards have limitations that make them less useful for devices with multiple biomaterials and functional components, or anatomic sites not specified in Annex E of ISO 10993-6:2016. 4 This is discussed further in Section 13 and Table 2.
7. Ancillary Imaging/Non-Routine Analyses Relevant to Pathology Evaluation
Implant studies often require additional methods of analysis to guide effective tissue sectioning and corroborate/compliment the gross and/or microscopic findings. Imaging studies often offer a more complete 3-dimensional (3D) representation of the medical device and its implantation site. Imaging studies may be helpful when performed in vivo, peri-mortem, and/or post-mortem. 13
The most common ancillary imaging methods include high-resolution radiography (i.e., Faxitron), CT or microCT, magnetic resonance imaging (MRI) or microMRI, SEM, optical coherence tomography (OCT), fluoroscopy, and angiography.15,23,34-41 Other imaging methods such as 3D electro-anatomical imaging may also be possible. The need to use an ancillary imaging method to support pathology evaluation of an implanted medical device should be determined on a case-by-case basis.
Not all test facilities will have the required equipment and trained personnel available for advanced imaging in vivo or at necropsy. With respect to medical device studies, the absence of imaging capabilities may complicate acquisition of macroscopic pathology data and also may affect histologic processing and efforts to correlate macroscopic observations and microscopic findings. A need for advanced imaging should be decided in advance of selecting the in vivo test site to ensure that the proper equipment and laboratory procedures are in place before the study commences.
For some devices (particularly biodegradable implants), 3D imaging to permit volumetric rendering can add additional information that enhances 2D histopathologic evaluation. This 3D imaging approach is quantitative and reduces or eliminates the need to produce many serial slide sections to capture similar volumetric information from 2D sections.
Histomorphometry of features in 2D sections may be performed if warranted to acquire quantitative data from 2D tissue sections.
8. Generation, Interpretation, and Communication of Histopathologic Data
The approach to the microscopic evaluation must be crafted on a case-by-case basis due to the wide variation in types of devices and study complexities, but certain design elements are common in analytical schemes for implanted devices.
Scoring protocols for medical device implants should be capable of evaluating the entire spectrum of complex cellular (inflammatory) and tissue responses associated with the implant as well as any associated regional responses (e.g., findings in draining lymph nodes). In general, semi-quantitative (ordinal) scores are preferred to qualitative (nominal) scores. Because of the inherent variability of these studies, pathologists are encouraged to provide a guide within the report narrative outlining the grading criteria used within their report and a complete narrative description integrating all aspects of the biological response, rather than relying on a simple numerical summary of tissue reactivity of test versus reference biomaterials.
The examples of semi-quantitative scoring protocols presented in ISO 10993-6:2016 Annex E are difficult to apply (either as is or with modifications) to all but the simplest implantation studies. Sponsors and regulatory agencies will often request a table based on this standard as well as use of the interpretive summary in the guidance for ranking the tissue response. However, in general, manipulation of numerical ordinal pathology data such as adding ordinal scores from two distinct and/or variably related biological parameters (e.g., “fibrosis” and “inflammation”) to generate a single composite value creates an artificial value without biological relevance and is not considered an accepted practice at most laboratories conducting medical device evaluation. For example, analyzing inflammation microscopically by enumerating individual cell types, then assigning an ordinal score for each, and subsequently combining ordinal scores for a composite value as a comparator to other treated animals or to control group animals often does not convey the phase and nature of local response as accurately as using appropriate histomorphological descriptions with a defined grading scale.
Due to the qualitative and context-based nature of microscopic data generated during a pathology evaluation, semi-quantitative (ordinal) grading criteria should typically be developed or applied for each study by the study pathologist. Building on grading from predicate studies is acceptable, when applicable, but terminology should not be dictated by standards or “made to fit” based on prior studies, published literature, and/or example criteria provided within a guidance or standard. Terminology should be based on the findings that are present within a study (i.e., “fatty infiltration” is not relevant in a subcutaneous implant) and applied to accurately characterize the findings within a study. The grading system from a previous study is often a good starting point, but the utility of prior grading schemes must be confirmed before they are used for a subsequent study.
Standardized grading rubrics as described in ISO 10993-6:2016, 4 may be appropriate for use or re-use for some simple devices and biomaterials if the design and conditions for the two studies were identical, but such equivalence is often not achievable for more complex study designs and intricate devices.
While it is completely appropriate to review a prior study to strive for consistency in diagnostic macroscopic and/or microscopic terminology, each study must be actively evaluated based on the actual microscopic findings present in the tissue sections within a given study.
Trends (i.e., general shifts in the trajectory of a biological change or difference) in microscopic findings between control and test groups should generally be evaluated based on incidence/severity tables for individual parameters, rather than combined parameters as a tissue or animal score.
Identification of microscopic reactions in draining lymph nodes may warrant a more detailed histopathologic evaluation, which generates separate scores for organ sub-compartments including the cortex, medulla, and germinal centers rather than the organ as a whole.42,43
The pathology report for device studies may be adjusted as necessary to effectively communicate key findings of the macroscopic and microscopic pathology evaluations. No one-size-fits-all structure is feasible for such reports due to the wide variation in types of devices, pathology endpoints, and sponsor expectations.
The narrative portion of the implant pathology report should strive for brevity. The narrative should communicate the important findings that occurred during the study and should be limited to descriptions of principal implant-related findings relevant to safety and, if warranted, efficacy assessments. Tabulation of key incidence and severity findings may also be considered within the report. Typically, the most important findings are presented first, followed by findings of lesser importance.
For complex study designs or where the microscopic finding can be better understood via visual depictions, it can be appropriate to include representative photographs (microscopic and often macroscopic) of the implant site and tissue reactions within a pathology and/or study report, with relevant annotations. Illustrative photomicrographs within the report are not raw data and should not be used to re-evaluate or supersede the study pathologist’s diagnoses. 28 The need for exhaustive photographic documentation (e.g., a photomicrograph for each implant site) is uncommon, and should be carefully considered based on such factors as the type of study, the use of images for data generation (e.g., morphometry performed on photomicrographs), the spectrum of findings observed within the study, and the value (in terms of increased data accuracy and sensitivity) to be gained by extensive imaging. Where feasible, exhaustive imaging should be avoided if other methods (e.g., whole-slide scanning, inclusion of representative photographs within the report) are suitable for ensuring that visual data are available. The study pathologist should have discretion to insert as many or as few images as they feel are necessary to communicate the pathology findings. A series of thoughtfully selected representative photomicrographs provided by the study pathologist is generally a more efficient means to provide important context for understanding microscopic findings, rather than an exhaustive series of photomicrographs of all implant sites.
In general, data tables giving scores for all histopathology findings (both those related to the test article and major incidental or procedure-related changes) and additional photographs should be provided within appendices unless all implant site data are necessary for discussion within the report narrative.
When making an observation for absence of findings, recording a finding of “not remarkable” or “no visible lesions” is highly preferable to cataloging numerous “absent” findings which are not relevant (such as lack of fibrosis in an acute study). Thus, when a finding is not recorded in the pathology data, it is assumed to be absent. This distinction is noted due to the cataloging of negative data in commonly utilized scoring rubrics (e.g., ISO 10993-6:2016) 4 . Utilizing the above approach (i.e., a finding not recorded in the pathology data is assumed to be absent) when it may be relevant to comment on the absence of findings in the report (e.g., lack of thrombosis associated with a coronary artery stent) is at the discretion of the study pathologist to bring the absence of the finding forward into the report narrative. Thus, information detailed as absent in the report narrative may be confirmed as absent by noting that it is not within the pathology data set. It is ultimately at the discretion of the study pathologist as to how and when they may diagnose and/or use negative data to communicate relevant findings, but it should not be a routine practice to record all absent findings. (Note: some electronic data entry methods do support entry of negative findings as part of the study database.)
INHAND microscopic terminology should be used where feasible for findings at and away from the implant site. 44 Other appropriate diagnostic and/or descriptive terminology outside the scope of INHAND may be employed for microscopic diagnosis if it is defined in the protocol and/or within the narrative or report glossary. Data sets in SEND (Standard for Exchange of Nonclinical Data) format are currently not required for medical devices to be evaluated by the FDA’s Center for Devices and Radiological Health (CDRH), but SEND-compliant data may be required for a combination (drug + device) product where the primary review by FDA will fall under either the Center for Biologics Evaluation and Research (CBER) or the Center for Drug Evaluation and Research (CDER).
Inferential statistics are not an appropriate means for testing the biological relevance of semi-quantitative (ordinal) microscopic data and should not be used. 45 For very large or unwieldy data sets, the study pathologist has the discretion to use basic descriptive statistics (e.g., mean, standard deviation [SD]) to facilitate identification and communication of major test article–related effects. Mean values must be evaluated in combination with microscopic data tables (using an incidence/severity format when possible) as interpretations based solely on group mean values commonly miss test article–related trends present in the incidence and/or severity tables.
Microscopic findings within a study can be directly compared among groups as the study pathologist will have used the same grading criteria throughout the study. In general, numerical grades should not be compared from one study with the next even if assigned by the same pathologist, although findings and trends can be compared if similar methods were used.
If data interpretations and trends must be assessed across multiple studies, an independent pathologist with experience in implant studies may provide an expert review of pathology reports from multiple finalized studies to help integrate the message across studies (Note: This practice is not the same as a pathology peer review.)
9. Pathology Peer Review of Pathology Reports for Medical Device Studies
Pathology peer review assists in confirming microscopic diagnostic terminology, avoiding “diagnostic drift” and assessment of the overall conclusions of the pathology report. The principals and activities of peer review recommended by the STP, including providing the details of the peer review methods in the study report and generation of a peer review certificate, can also be applied to peer review of medical devices. 22
The pathology peer review approach for medical device studies is selected on a case-by-case basis by the peer review pathologist, although input from the study pathologist and sometimes other study team members (e.g., engineer, surgeon) may be helpful due to the diverse nature of implant studies.
Peer review of ISO 10993-6:2016 4 generated tables generally requires that the peer pathologist review most or all implantation sites for the medical device and for the reference controls. This may involve one or multiple implantation sites in all animals and should be determined on a case-by-case basis by the peer review pathologist and study pathologist.
10. Regulatory Aspects Relevant to Implant Studies
Implant studies present a particular challenge due to the ever-evolving nature of medical devices and combination products as there is no standardized path-to-market. In addition, the designation of a medical device may vary by country and the review of products as combination drug-device products is variable by country as well.
In theory, this lack of regulatory guidance allows the implementation of tailored nonclinical development programs for a given medical device. However, the constant evolution in medical device development, and variations in the lead agency reviewing a product often introduces practical challenges when designing nonclinical safety studies for implants.
The onus for ensuring that individual studies and entire nonclinical programs provide sufficient safety and efficacy data for successful product registration ultimately rests with the sponsors who are developing the medical device. However, an understanding by the pathologist of what regulatory groups will be reviewing studies, and which criteria are likely to be applied and at what stage of development these are needed are particularly critical for complex combination products to successfully assist in design studies. It may require careful consideration to ensure studies are appropriately designed to accomplish this and avoid excessive animal use (thus following the principles of 3R).
All regulatory agencies benefit from the professional guidance and expertise of the device pathologist with their specialized knowledge and practical experience with implant studies, and the pathologist benefits from the broad perspective and deep experience of the regulatory agencies. This collaboration permits tailoring the implant study design to maximize the yield of information on tissue responses and biocompatibility.
11. Utilization of Early Regulatory Feedback on Device Studies
In the United States, medical device sponsors have the option to request feedback from the FDA on possible or planned nonclinical medical device or medical device–led combination product studies through their Q-submission (Q-Sub) program. 46 This interaction is managed by CDRH, CDER, or CBER (as appropriate depending on the primary mechanism of action and/or device type). While Q-Sub is an option for medical device–led combination products prior to opening an IND, there is no similar formal option for drug/biologic-led combination products (products that have a primary mechanism of action through the drug or biologic). Options to interact with regulatory agencies to both classify the primary mechanism of action and to get feedback on drug- or biologic-medical devices are available and should be pursued for complex combination products.
As part of the Q-Sub program, sponsors may submit their draft nonclinical implant study protocols for review ahead of the implant study. While a Q-Sub exchange may not be needed every time or for all medical device programs (particularly if a sponsor had a successful prior review and is making a submission for a very similar medical device), the MDISE strongly encourages sponsors to utilize the Q-sub option for implant studies to improve the likelihood that study designs will provide sufficient information to permit a regulatory review.
This early regulatory input is particularly helpful whenever novel or highly complex implantable medical devices are being developed. Early regulatory input is also very critical in designs for integrated drug or biologic combination products, as integrating the safety of the drug/biologic with the safety of the medical device must occur earlier in the process to support clinical trials as opposed to with a straight medical device.
Whenever possible, the study pathologist should be actively involved during any regulatory process or interaction. Participation by the pathologist is particularly critical for complex, novel, and/or very invasive devices as pathology data will be an essential part of the product registration package, and at least some medical device safety is required to inform and support early clinical trial development for combination products.
12. Post-Marketing Surveillance of Medical Devices
A method for post-market surveillance to identify serious adverse events (SAEs) not detected during clinical development generally is required for medical devices in use in the United States, especially Class II or Class III device failures that might reasonably have adverse health effects, as well as for devices with significant use in pediatric populations. 47 In the European Union (EU), it is incumbent on the manufacturer of the medical device to inform regulatory authorities of any medical device–related concerns identified during the post-market phase. 48
The role of the pathologist in post-market surveillance is dependent on the plan approved by the regulatory agency or system in place in the country in which the medical device is marketed. Experience with the nonclinical analysis and especially microscopic changes that occur at the tissue-device interface often inform the analytic approach that should be taken when investigating post-market device failures.
It may be beneficial to include a pathologist with relevant nonclinical implant study experience in a post-marketing surveillance study. Their knowledge of implant study design and device-related microscopic findings may be beneficial to the clinical trial design and a helpful resource for diagnostic pathologists in medical practice who may not have implant study experience.
13. How to Work with International Standards for Implant Studies
The ISO 10993-6:2016 standard for biomaterials is a common reference for evaluating implantation studies in multiple tissues, and pathologists typically are requested to assess tissue responses to implants using the scoring tables in Annex E of this standard. This standard was designed to apply to medical device materials but may also be applied to drug or biological combination products.
Annex E of the ISO 10993-6:2016 4 standard with the exemplar microscopic/histology scoring needs modification. The semi-quantitative scoring examples in this standard are not applicable to all biomaterials and tissues. For example, the pre-specified, semi-quantitative scoring rubrics of the standard are most applicable to scoring intramuscular implantation of polymers/simple biomaterials (Table E.3) and neural implants (Table E.4) only. Their inclusion in the standard makes these rubrics the most common scoring system requested by sponsors for a “biocompatibility assessment” of the FFF including for large or complex devices, or devices tested under ISO 10993-11:2017. 17 This rigid scoring system is difficult to apply and very often does not sufficiently describe the biological response when evaluating certain devices, or implants that are not in muscle or brain. These archetype scoring systems are especially ill-suited to all but very simple combination products, where both local and systemic impacts may be relevant and widespread and where a simple comparison with reference material may not be feasible.
The histology scoring rubrics in Annex E of ISO 10993-6:2016 4 are poorly suited for use in tissues other than muscle and neural tissue and/or for more complex materials when trying to ensure adequate evaluation of biological relevance (e.g., increased adipocytes/fatty infiltration is not recognized or appropriate with implants placed in many tissues, such as intraocular implants or implantation into subcutaneous fat depots). Accordingly, in designing the histopathology evaluation, the study pathologist should not rely upon prespecified scoring criteria. Instead, the pathologists should identify the relevant cellular change(s) for the implant tissue and biomaterial, establish grading criteria for each abnormal finding, grade them, and communicate the biological/clinical significance of the finding(s) within the narrative of the report.
While responses identified in ISO 10993-6:2016, 4 Annex E, Table E.1 (cellular response, stromal response) may be relevant endpoints, the location and composition of the implant and actual findings within the study should dictate the most relevant grading criteria. In addition, tissue-specific factors (e.g., articular cartilage parameters in a joint device) should be evaluated and documented as appropriate.
It is the experience of some MDISE members that agency reviewers may expect results to be formatted in a table similar to the ones in ISO 10993-6:2016, 4 Annex E, Tables E.1 to E.4, irrespective of the implant location, novelty of the biomaterial, and relevance of the parameters indicated for scoring by Annex E. While this ISO tabular formatting is not a strict requirement, if a study pathologist ensures that they have evaluated all appropriate parameters, including those listed in the ISO exemplar tables, data can be reformatted without having to re-evaluate the slides if it becomes necessary to provide these tables.
14. Navigating Differences between ISO 10993-6:2016 4 and Standard Toxicologic Pathology Practices for Pharmaceuticals and Biologics
The standard pathology modules in common data entry systems are often not suitable for implant study evaluation; thus, it may be necessary to use other systems such as word processing, spreadsheet, and document files for implant study pathology data capture and tabulation. A combination of systems may also be utilized when relevant, that is, using a standard pathology module data entry system for systemic tissue evaluation, and a word processing spreadsheet for implant site findings. As there is a GLP compliance requirement for device studies, awareness of the GLP compliance (or non-compliance) of different data capture systems is important.
Where permitted by the sponsor, the study pathologist should prepare data tables that best communicate the findings and their significance rather than uncritically adopting a less suitable format listed in an insufficiently flexible standard.
Inclusion of an ISO-10993-6:2016 4 Annex E-style table in a pathology report may be requested or required to meet a specific sponsor’s expectations for a device development program or a regulatory submission. However, the Annex E tables are illustrative examples and study data should not be forced into the ISO-10993-6:2016 4 Annex E tabulated categories when not appropriate for the medical device, tissue, or implantation method. Generally, Annex E tables, as published, are only appropriate for very simple devices and biomaterials implanted in muscle and neural tissue.
Recommendations for how to navigate the ISO 10993-6:2016 4 standard when requested for implant studies other than biomaterials/simple implants are summarized in Table 2.
MDISE practical applications, limitations, and advisable modifications of the ISO 10993-6:2016 standard for implant study pathology assessment.

Abbreviations: FFF, Finished Final Form (clinically relevant form of a medical device under development); GLP, Good Laboratory Practice; HDPE, High Density Polyethylene; ICH, International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use; ISO, International Organization for Standardization; MALT, mucosa-associated lymphoid tissue; MDISE, Medical Device Implant Site Evaluation working group of the Society of Toxicologic Pathology; SOP, standard operating procedure.
Conclusions
Complex biological responses are induced by implanted medical devices through a combination of topographical, physiochemical, and sometimes pharmacologic (i.e., drug-containing medical devices) influences. These influences paired with the constantly evolving nature and often highly technical engineering of biomaterials makes implantable medical devices some of the most challenging products that a pathologist may encounter. Historically, methods for macroscopic and microscopic pathology evaluation of devices have emphasized inflexible sampling and analytical approaches. The more flexible principles of macroscopic and microscopic evaluation established by toxicologic pathologists for assessing other biomedical products (e.g., pharmaceuticals and biologics) can be used to greatly improve the scientific quality and clinical applicability of device pathology evaluations in medical implant studies. Attention to these practical points to consider when designing and performing implant studies will improve the consistency and standards for evaluation of implanted medical devices.
Supplemental Material
sj-docx-1-tpx-10.1177_01926233221103202 – Supplemental material for Scientific and Regulatory Policy Committee Points to Consider for Medical Device Implant Site Evaluation in Nonclinical Studies
Supplemental material, sj-docx-1-tpx-10.1177_01926233221103202 for Scientific and Regulatory Policy Committee Points to Consider for Medical Device Implant Site Evaluation in Nonclinical Studies by Maureen T. O’Brien, JoAnn C. L. Schuh, Lyn M. Wancket, Sarah D. Cramer, Kathleen A. Funk, Nicolette D. Jackson, Kamala Kannan, Kevin Keane, Abraham Nyska, Serge D. Rousselle, Adrienne Schucker, Valerie S. Thomas and Stefan Tunev in Toxicologic Pathology
Footnotes
Acknowledgements
We thank individuals who provided key input during the manuscript process (Natalie Keirstead). In addition, the working group acknowledges companies where authors worked during part of the manuscript process (NAMSA, Novo Nordisk), as well as the organizations that became part of StageBio (Alizée, Vet Path Services, Inc., Tox Path Specialists, LLC), Charles River (AccelLAB), and NAMSA (American Preclinical Services).
This manuscript has been reviewed and endorsed by the leadership of the European Society of Toxicologic Pathology, the British Society of Toxicologic Pathology, The Société Française de Pathology Toxicologique (the French Society of Toxicologic Pathology), and the Society of Toxicologic Pathology-India.
Declaration of Conflicting Interests
All author(s) are pathologists with experience in device evaluation studies employed by pharmaceutical or medical device companies conducting nonclinical studies, contract/academic research organizations, or independent consultants providing services for nonclinical implant studies.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.
ORCID iDs
Supplemental Material
Supplemental Material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.