
Abstract
Multiple applications of nanomaterials have raised concern with regard to their toxicity. With increasing research into nanomaterial safety, mechanisms involved in the toxic effects of nanomaterials have begun to emerge. The importance of nanomaterial-induced lysosomal membrane permeabilization through overloading or direct damage of the lysosomal compartment, resulting in the blockade of autophagosome–lysosome fusion and autophagy dysfunction, as well as inflammasome activation were cited as emerging mechanisms of nanomaterial toxicity. It has recently been proposed that these very mechanisms leading to nanomaterial toxicity may be utilized in nanotherapeutics. This review discusses these nanomaterial-induced mechanisms in detail and how it has been exploited in cancer research. This review also addresses certain considerations that need to be kept in mind when using nanomaterials in therapeutics.
Keywords
The involvement of lysosomes and their membrane permeabilization, autophagy dysfunction, and inflammasome activation in the toxicity of nanomaterials are now well established. Nanomaterials can have detrimental effects on the autophagy and lysosomal pathways, resulting in toxicological consequences. Lysosomal dysfunction through lysosomal membrane permeabilization (LMP) and autophagy dysfunction by overloading or directly damaging the lysosomal compartment induced by specific nanomaterials has been used in cancer therapies (W. Li et al. 2015).
LMP, Lysosomal Dysfunction and Autophagy, and Nucleotide-binding Oligomerization Domain–like Receptor Family, Pyrin Domain–containing 3 (NLRP3) Inflammasome Activation Pathways: Mechanisms of Toxicity by Nanomaterials
Cellular Entry Mechanisms and Cellular Fate of Nanomaterials
The importance of the mechanisms of cellular entry of nanocarriers for therapeutic applications was reviewed earlier (Hillaireau and Couvreur 2009) and was further emphasized later by others (Stern, Adiseshaiah, and Crist 2012). The physicochemical properties of nanoparticles identified to affect their uptake through the endocytic pathway included size, surface charge, surface coating, shape, and surface multifunctionalization and their impact varied upon the type of cells (Hillaireau and Couvreur 2009). The mechanisms described in these reviews for cell entry of nanocarriers included phagocytosis and endocytosis with the latter being effected by macropinocytosis, clathrin-mediated endocytosis, and caveolae-mediated endocytosis. During caveolae-mediated endocytosis, the contents are delivered to endosomes to form caveosomes, which in turn avoid lysosomal enzymatic degradation and are transported along the cytoskeleton to the endoplasmic reticulum/Golgi apparatus (Stern, Adiseshaiah, and Crist 2012). The most relevant and important aspect of these endocytic pathways is the realization that, although most of the nanocarriers entering the cell may end up in the lysosomes, caveolae-mediated endocytosis of a nanocarrier is delivered to caveosomes, avoiding a degradative acidic and enzyme-rich environment. Hence, it is emphasized that exploiting caveolae-mediated endocytosis may therefore be advantageous to bypass the lysosomal degradation pathway when nanocarriers are tailored, particularly when the carried drug (e.g., peptides, proteins, nucleic acids) is highly sensitive to enzymes (Hillaireau and Couvreur 2009). Control and interplay between these different entry mechanisms for nanomaterials are critical for their intracellular targeting for cancer therapy (Gary-Bobo et al. 2013).
Lysosomes, LMP, and Nanomaterials
Lysosomes are critical subcellular acidic, membrane-bound organelles that play a crucial role in protein degradation, endocytic receptor recycling, energy metabolism, cell signaling, and cellular homeostasis (Blott and Griffiths 2002; Futerman and van Meer 2004; Appelqvist et al. 2013). Lysosomes are intracytoplasmic organelles derived from Golgi elements that form a single-membranous vesicle, of multiple possible sources, the center of which has an acidic milieu (pH around 4.5–5.0; Puri et al. 2013; Piao and Amaravadi 2016). Lysosomes contain about 40 types of hydrolytic enzymes including proteases, nucleases, glycosidases, lipases, phospholipases, phosphatases, and sulfatases that usually exert their maximal enzymatic activity at low pH (Luzio et al. 2014). These hydrolytic enzymes are used for the controlled intracellular degradation of macromolecules delivered through endocytic pathways with fusion of endosomes or phagosomes to form endolysosomes or phagolysosomes, respectively, or through the autophagic pathway with fusion of autophagosomes to form autophagolysosomes (Piao and Amaravadi 2016; Repnik, Cesen, and Turk 2013). The lysosomal membrane is protected from the acidic hydrolases by lysosome-specific expression of membrane proteins such as lysosomal-associated membrane protein-1 and -2 (LAMP-1 and LAMP-2) which are heavily glycosylated and hence resist digestion (Eskelinen 2006). However, in response to some lethal stimuli including reactive oxygen species (ROS; Blomgran, Zheng, and Stendahl 2007) and lysosomotropic detergents (Ostenfeld et al. 2008), LMP occurs (Johansson et al. 2010; Oberle et al. 2010) with larger lysosomes being more susceptible to such permeabilization (Ono, Kim, and Han 2003). LMP may then lead to apoptosis or necrosis, where a complete breakdown of the organelle may cause the release of high concentrations of lysosomal proteases into the cytosol to produce necrotic cell death, while partial, selective permeabilization may lead to apoptotic cell death (Boya and Kroemer 2008; Serrano-Puebla and Boya 2016).
Cathepsin hydrolases are subdivided into 3 subgroups on the basis of the active site of amino acids, which confers the catalytic activity: cysteine cathepsins, serine cathepsins, and aspartic cathepsins (Repnik et al. 2012; Piao and Amaravadi 2016). Lysosomal cathepsins such as aspartic protease cathepsin D, and cysteine cathepsins including B, H, and L, are known as lysosomal proteases, which are responsible for the degradation of intracellular and endocytosed proteins and thus maintain cellular homeostasis and differentiation (B. Turk and Turk 2009). Acidification is crucial in cathepsin activation and the efficient degradation of their substrates in lysosomes. Lysosomal hydrolases are also referred to as acid hydrolases because they have optimal activity at the acidic pH 4 to 5 in the lysosome. But, in some cases, they can also function at a neutral pH outside of the lysosomes to initiate cell death following LMP (Kirkegaard and Jaattela 2009). Lysosomal cathepsins have also been implicated in cell death, which in turn may activate apoptotic effectors including mitochondrial cytosolic cytochrome c and/or caspases (Kagedal, Johansson, and Ollinger 2001; Repnik et al. 2012; V. Turk et al. 2012; Bidere et al. 2003; Zhao et al. 2003; Michallet et al. 2004) through mitochondrial outer membrane permeabilization, decrease in mitochondrial membrane potential, increase in ROS level, and subsequent apoptosome-dependent caspase activation (Gillies and Kuwana 2014). The contribution of lysosomal damage and LMP to the pathogenic features of several disease states (other than cancer), for example, lysosomal storage disorders, neurodegenerative diseases (e.g., Alzheimer’s and Parkinson’s diseases), and infectious diseases, has also been discussed recently (Serrano-Puebla and Boya 2016). In addition, the role that lysosomes and LMP play in the final steps of homeostatic catabolic processes and cell death and their role as upstream modulators of autophagy and NLRP3 inflammasome activation have also been presented in the literature (Serrano-Puebla and Boya 2016).
It seems now that nanomaterial-induced LMP is an important mechanism in their toxicity (Stern, Adiseshaiah, and Crist 2012; Figure 1), since numerous toxic nanomaterials have been shown to produce LMP including TiO2 (Hamilton et al. 2009; Kenzaoui et al. 2012), zinc oxide (Cho et al. 2011), iron oxide (Kenzaoui et al. 2012), and gold nanoparticles (Bhattacharyya, Mehta, and Viator 2012) as well as gold nanorods (W. Zhang et al. 2013), multi-walled carbon nanotubes (MWCNTs; W. Zhu et al. 2016), and graphite carbon nanofibers (Mittal et al. 2017).
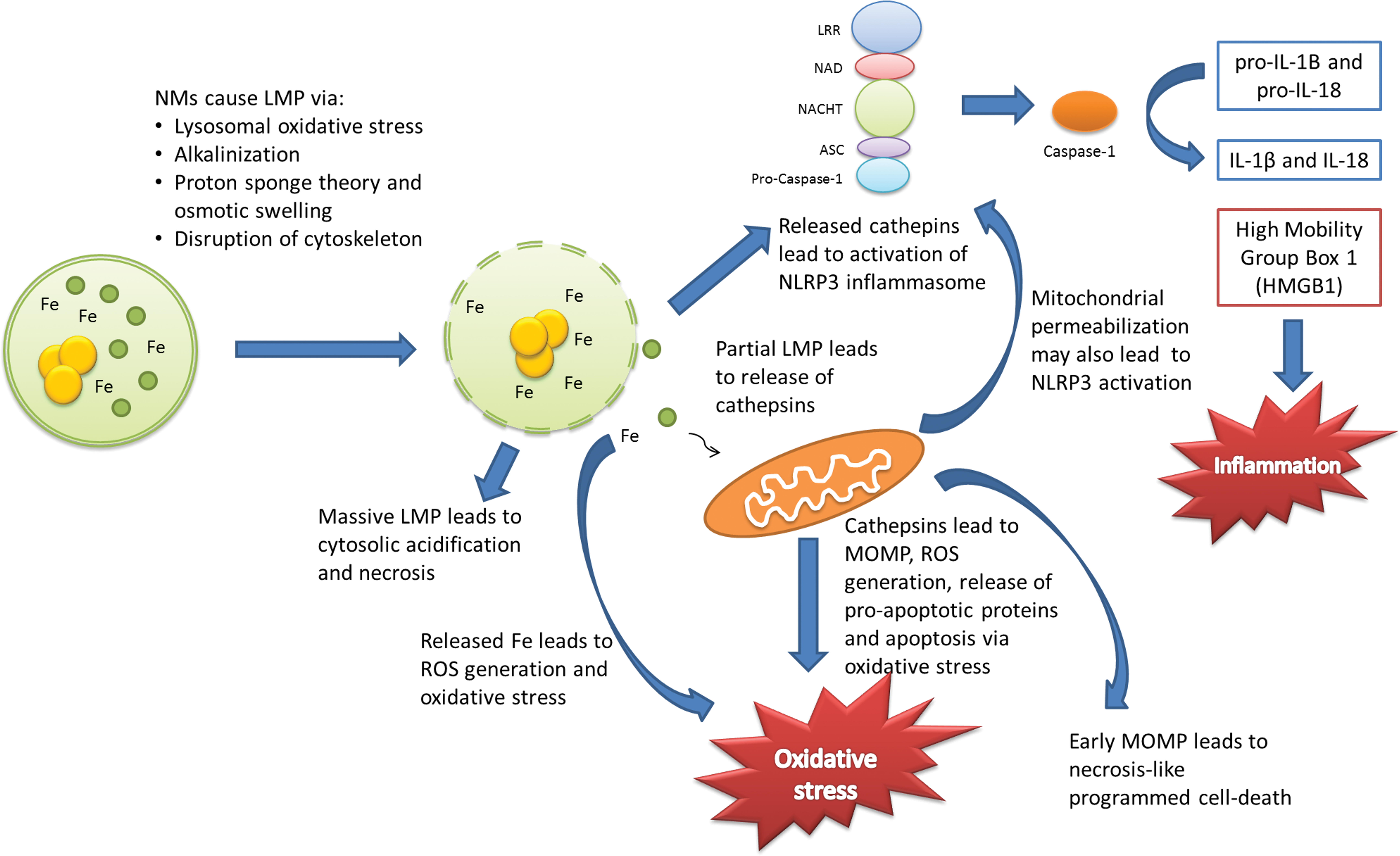
Nanomaterial-induced lysosomal membrane permeabilization and subsequent mechanisms leading to their toxicity.
Autophagy and Nanomaterials
Autophagy is a process that involves the formation of double-membrane vesicles that capture cellular material in autophagosomes, which then fuse with lysosomes for degradation. The process consists of several different mechanisms including microautophagy, macroautophagy, and chaperone-mediated autophagy, all of which are involved in the delivery of autophagosomes to lysosomes (Mizushima 2007). Factors such as stress and starvation trigger autophagy where accelerated turnover of cellular components sustains energy homeostasis (Kroemer, Marino, and Levine 2010). The process therefore confers stress tolerance, limits damage, and sustains viability under adverse conditions such as hypoxia and poor nutrition (White and DiPaola 2009; Mizushima 2007). Thus, autophagy is a cell biological process that is a central component of the integrated stress response (Kroemer and Jaattela 2005) and as such plays a crucial cytoprotective homeostatic role against diverse pathologies (Levine and Kroemer 2008).
The prosurvival function of autophagy in some circumstances of stress may paradoxically lead to apoptotic cell death (Gonzalez-Polo et al. 2005), when autophagy is inhibited either pharmacologically or by depletion of the essential autophagy-related gene products responsible for this phenomenon (Zavodszky, Vicinanza, and Rubinsztein 2013; Boya et al. 2005). It has thus been argued in the literature whether autophagy represents an independent mode of programmed cell death (Maiuri et al. 2007). The presence of extensive autophagy commonly observed in dying cells has led to its classification as an alternative form of programmed cell death (Debnath, Baehrecke, and Kroemer 2005), while others argued that accumulation of autophagic vacuoles can precede apoptotic cell death, thus arguing against the clear-cut distinction between type 1 (apoptotic) and type 2 (autophagic) cell death (Gonzalez-Polo et al. 2005). Yet, others have argued that autophagy may be cytoprotective, at least under conditions of nutrient depletion, and point to an important cross talk between type 1 and type 2 cell death pathways (Boya et al. 2005).
A variety of nanomaterials have also been shown to induce dysfunction of the autophagy pathway (Zabirnyk, Yezhelyev, and Seleverstov 2007; Stern et al. 2008; L. Wang et al. 2010) either through autophagy induction or blockade of autophagy flux (Figure 2A and B), both of which lead to autophagosome accumulation (Johnson-Lyles et al. 2010; Ma et al. 2011). It has also been shown that aggregated nanoparticles could induce significant autophagic effects compared to well-dispersed nanoparticles (D. Huang, Zhou, and Gao 2015).
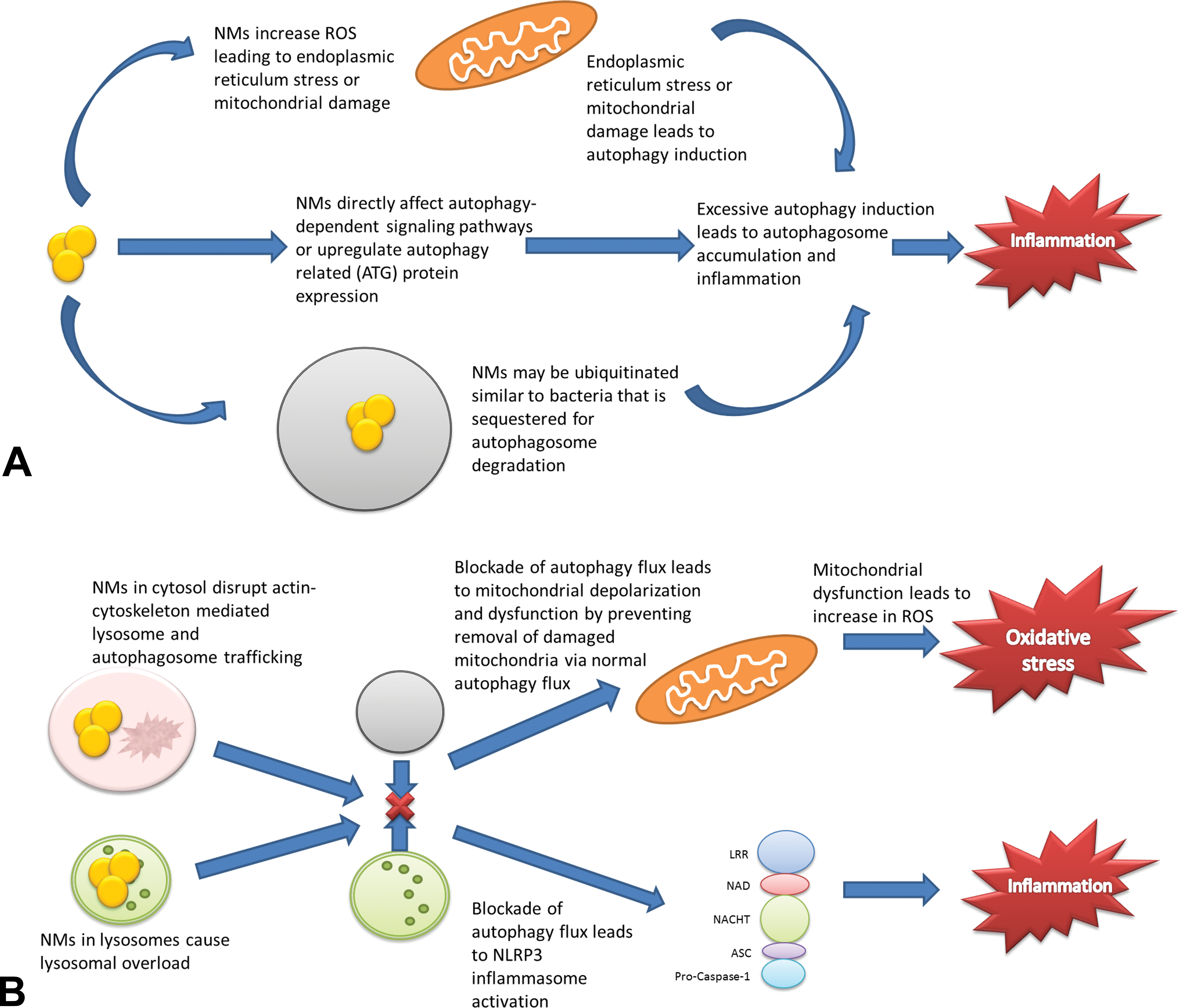
Mechanisms involved in autophagy induction (A) and autophagy blockade (B) by nanomaterials.
NLRP3 Inflammasome Activation and Nanomaterials
The NLRP3 inflammasome is a protein scaffolding complex consisting of NLRP3, caspase 1, and the adaptor molecule ASC (apoptosis-associated speck-like protein containing a caspase recruitment domain; Pycard). The processing of interleukin-1B (IL-1B) from biologically inactive precursor to the active form requires cleavage by caspase 1, which is recruited to the NLRP3 inflammasome complex by binding ASC (Martinon, Burns, and Tschopp 2002; Agostini et al. 2004). NLRP3 inflammasome activation also facilitates the release of other inflammatory cytokines thereby producing inflammatory response (Martinon, Burns, and Tschopp 2002; Jo et al. 2016). NLRP3 inflammasome activation has also been reported to produce actions beyond inflammation, which may culminate in pyroptosis, a form of cell death that combines the characteristics of apoptotic and necrotic death pathways (Kepp et al. 2010; Fernandes-Alnemri et al. 2007).
It was demonstrated that LMP and the release of lysosomal proteases precede and facilitate NLRP3 inflammasome activation (Biswas, Hamilton, and Holian 2014). Most recently, it has been shown that signaling for NLRP3 inflammasome activation is initiated by low-level lysosome disruption but inhibited by extensive lysosome disruption (Katsnelson et al. 2016).
Different types of nanomaterials have also been shown to induce the activation of NLRP3 inflammasomes including carbon nanotubes (CNTs; M. Yang et al. 2013), silica (Peeters et al. 2013; Hornung et al. 2008; Yazdi et al. 2010; Baron et al. 2015), TiO2 (Yazdi et al. 2010; Baron et al. 2015), and silver nanoparticles (E. J. Yang et al. 2012; Simard et al. 2015). A summary of the 3 mechanisms described above and shown to be involved in the toxicity of nanomaterials is illustrated in Figure 3.
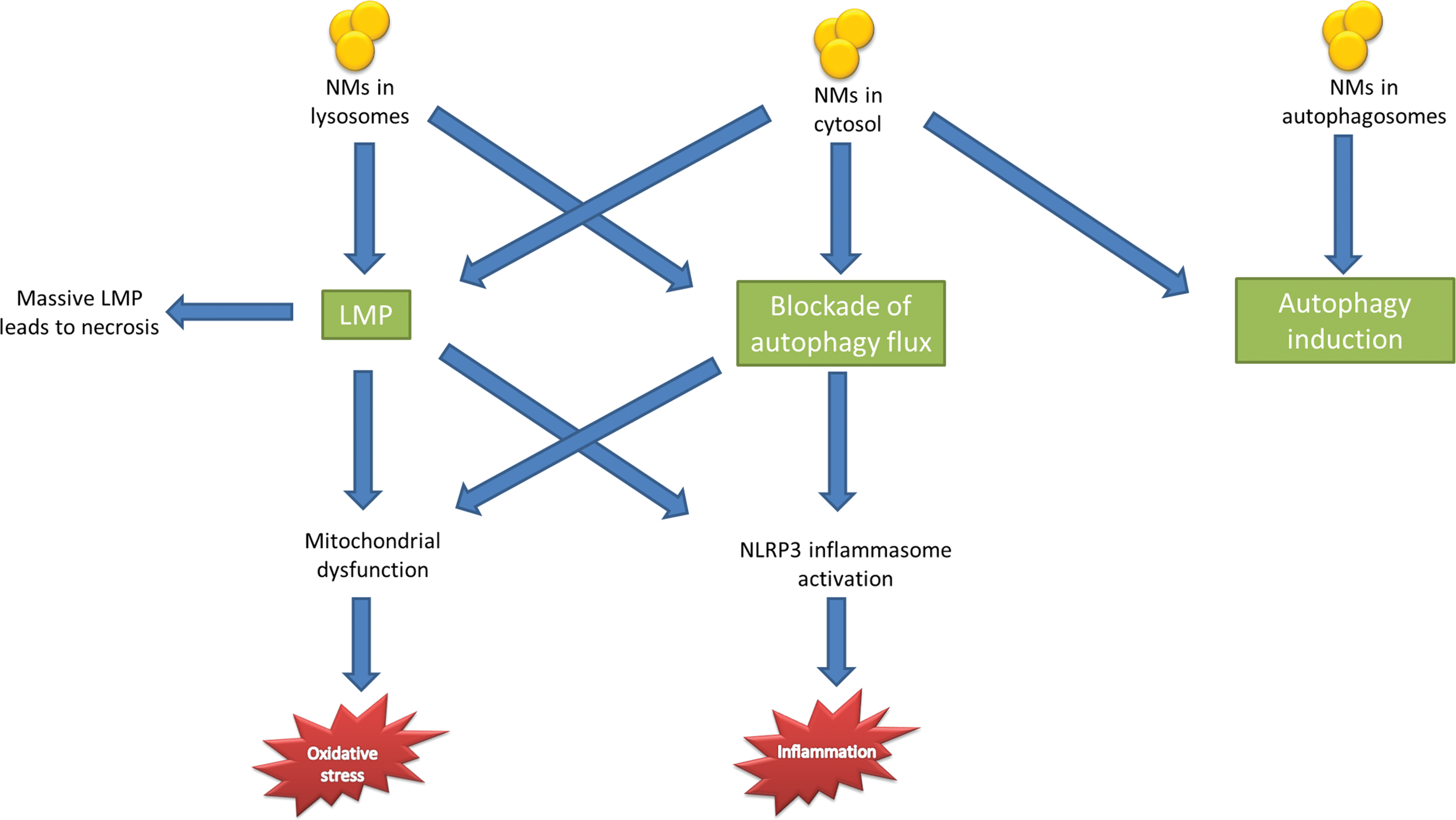
Summary of mechanisms involved in the toxicity of nanomaterials.
LMP, Autophagy, and NLRP3 Inflammasome Activation in the Initiation or Prevention of Cancer
Lysosomal death pathways, autophagy, and NLRP3 inflammasome activation (Lamkanfi 2011) may act as mediators for different diseases (Gattorno and Martini 2013) including autoinflammatory diseases (Wilson and Cassel 2010), neurodegenerative diseases (H. Guo, Callaway, and Ting 2015), lung (Sayan and Mossman 2016) and liver fibrosis (Mridha et al. 2017), and cancer (Kolb et al. 2014).
Lysosomal Cathepsins and Cancer
Lysosomal hydrolases participate in the digestion of endocytosed and autophagocytosed material inside the lysosomal/autolysosomal compartment (Kroemer and Jaattela 2005). However, the involvement of cathepsins in the field of oncology has become increasingly apparent (Kirkegaard and Jaattela 2009). Interestingly, depending on the location of cathepsins, they can either suppress or promote tumor growth, where the former is achieved when they are located in the cellular cytosol while the latter is achieved when they are located extracellularly (Repnik et al. 2012). In acute cell death, they are released into the cytosol and they contribute to cancer progression, following their release into the extracellular space (Kroemer and Jaattela 2005) where, for example, cathepsins B, S, and E are all shown to be associated with cancer progression and metastasis in various cancer types (Withana et al. 2012; Small et al. 2013; Keliher et al. 2013). It has also become apparent that, in addition to these tumor-promoting effects of cathepsins, they may have tumor suppressor functions through induction of apoptotic cell death (Lopez-Otin and Matrisian 2007). Thus, therapeutically induced LMP in cancer cells and subsequent release of cathepsins in the cytosol may represent a strategy in cancer therapy (Erdal et al. 2005; Fehrenbacher and Jaattela 2005; Piao and Amaravadi 2016; Boya 2012; Maynadier et al. 2013; Kos, Mitrovic, and Mirkovic 2014).
Autophagy and the Promotion of Cancer
Paradoxical roles of autophagy are also illustrated in normal and cancerous cells in which it plays a key role in the maintenance of cellular homeostasis and therefore acts as a robust barrier against malignant transformation, while defective autophagy—excessive autophagy induction or blockade of autophagy flux—predisposes healthy cells to undergoing malignant transformation. Yet, it plays a prosurvival role for cancerous cells to grow with intracellular and environmental stress, thus favoring tumor progression (Galluzzi et al. 2015; Galluzzi, Bravo-San Pedro, and Kroemer 2016a, 2016b; Galluzzi et al. 2017; White and DiPaola 2009; Livesey et al. 2009; Auberger and Puissant 2017).
The realization that autophagy dysfunction contributes to the onset of many diseases has prompted efforts to identify the autophagy machinery, the molecular processes involved, the regulatory pathways, and the means to modulate autophagy in disease settings (Kroemer and White 2010).
Autophagy and Prevention of Cancer
Mechanisms by which autophagy may prevent oncogenesis have recently been elucidated. Autophagy plays a similar role in both tumor and normal cells, tumor cells show a greater dependence on autophagy due to the fact that the inherent stress that tumor cells encounter is greater than that of normal cells. This very aspect of greater dependence of cancerous cells on autophagy has been proposed as a useful mechanism in cancer therapy through modulation of autophagy (White and DiPaola 2009; Morselli et al. 2011). Inhibition of autophagy or excessive induction of autophagy has been proposed as strategies to kill cancer cells or sensitize them to therapy. Excellent reviews have therefore explored the paradoxical roles autophagy plays in cell death, cancer development, and cancer treatment, where in one hand, it plays a key role in the maintenance of cellular homeostasis therefore acting as a robust barrier against malignant transformation, while on the other hand, it presents a means to cope with intracellular and environmental stress, thus favoring tumor progression (Galluzzi et al. 2017; Pietrocola et al. 2016; L. Lin and Baehrecke 2015; Anding and Baehrecke 2015; Lindqvist, Simon, and Baehrecke 2015; Q. Li and Wang 2015).
NLRP3 Inflammasome, Inflammation, and Cancer
The activation of inflammasomes has been documented and the role of inflammasome-related cytokines such as IL-1β is also implicated in many human cancers in in vivo experiments. It is indicated that inflammation, especially with the participation of inflammasomes, plays a key role in carcinogenesis as well as the promotion of cancer (Kundu and Surh 2012; Y. Wang et al. 2016; Zitvogel et al. 2012; C. Lin and Zhang 2017). Conversely, anticancer effects of inflammasomes are also recorded, where it is proposed that inflammasomes may remove tumor cells through pyroptosis (Zitvogel et al. 2012; C. Lin and Zhang 2017). Therefore, once again the paradoxical roles of inflammasomes and their components, on the one hand, are important regulators of internal homeostasis, protecting healthy tissues against cancers, and on the other hand, they may exhibit distinct and even contrasting effects in cancer development, where they may initiate carcinogenesis through the extrinsic pathway and maintain the malignant cancer microenvironment through the intrinsic pathway. It was proposed that targeting the inflammasome/IL-1 pathway in tumor microenvironments may provide a novel approach for the treatment of cancer (H. Guo, Callaway, and Ting 2015; B. Guo et al. 2016; Zitvogel et al. 2012; Hu, Elinav, and Flavell 2011). A summary of the 3 mechanisms described and shown to be involved in carcinogenesis as well as their involvement in cancer therapy is illustrated in Figure 4.
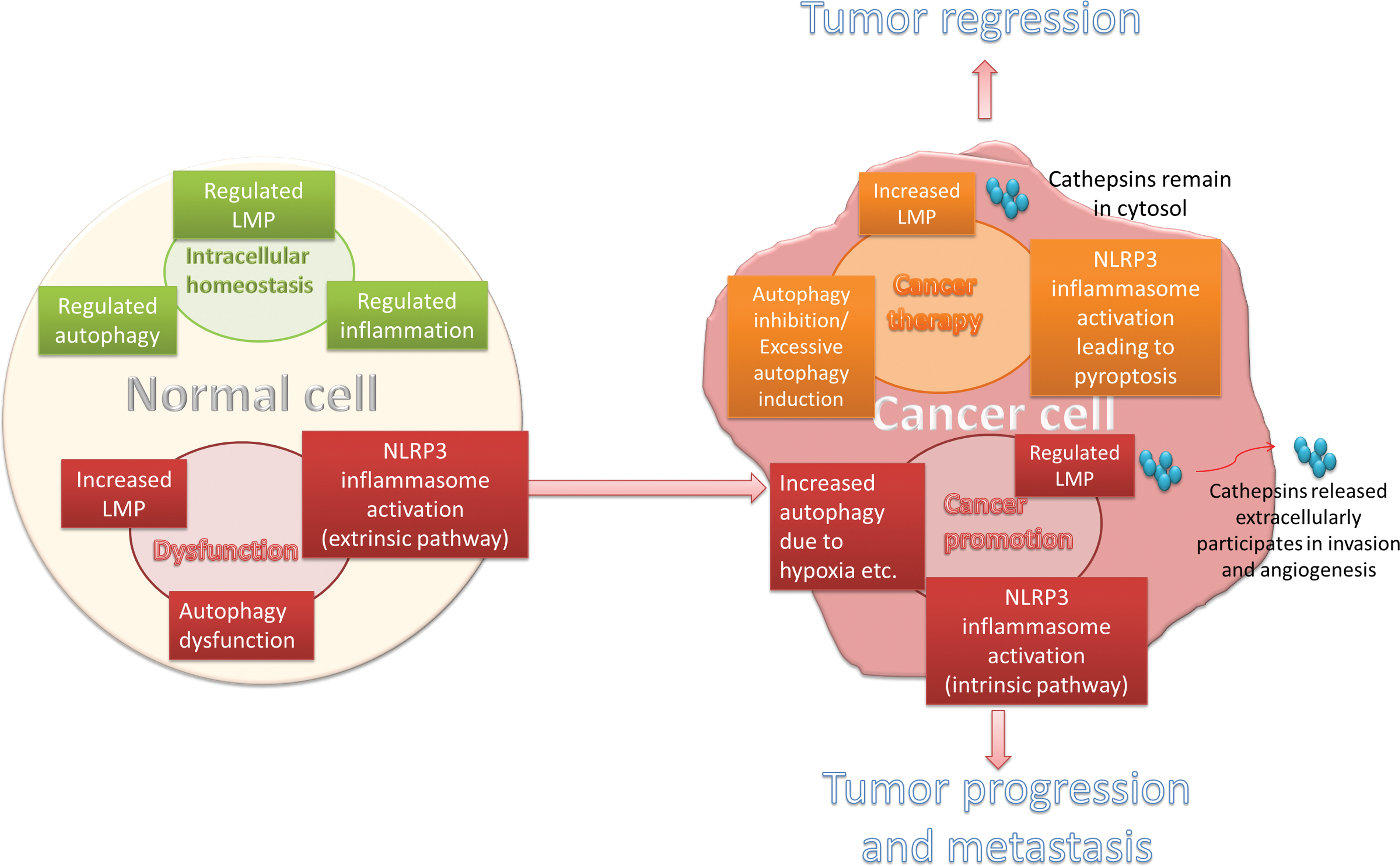
Summary of the involvement of lysosomal membrane permeabilization, autophagy, and nucleotide-binding oligomerization domain–like receptor family, pyrin domain–containing 3 inflammasomes in the initiation of cancer as well as their involvement in cancer therapy.
LMP, Autophagy, and NLRP3 Inflammasome Activation in the Treatment of Cancer Using Nanomaterials
It seems that the interaction of nanomaterials either with autophagy blockade or induction as well as lysosomal pathways, and resulting dysfunction, or their involvement in NLRP3 inflammasome activation may induce toxicity. Moreover, the existence of an interrelationship between LMP, autophagy, and inflammasome activation has already been elucidated for silver nanoparticles (Mishra et al. 2016). Subsequently, development of nanotherapy has very much utilized the autophagy dysfunction, LMP, or NLRP3 inflammasome activation as therapeutic targets for cancer, and therefore nanomaterials are proposed to be considered in cancer chemotherapy (Peynshaert et al. 2014; W. Li et al. 2015; Zarschler et al. 2016).
For example, it was shown that iron oxide magnetic nanoparticles targeted to the epidermal growth factor receptor (EGFR) could selectively induce LMP in cancer cells overexpressing the EGFR under the action of an alternating magnetic field (AMF), where LMP was observed to correlate with the production of ROS and a decrease in tumor cell viability (Domenech et al. 2013). It was also shown that these nanoparticle rotations under a weak AMF were able to induce specific mechanical lysis of cancer cells through LMP, causing extravasation of lysosomal content and inducing apoptosis of head and neck squamous cell carcinoma cell culture (Vegerhof et al. 2016) and in human breast cancer MDA-MB-231 cells (Hapuarachchige et al. 2016). Other nanoparticles that could act through this mechanism in causing cancer cell demise also included gold-zinc oxide (Au-ZnO) hybrid (Gao et al. 2014), tungsten oxide/platinum (WO3/Pt; Clark et al. 2016), and titanium dioxide (TiO2; Y. Zhu, Eaton, and Li 2012) nanoparticles. Similarly, C60 nanocrystals (Q. Zhang et al. 2009), water-dispersed neodymium fullerene derivative (Wei et al. 2010), manganese oxide (MnO; Lu et al. 2013), gold nanoparticles (AuNPs; Ma et al. 2011; Ding et al. 2014; Bhattacharyya, Mehta, and Viator 2012), iron oxide (Khan et al. 2012), iron core-gold shell (Wu et al. 2011), and alumina nanoparticles (H. Li et al. 2011), alone or in combination with different drugs, were shown to induce autophagy and autophagosome accumulation and were used in cancer therapy. Finally, the ability of NLRP3 inflammasome activation by nanomaterials has become a promising therapeutic target in cancer-related clinical conditions (Zitvogel et al. 2012; Hu, Elinav, and Flavell 2011).
Considerations in Nanoparticle Toxicity and Cancer Therapy
The first consideration should be given to the presence of endotoxins, the lipopolysaccharide product of Gram-negative bacterial contamination, in nanoparticle samples, when assessing their toxicity or during their use in nanotherapy, since it has been shown that endotoxins may interfere with the very mechanisms described above that are being utilized in nanomedicine and chemotherapy strategies. For example, it has been shown that endotoxins may lead to NLRP3 inflammasome activation (Y. Li and Boraschi 2016) or inhibition (Gurung et al. 2015) as well as autophagy induction (Yuan et al. 2009) and inhibition of autophagosome–lysosome fusion (Baker et al. 2015). The second consideration involves attempts in the reduction of toxicity of nanoparticles. With the potential of the use of nanomaterials as vehicles for drug delivery, some reviews have stressed the importance of the assessment of their safety prior to their use in nanomedicine, with the conclusion that changes in surface properties with coating and functionalization may reduce their toxicity (Yamashita et al. 2012; Wolfram et al. 2015; Stern and McNeil 2008). In fact, the very effect of functionalization, for example, with carboxyl group of CNTs was shown to reduce LMP and inflammasome activation (Hamilton et al. 2013). Since damage to lysosomes in lysosome-based cancer therapeutics with LMP is central in the application of nanomaterials in cancer therapy (W. Li et al. 2015), reduction in their toxicity, for example, through functionalization should be considered in a manner that may not interfere in this aspect of particle toxicity. This strategy has recently been used to kill HeLa cells in vitro and also in vivo in HeLa tumor-bearing mice with directed delivery (L. Huang et al. 2017). However, considerations should be given to the biopersistency of the nanomaterials used once they achieve the task of killing off cancer cells. Therefore, the rapid removal of nonbiodegradable nanomaterials from the body through renal elimination by reduction in size to minimize exposure time is suggested to be an effective strategy worthwhile of consideration (Zarschler et al. 2016).
In conclusion, it can be said that expanding knowledge in the mechanisms involved in nanotoxicity will have great potential to aid in understanding the risks associated with nanotechnology and design of safer nanomaterials and even nanomedicines. Moreover, nanomaterial therapeutic efficacy potential for cancer therapy may in fact very much rely on the very mechanisms that induce their toxicity. Therefore, a therapeutic window and a very fine/precise mechanistic understanding need to be achieved through additional studies before these approaches are approved for therapeutic interventions in humans.
Footnotes
Author Contribution
All authors (MG, CA) contributed to conception or design; data acquisition, analysis, or interpretation; drafting the manuscript; and critically revising the manuscript. All authors gave final approval and agreed to be accountable for all aspects of work in ensuring that questions relating to the accuracy or integrity of any part of the work are appropriately investigated and resolved.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.