
Abstract
An Innovation and Quality (IQ) Consortium focus group conducted a cross-company survey to evaluate current practices and perceptions around the use of animal models of disease (AMDs) in nonclinical safety assessment of molecules in clinical development. The IQ Consortium group is an organization of pharmaceutical and biotechnology companies with the mission of advancing science and technology. The survey queried the utilization of AMDs during drug discovery in which drug candidates are evaluated in efficacy models and limited short-duration non-Good Laboratory Practices (GLP) toxicology testing and during drug development in which drug candidates are evaluated in GLP toxicology studies. The survey determined that the majority of companies used AMDs during drug discovery primarily as a means for proactively assessing potential nonclinical safety issues prior to the conduct of toxicology studies, followed closely by the use of AMDs to better understand toxicities associated with exaggerated pharmacology in traditional toxicology models or to derisk issues when the target is only expressed in the disease state. In contrast, the survey results indicated that the use of AMDs in development is infrequent, being used primarily to investigate nonclinical safety issues associated with targets expressed only in disease states and/or in response to requests from global regulatory authorities.
The value of animal models of disease (AMDs) to evaluate efficacy and proposed modes of action for potential therapeutics has been widely accepted in the pharmaceutical industry, encompassing development of both small molecules and biotherapeutics. In contrast, AMDs are less frequently utilized in nonclinical safety testing paradigms, where conventional (healthy) rodent and nonrodent models represent the most common approach.
Recommendations for the use of AMDs in nonclinical safety testing have been outlined previously (Morgan et al. 2013; Cavagnaro and Silva Lima 2015). Salient features to consider when utilizing an AMD in such investigations include robust characterization of the disease model with subsequent evaluation of a specific hypothesis-driven question and focusing on hazard identification/understanding rather than on establishing a safety margin (although there are circumstances where approximations or rank ordering of safety margins from AMDs can be useful). When appropriate, traditional studies in conventional animal models are advised in conjunction with studies in AMDs (Morgan et al. 2013).
A lack of complete concordance between nonclinical and clinical toxicities, particularly for cutaneous, neurological, and hepatobiliary toxicities (Olson et al. 2000; Thiele 2007), has resulted in an increased interest in the potential utility of AMDs to support the evaluations of safety. Their potential utility is particularly appealing to evaluate the likelihood of some toxicities that are known to have low predictability or the toxicity of certain drug classes that are associated with clinical effects not readily predicted by conventional nonclinical toxicity studies. However, two of the limitations associated with utilizing an AMD include (1) a lack of consistent concordance between the AMD and the human disease, and (2) a lack of well-characterized toxicology/pathology background information in AMDs relative to conventional models utilizing healthy animals (Bolon and Galbreath 2002; Cordaro 1989). Selection of an inappropriate or poorly understood model of human disease may result in substantial risks to the development of a compound including inappropriate termination of a potentially promising therapeutic candidate based on misleading information from the AMD (Morgan et al. 2013). However, selection of an appropriate model (considering both sufficient concordance with human disease and proper characterization of the disease model) along with careful development of a targeted study plan may result in an enhanced understanding of the actual risks to the human population under consideration. Examples of potential useful applications for AMDs may include: (1) allowing for more robust evaluation of toxicity in cases where the intended pharmacologic effect (e.g., lowering of glucose) may result in significant dose-limiting toxicity at low multiples of therapeutic efficacy in healthy animals, (2) differentiation of an on-target versus off-target effect such as what has been employed to determine whether peripheral neuropathy was a manifestation of hypoglycemia (on-target effect) rather than an off-target effect (Ozaki et al. 2010), (3) evaluation of target toxicity when the target is not expressed in a conventional animal model, and (4) follow-up investigations of unexpected toxicity evident in clinical investigations but not found in conventional animal models (Morgan et al. 2013).
As noted, recent publications (Cavagnaro and Silva Lima 2015; FDA 2015; Morgan et al. 2013) have outlined recommendations and regulatory considerations for the use of AMDs. Several other publications have also described specific advantages/disadvantages of various AMDs, particularly with respect to their utility in pharmacology and/or efficacy studies (Bolon and Galbreath 2002; Cordaro 1989; Murray et al. 2015; Ozaki et al. 2010; Racke et al. 2005). However, there are no publications addressing the current practices and applications of AMDs across the pharmaceutical industry with respect to specific stages of drug development. This article summarizes a recent cross-company survey conducted to evaluate currently employed approaches for the use of AMDs in nonclinical safety assessment testing strategies, during both discovery and development stages of a potential therapeutic.
Methods
For the purpose of the survey, an AMD was defined as modified animals (transgenic, knockout naturally occurring or induced) that emulate the human disease phenotype and/or other laboratory-generated models of efficacy, as previously described (Morgan et al. 2013).
DruSafe, the Preclinical Safety Leadership Group within the International Consortium for Innovation and Quality (IQ) in pharmaceutical development, represents a broad cross section of the pharmaceutical industry including large, midsize, and small companies. A survey on the current use of AMDs in nonclinical safety testing was designed by a cross-functional team composed of subject matter experts from several member companies. The survey covered a range of topics including general questions on whether a company had experience with the use of AMDs as part of nonclinical safety testing, the types of models used, the frequency, timing (discovery vs. development), and motivation for their use. The survey was then distributed to the 32 member companies participating in DruSafe at the time (Table 1).
Invited Survey Participants.

Twenty-seven companies (84% response rate) responded to the survey. All responses were anonymized before review by the authoring team. All survey responses were received by May 2015. The response results for each question are presented based on the number of respondents to each specific question or set of questions; not all of the 27 companies responded to each question.
Results
Of the 27 respondents, 18 (67%) indicated that they have collected nonclinical safety data from AMDs, while 9 (33%) had no such experience. The most often cited concern with using AMDs for the collection of safety data was lack of confidence in the models and/or in their utility for decision-making. Additional information on the type of AMDs used in their institutions was provided by 17 of the 18 (94%) respondents who had collected safety data in AMDs. Specifically, the use of laboratory-generated and transgenic models was most common, based on participant response of 16 of 18 (89%) and 15 of 18 (83%) respondents, respectively, followed by naturally occurring disease models of 7 of the 18 respondents (39%; Figure 1). These data suggest that respondents applying AMDs often use more than one approach. Examples of AMDs employed by participating respondents and the rationale for their selection are listed in Table 2. Of the 18 companies that had experience with AMDs, 16 (89%) used AMDs for testing small or large molecules; 2 of the 18 respondents (11%) did not respond to this question. The AMDs were generally equally applied by these companies to collect safety data in support of small and large molecules. Of the 16 respondents, 10 (63%) utilized AMDs for both large and small molecules whereas 2 of the 16 (13%) used them only for small molecules and 4 of the 16 (25%) used them only for large molecules. Based on the information provided by the 17 respondents, the frequency of AMD use for safety evaluations across therapeutic areas was in the order of oncology > neuroscience > metabolic/endocrine/immunology > cardiovascular/other > infectious diseases (Figure 2). Therapeutic areas listed in the other category included ophthalmology, respiratory, renal, and dermatology.
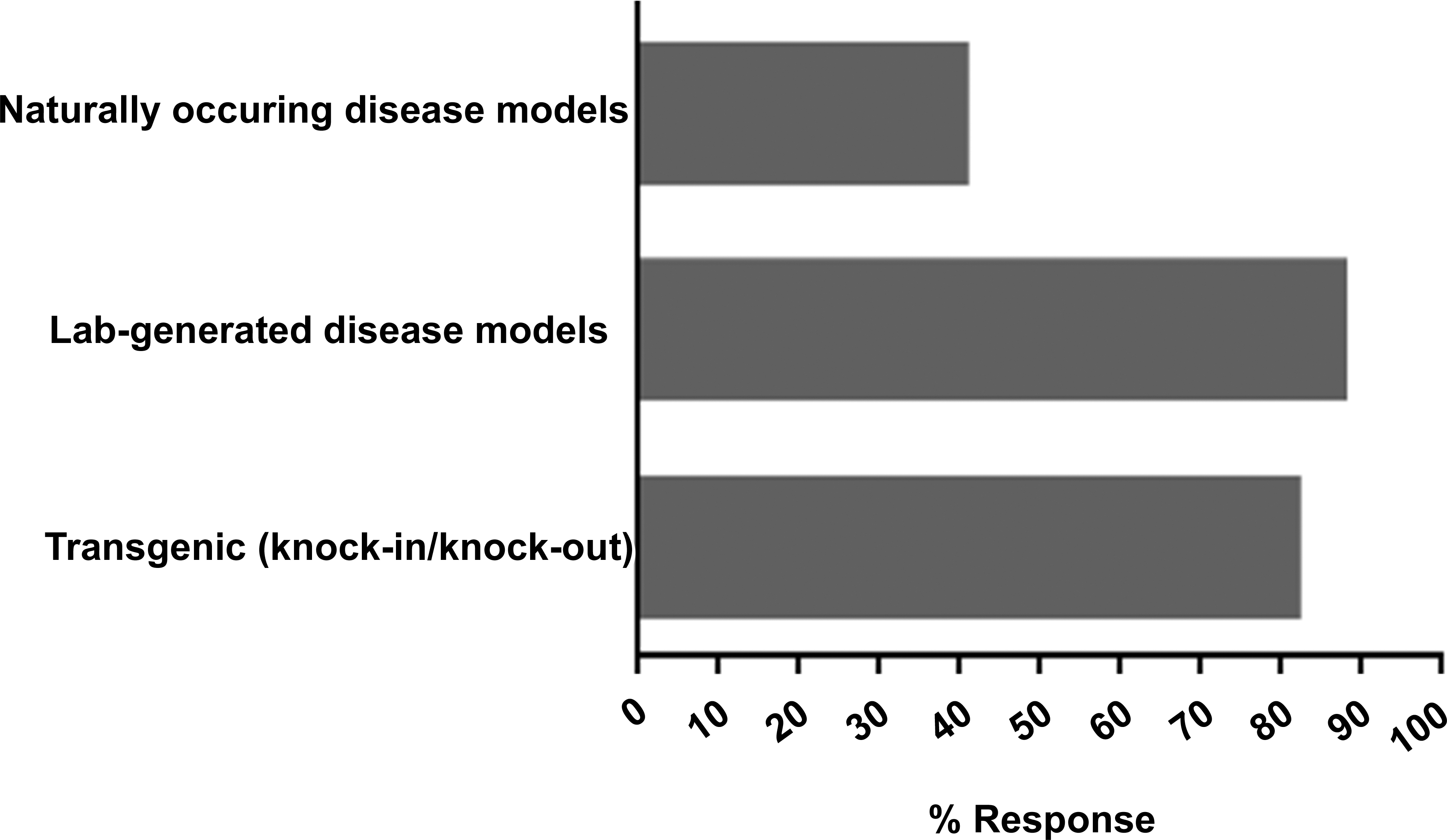
Types of animal models of disease used for safety evaluation by participating companies. Of 18 companies that have experience with the model, 17 companies responded.
Examples of AMDs and Rationale for Their Use in Safety Evaluation.

Note. AMD = animal model of disease; APPSL = amyloid precursor protein transgenic.
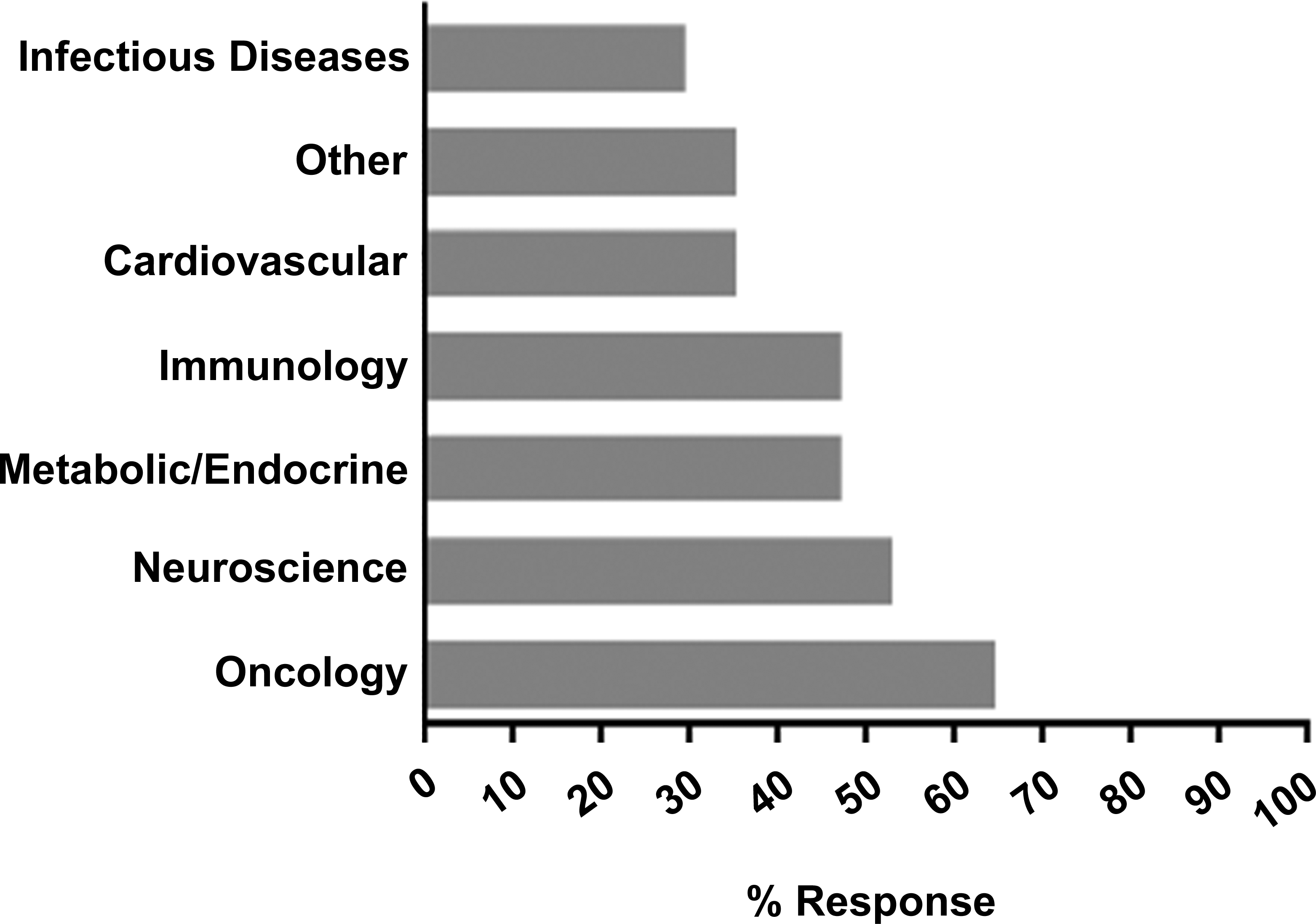
Frequency of use of animal model of disease for safety evaluation by therapeutic areas. Of 18 companies that have experience with the model, 17 companies responded.
In general, the use of AMDs for safety data collection occurred with the greater frequency during drug discovery compared to development (Figure 3). Frequency was assessed based on participant responses as to whether these data were collected routinely, occasionally, rarely, or never. A very small number of companies collected safety data routinely from AMDs during drug discovery (2 of 17 respondents, 12%), and as expected none reported using AMDs routinely during development. The majority of respondents reported the collection of AMDs data occasionally (12 of 17 respondents, 71%) during drug discovery and rarely (11 of 17 respondents, 65%) in development (Figure 3). Occasional use of AMDs during discovery is expected because the studies are often hypothesis driven based on a toxicologic concern for a given target/disease indication. Similarly, rare use of AMD in development is consistent with their use on an as-needed basis to address toxicologic and/or regulatory questions of specific concern.
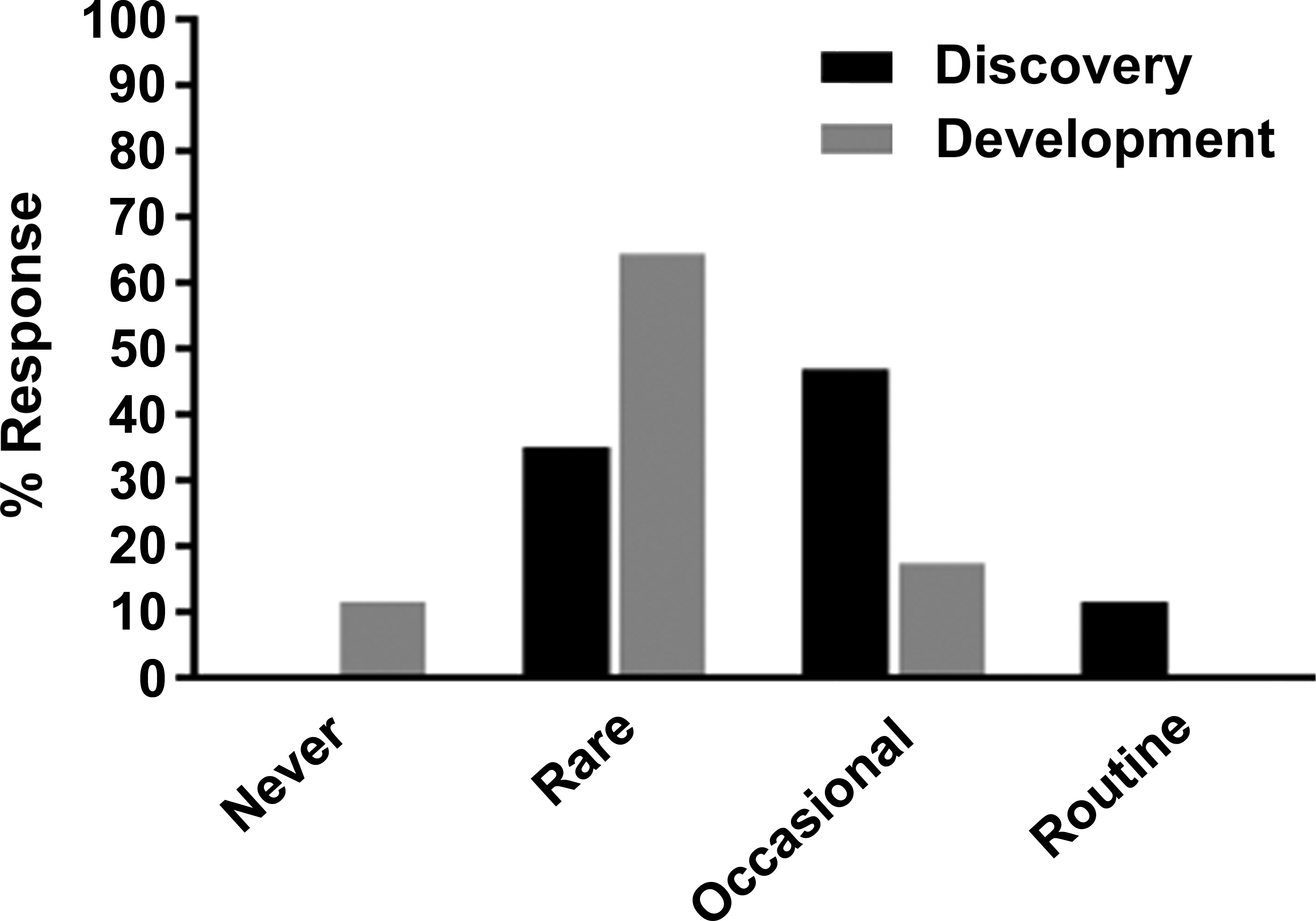
Frequency of animal model of disease use for safety evaluation in drug discovery (black bars) and development (gray bars). Of 18 companies that have experience with the model, 17 companies responded.
The survey also revealed that the majority of companies use AMDs during drug discovery as a means for proactively assessing potential safety issues prior to the conduct of toxicology studies, followed closely by their use to better understand toxicities associated with exaggerated pharmacology in traditional toxicology models or to derisk issues when the target is only expressed in the disease state (Table 3). Thirteen of the 18 (72%) respondents using AMDs that answered the development-related question indicated that the primary drivers for the use of AMD in development were exploring safety issues associated with targets expressed only in disease states and requests from global regulatory authorities (9 of 13 respondents for each, 69%; Table 4).
Drivers for AMD Use during Drug Discovery.

Note. AMD = animal model of disease.
Drivers for AMD Use during Development.
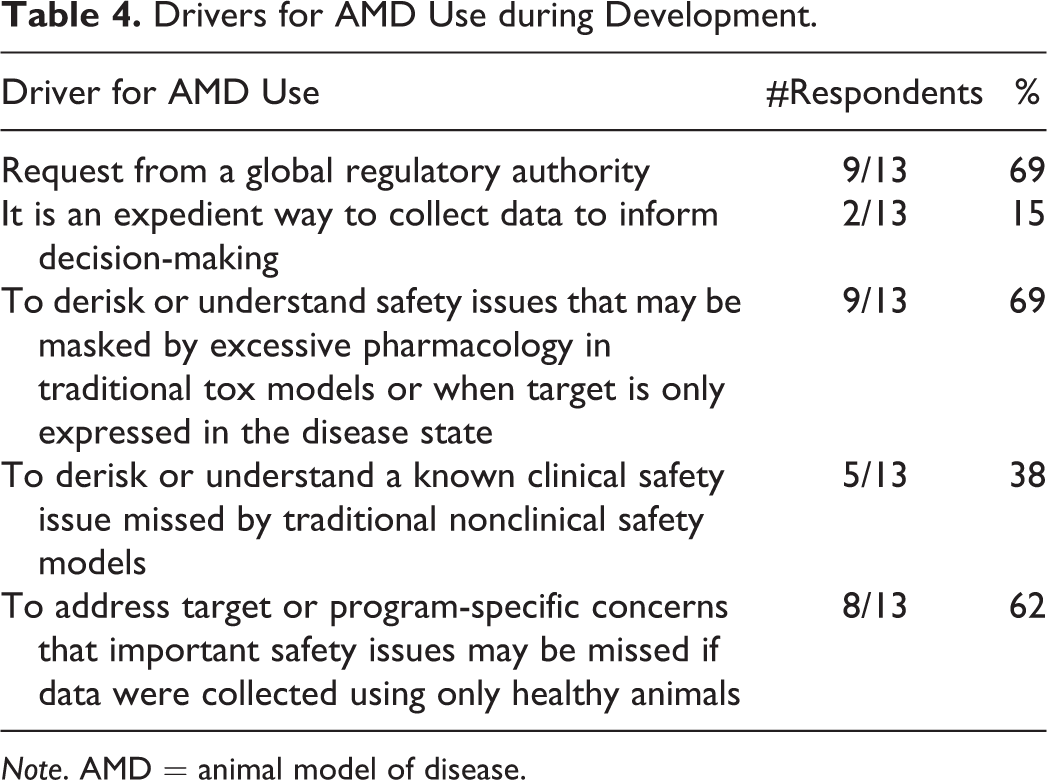
Note. AMD = animal model of disease.
Questioning on the logistics and timing for safety data collection during drug development indicated that all of the 13 respondents that used AMD during development utilized the models during early development (preclinical—phase 2). The use of AMD was reported by 5 of the 13 respondents (38%) in late (phase 3) development and by 3 of the 13 respondents (23%) during the postmarketing.
In response to questions regarding how safety data were collected using AMDs during drug discovery and development, the common practice for both phases was to add safety endpoints onto pharmacology/efficacy studies (15 of 17 respondents or 88%), followed by the conduct of dedicated studies to assess safety in an AMD as a supplement to a traditional nonclinical safety package. The least common practice was the use of a dedicated AMD for safety evaluation as an alternative to traditional nonclinical models (Figure 4). Drug development studies using AMDs were mostly conducted in-house (10 of 13 respondents, 77%) and to a lesser extent at a contract research organization (CRO; 3 of 13 respondents, 23%) or at an academic institution (1 of 13 respondents, 8%). A similar trend was noted during drug discovery, where the majority of studies using AMDs were also conducted in-house (14 of 16 respondents, 88%).
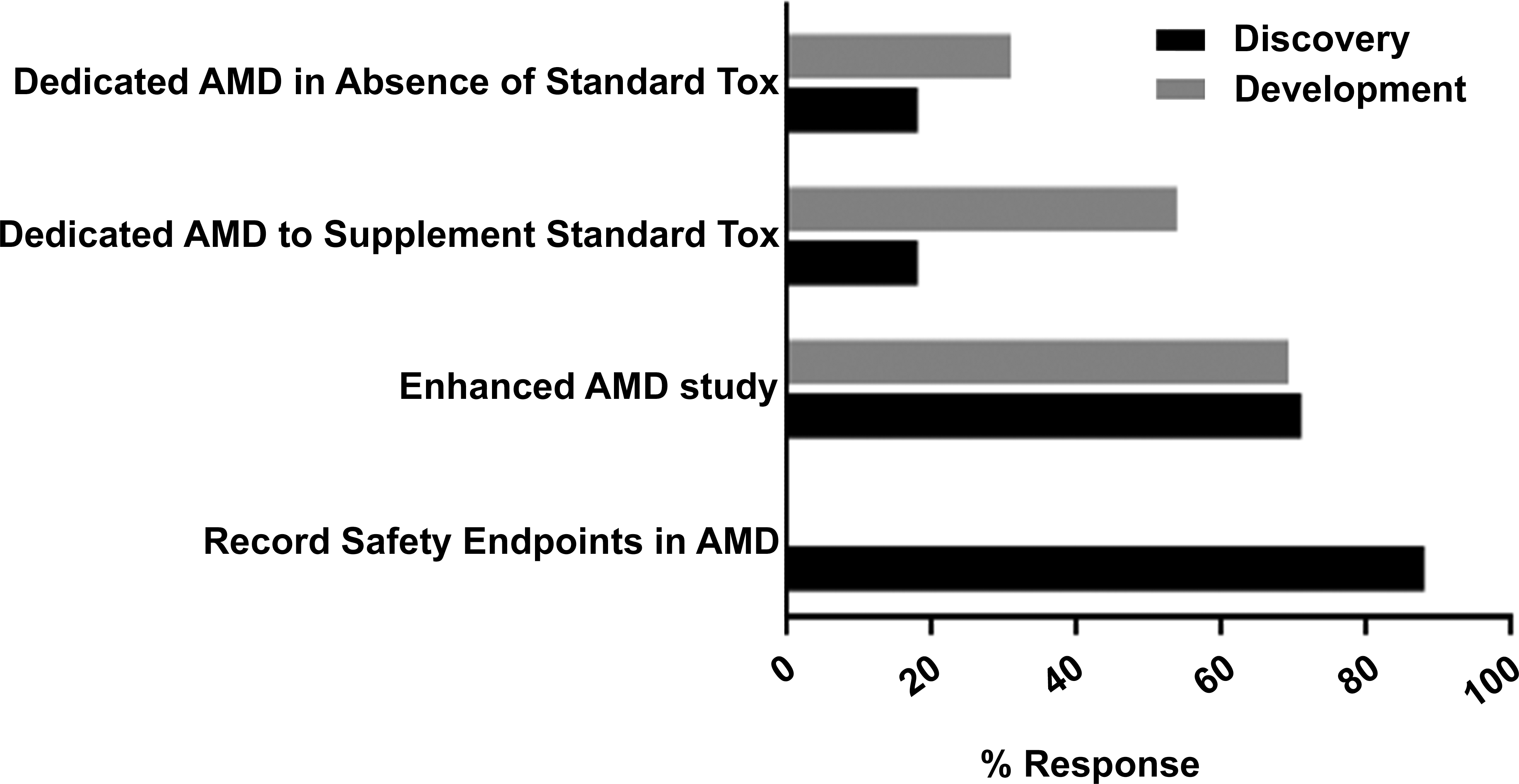
Participating companies’ practice of collecting safety data in drug discovery (black bars) and development (gray bars). Of 18 companies that have experience with the model, 17 companies responded for use in discovery and 13 companies for use in development.
AMDs were characterized to some extent prior to use, mostly to confirm the disease phenotype, but robust information on historical controls was generally not available. In some cases, thorough characterization of the disease models prior to conduct of definitive studies provided information on the inherent model variability and proved useful toward interpretation of the toxicity endpoints. In other cases, a weight-of-evidence approach was taken to interpret data, with consideration given to interanimal variability within a given study. Collection of toxicity endpoints varied, but generally included standard toxicology assessments of clinical observations, clinical chemistry, biomarkers, and histopathological assessments. However, there were also examples of more specialized endpoints collected in an AMD, such as a request to conduct a fertility study as a postapproval commitment and another model that was used to assess genotoxicity. Most respondents (8 of 13, 62%) indicated that use of an AMD during development achieved their intended purpose while others (4 of 13, 31%) had mixed results. Response from regulatory authorities was generally favorable for those who received comments. In other cases, there was no opportunity for regulatory comment because the compounds were terminated or the health authorities did not provide an opinion. In some cases, these data ultimately proved important for compound registration.
Discussion
The clinical development of novel pharmaceutical agents is supported by nonclinical safety testing in multiple animal species, typically young healthy animals. However, these standard nonclinical general toxicology safety testing paradigms do not always anticipate human safety issues that may arise during clinical development or following marketing authorization (Olson et al. 2000). While it is understood that nonclinical safety general toxicology testing paradigms cannot feasibly predict the range of effects that may become evident in a given clinical population due to a variety of factors, including logistics of clinical compared to nonclinical evaluation (e.g., general toxicology studies may involve up to 20 rodents/sex/dose and 5 nonrodents/sex/dose, compared to a much larger population of humans in clinical trials and ultimately postmarketing), AMDs may still offer advantages relevant to nonclinical safety testing in specific cases. For example, the use of AMDs may either assist in prediction of nonclinical safety events that would not be anticipated using standard nonclinical safety general toxicology safety testing paradigms or, conversely, may demonstrate that findings in standard nonclinical general toxicology safety testing paradigms are associated with the intended pharmacology of the drug candidate and may not result in a safety concern in the relevant patient population.
A previous publication (Morgan et al. 2013) provided an outline of some of the caveats to be considered when contemplating the use of AMDs in a nonclinical safety testing scheme and provided recommendations regarding when and how these models should be employed. Some of these key considerations for AMDs include validation—in terms of representing the intended human disease state; characterization—in terms of stability of the model and associated background findings; and performance—in terms of human outcome prediction.
While there have been numerous publications focusing on the use of AMDs in specific indications/therapeutic areas, there is a gap for a publication outlining actual industry practices related to broader use of AMDs in nonclinical safety evaluations. In considering approaches to assess the utility (and limitations) of AMDs in nonclinical safety testing, it was felt that a reasonable first step was to understand how these models are currently being applied across the industry.
In analyzing the results from the cross-company survey, it became apparent that there are a few common themes supporting the rationale for the use of AMDs in safety evaluations. These comprise (1) as a model to optimize early data gathering and issue/hazard identification, (2) to provide a prospective approach to nonclinical safety assessment when it is known that the target is not expressed or that the secondary effects associated with target engagement will not occur in the absence of the disease state, (3) to support requests/recommendations from regulatory authorities and/or internal medical experts, and (4) to support follow-up investigations where standard nonclinical evaluations failed to demonstrate toxicity signals that were identified in a clinical setting.
Consideration of AMD as a Model to Optimize Early Decision-making
The drivers to employ AMDs in discovery phase safety evaluations to support early decision-making on candidate selection may be motivated by a variety of factors. Perhaps the most obvious among these is the desire to obtain information on the potential safety liabilities of a compound or chemical series as early as possible in the project life cycle to enhance optimal alignment of resources. Early target engagement and efficacy studies are generally initiated during the earliest phases of a therapeutic discovery program and, along with pharmacokinetic studies, represent the first opportunities to observe potential safety signals in vivo. In many cases, efficacy models are based on internally generated and/or genetically modified mouse models, requiring relatively low amounts of compound. At pharmacologic doses, safety investigators can potentially begin to discern safety concerns related to the target, particularly when findings are seen in cells or tissues that are not related to the intended therapeutic benefit. Discovery teams can easily add an additional higher dose group to such studies with minimum impact on drug substance and animal requirements and thus gain information on possible adverse effects at therapeutic as well as supratherapeutic exposures, with an additional benefit of minimizing animal use in the future. The results of the current survey suggest that nearly all of the responding companies that use AMDs have used this discovery “tag-on” or a similar approach. Another case in which efficacy studies represent an attractive opportunity to collect early safety information is when such models are difficult or time-consuming to generate or when the duration of efficacy studies is protracted. In these situations, the efficacy model itself may be rate limiting in the compound profiling cycle, and the ability to obtain information on safety concurrent with efficacy data may significantly expedite throughput during lead optimization. A third case in which it may be highly desirable to collect data on safety endpoints from pharmacology models is when the model represents a unique opportunity to investigate the safety of a therapeutic candidate under the conditions of clinical use. One example might be intended combination of a novel chemotherapeutic agent with ionizing radiation in the clinical population, which could result in unique or exacerbated toxicity that may not be observed in conventional single-agent safety studies.
Although the collection of safety information from early pharmacology studies may be convenient and attractive for a variety of reasons, caution must be exercised when using these data to drive safety-related decisions for discovery programs. In addition to the issues cited previously concerning relevance of the AMDs to the actual human disease state (absence of historical control data, lack of optimized reagents, etc.), another caveat is that the chemical matter for small molecule programs may be insufficiently optimized to draw clear conclusions about potential nonclinical safety signals seen in disease models, particularly for early programs. Similarly, early compound batches are often of lower purity than would ordinarily be used for later nonclinical safety studies, and impurity-related toxicity could potentially confound interpretation of the data. All of these factors should be carefully considered when using safety data from AMDs to drive decision-making on early programs.
In recent years, the pharmaceutical industry has continued to focus on the early selection of new molecular entities that are both safe and efficacious prior to committing to more costly and time-consuming development research. Early evaluation of nonclinical safety in the discovery phase has been one way in which the industry has moved in an attempt to reduce later stage attrition of new candidates. In addition, with an industrywide focus on reduction of animal use in research, new paradigms have evolved to maximize the information collected from nonclinical efficacy and safety studies, and toxicology/pathology experts now typically work alongside pharmacologists and other team members on early discovery teams. As a starting point for generating hypotheses around potential toxicities that may occur, toxicology/pathology experts rely heavily on information related to the intended target of the therapeutic—including pathway and disease biology, identification of potential target organs from knockout animals, prior safety information on lead compounds in development, and competitor information from similar targets when available. Hence, to test safety-related hypotheses and begin building safety derisking strategies, it may be helpful to assess select endpoints of toxicity while conducting in vivo studies for efficacy using AMDs that more closely simulate the physiology and target distribution of patients with the disease(s) of interest. The data gathered from this cross-company survey have demonstrated an increasing consideration of safety data from AMDs as part of a compound’s discovery strategy. For the companies that reported the use of AMDs for toxicity assessments, a majority (14 of the 17 respondents; 82%) did so at least occasionally in order to screen for potential safety concerns before initiating stand-alone toxicity studies using standard toxicology species.
Prospective Use of AMD When the Target or Disease Pathway Is Absent in Normal Animals
Other significant drivers for using AMDs while in the early discovery phases include when the intended pharmacological target is only thought to be expressed in the disease state or when the disease condition itself could modulate target engagement. In either case, the target and any associated adverse effects would not be expected to occur in standard nonclinical general toxicology safety testing paradigms and may underlie situations where toxicity was observed in the clinic but was not detected using a traditional nonclinical safety testing paradigm. Several companies have indicated these concerns as potential reasons for using AMDs for early exploration of toxicity before employing the standard safety models needed to support regulatory requirements.
Most of the companies (15 of 17, 88%) that have explored toxicity in an AMD during discovery have done so by simply recording safety-related findings during the conduct of the pharmacology studies. Animal studies of pharmacology and efficacy are typically conducted at relatively low dosages sufficient to bind to the target and elicit the intended pharmacological response, in contrast to safety studies that require relatively high levels of exposure in order to demonstrate target organ toxicity. Therefore, in some (12 of 17, 70%) cases, companies have added higher dose groups and/or have incorporated additional safety endpoints in efficacy studies using AMDs. In rarer cases (3 of 17, 18%), companies have either used pharmacology/efficacy models to conduct dedicated nonclinical safety evaluation studies in AMDs or have used AMDs distinct from pharmacology/efficacy models to conduct dedicated nonclinical safety evaluation studies. It was not clear if these dedicated nonclinical studies in AMDs served as an additional study to follow up on findings seen in a routine pharmacology study or if this was standard practice for those companies.
Requests/Recommendations from Regulatory Authorities and/or Internal Medical Experts
The use of AMDs during the development stages of a potential therapeutic is relatively rare, as conventional regulatory guidelines call for the use of healthy animals in toxicity testing. However, in some instances, there is a need to assess toxicity in a model that replicates the human disease due to some unique characteristics of the disease that could predispose the patient to an adverse event or alternatively if the target is not expressed in healthy animals. These studies can be proactively initiated by the drug sponsor company or conducted in response to a request from a regulatory agency.
In the recombinant human acid sphingomyelinase (rhASM) program (Murray et al. 2015), an observation of unexpected toxicity with escalation of dose in pharmacology studies with acid sphingomyelinase knockout mice led to the use of these KO mice in toxicity studies. While low levels of the substrate for the therapeutic enzyme are present in the brain of normal mice, considerably higher levels of the substrate accumulate in the brain of the AMD. Thus, the AMD is a useful model to evaluate the toxicity associated with pharmacologic effects of the products of enzyme activity. Results from these studies successfully guided conduct of phase 1 clinical trials so that efficacy was achieved without undue toxicity due to substrate metabolism. While transgenic mouse models are most frequently utilized for the evaluation of efficacy and potential safety of compounds intended as therapy for lysosomal storage diseases, naturally occurring large animal models also exist and have been utilized (Vuillemenot et al. 2011). A recent FDA draft guidance (FDA 2015) outlines considerations for the design and conduct of nonclinical studies during development of investigational enzyme replacement products (primarily lysosomal storage diseases) and provides specific recommendations for the use of AMDs. Other examples of proactive use of AMDs exist, particularly where the drug target is not present in normal animals and/or the pharmacodynamics are altered in the disease state.
Examples such as that of rhASM development show a clear utility for AMDs in protecting patients in early clinical trials by identifying risks that are not possible to determine in normal animals. This can be due to the presence or absence of the target in the AMD, physiological changes in animals and humans due to diseases states such as diabetes, altered pharmacodynamics in the AMD, and so on. The use of AMD and their potential to inform clinical trial safety should be considered on a case-by-case basis by sponsor internal medical and drug safety experts as well as by regulators with oversight over many drug development programs in a specific disease area. Recognition of the utility of an AMD can occur anywhere along the development process, with examples of application not only in early-stage development but also via regulatory-driven requests based on postmarketing adverse event reports.
An example of AMD use occurring in late stage development based on a regulatory request to conduct toxicity studies is the use of an AMD of type 2 diabetes in an attempt to assess potential pancreatic injury risk. Following approval of several drugs in the GLP-1 agonist class, wider use in the human population led to a postmarketing observation of injury to the exocrine pancreas. As several GLP-1 agonists were in development at that time, the FDA requested that the sponsors conduct studies in a diabetic AMD, based on the hypothesis that the mechanisms leading to pancreatic injury would be more likely to occur in the diabetic state than in normal animals. Subsequent evaluation in various rat models (Sprague-Dawley, Zucker diabetic fatty, and rats expressing human islet amyloid polypeptide) indicates that the pancreatic findings of exocrine atrophy/inflammation attributed to GLP-1 compounds are common background findings in rats, observed without drug treatment and independent of diet or glycemic status, thus not likely relevant for human safety (Chadwick et al. 2014).
Regulators have also previously requested studies in animals containing amyloid plaques in the brain as an AMD for Alzheimer’s disease to assess the potential for test article–related side effects (microhemorrhages) that could occur in the presence of the plaques (which do not occur in animals typically utilized in standard general toxicity studies). As is the case with many models of human disease, particularly those with an incompletely understood and complex pathogenesis, there is no single ideal model that recapitulates all of the characteristics of the human disorder (Wilcock and Colton 2009). However, the PDAPP mouse (overexpresses human mutant amyloid precursor protein V717F) has been useful in prediction of emergence of test article–related microhemorrhages, and its use may be helpful in rank ordering compounds from a clinical safety perspective (Racke et al. 2005). Serial evaluation of mice administered β-amyloid (Aβ) immunotherapy provided insight into the potential pathogenesis and susceptibility period for microhemorrhages as well as a relationship of the microhemorrhages to amyloid-related imaging abnormalities as detected in a clinical setting (Zago et al. 2013). Special care must be taken to ensure that the correct age of animals is utilized in the intended study (e.g., microhemorrhages are likely to occur in the age of animals utilized and at least a high proportion of animals are likely to survive to the end of the planned study).
Follow-up on Standard Nonclinical Evaluation That Failed to Demonstrate Toxicity Signal
An AMD may be utilized to further understand a human safety finding that was not anticipated by standard nonclinical safety models. This would take the form of investigative studies using AMDs in an attempt to recapitulate a clinical safety event. This is a retrospective exercise (applied after standard nonclinical safety testing and human outcome data were available) but could form the basis for future prospective screening to avoid the clinical safety issue with other molecules.
An example of the use of retrospective investigative studies using AMDs includes those completed for fialuridine. Fialuridine was an antiviral nucleoside analog being considered as a therapy for chronic hepatitis B infection. Despite the lack of safety signals in standard nonclinical species, patients developed severe multisystemic toxicity in clinical trials beginning at the 13th week of investigation. The toxicity was characterized by hepatic failure, lactic acidosis, pancreatitis, neuropathy, and myopathy. Based on the constellation of findings in conjunction with electron microscopy, the toxic reaction was considered to be secondary to widespread mitochondrial damage that may infrequently be seen with nucleoside analogs (McKenzie et al. 1995). Since the evaluation of the compound in standard nonclinical species was not predictive of the clinical toxicities, subsequent investigations involved the use of an AMD. The eastern woodchuck (Marmota monax) infected by the hepadnavirus woodchuck Hepatitis B virus (WHV) has been applied as a predictive model to support the development of new HBV therapies (D’Ugo et al. 2010) and in some cases has exhibited hepatic toxicity that is similar to that observed in humans with some therapeutic agents (Tennant and Gerin 2001). Studies revealed that woodchucks with chronic hepatitis secondary to WHV developed a similar (albeit less severe) syndrome after chronic (12 weeks) administration of fialuridine (Lewis et al. 1997), resulting in widespread mitochondrial toxicity (heart, liver, diaphragm, skeletal muscle, and kidney). While these results suggest that the coexistence of the virus and/or inflammatory changes plays a role in the pathogenesis of the mitochondrial toxicity, additional studies have revealed no differences in hepatic effects with chronic (≥6 weeks) fialuridine administration between woodchucks that are the carriers of the virus compared to those that are not carriers (Tennant et al. 1998). Thus, it was apparent that the different effect was related to species sensitivity not the disease state. In order to further support the role of a species rather than a disease-state effect, fialuridine was administered to the chimeric mouse model (humanized liver). Administration of fialuridine to the chimeric TK-NOG mouse for 14 days resulted in a spectrum of hepatic findings that was similar to that observed in humans (Xu et al. 2014). Thus, it should be remembered that investigative studies in multiple AMDs and other models (e.g., chimeric mouse models) may be necessary to elucidate the pathogenesis of unique toxicities in humans.
Conclusions
The majority of companies surveyed the use of AMD during drug discovery as a means for proactively assessing potential safety issues prior to the conduct of toxicology studies, followed closely by their use to better understand toxicities associated with exaggerated pharmacology in traditional toxicology models or to derisk issues when the target is only expressed in the disease state. In contrast, the surveyed companies indicated that the use of AMDs in development is relatively infrequent. Exploring nonclinical safety issues associated with targets expressed only in disease states and/or requests from global regulatory authorities remained the primary drivers for the use of AMD in development.
Footnotes
Author Contribution
Authors (SM, JC, PG, DK, RK, MO, RS, TJ) contributed to conception or design; data acquisition, analysis, or interpretation; drafting the manuscript; and critically revising the manuscript. All authors gave final approval and agreed to be accountable for all aspects of work in ensuring that questions relating to the accuracy or integrity of any part of the work are appropriately investigated and resolved.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.