
Abstract
Drug-induced valvular heart disease (VHD) is a serious side effect linked to long-term treatment with 5-hydroxytryptamine (serotonin) receptor 2B (5-HT2B) agonists. Safety assessment for off-target pharmacodynamic activity is a common approach used to screen drugs for this undesired property. Such studies include in vitro assays to determine whether the drug is a 5-HT2B agonist, a necessary pharmacological property for development of VHD. Measures of in vitro binding affinity (IC50, Ki) or cellular functional activity (EC50) are often compared to maximum therapeutic free plasma drug levels (fCmax) from which safety margins (SMs) can be derived. However, there is no clear consensus on what constitutes an appropriate SM under various therapeutic conditions of use. The strengths and limitations of SM determinations and current risk assessment methodology are reviewed and evaluated. It is concluded that the use of SMs based on Ki values, or those relative to serotonin (5-HT), appears to be a better predictor than the use of EC50 or EC50/human fCmax values for determining whether known 5-HT2B agonists have resulted in VHD. It is hoped that such a discussion will improve efforts to reduce this preventable serious drug-induced toxicity from occurring and lead to more informed risk assessment strategies.
Since the initial descriptions in the 1960s linking development of valvular heart disease (VHD) with the use of the ergot derivatives methysergide and ergotamine to treat migraine (Bhattacharyya et al. 2009; Cavero and Guillon 2014), additional drugs have been identified with having VHD as a possible side effect associated with their long-term use. Although most of these drugs have since been withdrawn, some (e.g., cabergoline, dihydroergotamine, lorcaserin, pergolide) are still on the market with appropriate warnings in their labels. The one common feature linking these drugs to this serious and sometimes fatal outcome is their agonist activity for the 5-hydroxytryptamine (serotonin) receptor 2B (5-HT2B; Rothman et al. 2000; Fitzgerald et al. 2000; Setola et al. 2003; Huang et al. 2009; Cavero and Guillon 2014). The 5-HT2B is a G-protein-coupled receptor (GPCR) expressed by a wide variety of cell types, and its expression on cardiac valve leaflets is important for cardiac development and in maintaining normal cardiac valve structure and function. However, the sustained overstimulation of 5-HT2B either by high plasma levels of serotonin or by drugs with 5-HT2B agonist properties can lead to development of VHD by stimulating myofibroblast mitogenesis and extracellular matrix deposition that eventually result in thickened valve leaflets, regurgitation due to improper closing during diastole, pulmonary hypertension, and eventually heart failure (Elangbam 2010; Hutcheson et al. 2011; Cavero and Guillon 2014).
Most new drugs submitted to regulatory agencies generally undergo an assessment for secondary or off-target pharmacodynamic activity in vitro against a broad range of potential targets that are related to or distinct from the intended or primary therapeutic target (Whitebread et al. 2005; Bass et al. 2009; Bowes et al. 2012; Papoian et al. 2015). Initial assessments include receptor binding studies; but if significant binding activity is detected, then follow-up cell-based functional assays are usually conducted to determine whether the drug acts as an agonist or antagonist for that target. In the case of the 5-HT2B, such functional assays are important to determine whether the drug possesses 5-HT2B agonist activity, a necessary pharmacological property for development of VHD. When in vitro data suggest such activity, the likelihood of risk under clinically relevant conditions and drug exposures is carefully assessed in a regulatory context, and safety recommendations made for the proposed clinical studies, or additional nonclinical studies requested to help better understand or reduce the risk.
A common approach for assessing risk to humans for drugs with possible off-target activity is to compare measures of in vitro binding or functional activity using human-specific targets to therapeutic free plasma drug levels seen in animals or to levels seen in humans once that data become available. From such a comparison, a safety margin (SM) can be derived, which is defined as the ratio of values of activity in vitro (IC50, Ki , or EC50) to the maximum therapeutic free plasma drug concentration in vivo (fC max; Bowes et al. 2012; Muller and Milton 2012). Such an approach has been described previously for several targets critical for proper cardiac function. These include (1) the human ether-a-go-go-related gene-encoded voltage-dependent potassium channel (hERG or IKr), whose inhibition has been linked to development of drug-induced QT interval prolongation and the appearance of torsade de pointes (Redfern et al. 2003), (2) the human cardiac Na+ channel (hNav1.5), whose inhibition can prolong the QRS interval and slow cardiac conduction (Harmer, Valentin, and Pollard 2011; Bowes et al. 2012), and (3) the human 5-HT2B receptor whose activation has been associated with development of VHD (Whitebread et al. 2005; Cavero and Guillon 2014). Generally, for hERG and hNav1.5a, a 30- to 100-fold ratio of IC50 values to maximum human free plasma drug concentrations (fC max) appears to confer an acceptable margin of safety for most drugs, whereas a 10-fold SM may be acceptable for drugs intended to treat some life-threatening indications. However, in the case of the 5-HT2B, there still appears to be no clear consensus on what constitutes an appropriate SM under various therapeutic conditions of use. This is further complicated by the existence of multiple cell-based assay end points for measuring agonist activity (Huang et al. 2009) and lack of standardized assay protocols (Cavero and Guillon 2014).
The purpose of this article is to provide a regulatory perspective on the current utility of in vitro secondary pharmacology data for the 5-HT2B receptor to assess risk of drug-induced VHD in humans. It focuses on the current assay methodology, strengths and limitations of SM determinations, and approaches used by the various stakeholders for assessing risk for development of VHD with 5-HT2B agonists. It is our hope that such a discussion will improve efforts to reduce the likelihood of this preventable and serious drug-induced toxicity from occurring and lead to more informed risk assessment strategies.
Roles of 5-HT (serotonin) and 5-HT2B Signaling in Development of VHD
In mammalian species, vascular (semilunar aortic and pulmonary) and atrioventricular (mitral and tricuspid) heart valves maintain unidirectional blood flow across the heart chambers.
Valvular abnormalities, either genetic or acquired through a variety of etiologies, can lead to hemodynamic overload of the ventricles, myocardial dysfunction, congestive heart failure, and sudden death.
Early evidence implicating 5-HT2B receptors in some acquired forms of VHD came from the association of certain carcinoid tumors secreting a variety of bioactive products, including 5-HT (serotonin), with subsequent development of VHD. Later, valvulopathy was shown to occur following long-term exposure to various drugs with 5-HT2B agonist activity (Bhattacharyya et al. 2009; Huang et al. 2009; Rothman and Baumann 2009; Elangbam 2010; Hutcheson et al. 2011; Unett et al. 2013; Cavero and Guillon 2014). These drugs have been used to treat migraine (methysergide, ergotamine), obesity (fenfluramine, dexfenfluramine), diabetes (benfluorex), Parkinson’s disease (pergolide), and hyperprolactinemia (cabergoline). There have also been reports of VHD with the use of recreational drugs (3,4-methylenedioxy-N-methylamphetamine; ecstasy; Setola et al. 2003; Droogmans et al. 2007). In drug-induced valvulopathy, there is an increase in the thickness of the valves due to an abnormal deposit of fibrous tissue and glycosaminoglycans within the valve leading to valvular leakage and regurgitation (Rothman and Baumann 2009; Elangbam 2010; Hutcheson et al. 2011).
5-HT has a complex role in both normal and pathological processes (Hutcheson et al. 2011). 5-HT produces its effects through a variety of ion channels and receptors that are found in the cardiovascular system, blood, gastrointestinal tract, and central and peripheral nervous systems. Up to fifteen 5-HT receptor subtypes have been characterized (Cavero and Guillon 2014). Other than the 5-HT3 receptor, which is a ligand-gated ion channel, the remaining 5-HT receptors belong to the GPCR superfamily. The 3 major 5-HT receptor subtypes (A, B, and C) are coupled primarily to the Gq protein family. Activation of the 5-HT receptor results in dissociation of the heterotrimeric G protein complex into a Gα subunit that then activates phospholipase C. This leads to generation of intracellular signaling pathways involving inositol trisphosphate/calcium and diacyl glycerol/protein kinase C (PKC).
The development of drug-induced VHD is likely to be a manifestation of complex interactions among various factors, including endogenous 5-HT levels, 5-HT2B density on valve leaflets, the serotonin transporter, and polymorphisms in individual gene expression. Often patients have comorbidities and receive concomitant therapies that, through metabolic or other interactions, can increase the normally low exposure of a 5-HT2B agonist to a toxic level. Also, other drugs, such as those that increase 5-HT levels through impaired 5-HT metabolism or inhibition of the transmembrane 5-HT transporter, can become a reasonable safety concern when combined with a 5-HT2B agonist.
The clinical diagnosis of drug-induced VHD is most commonly assessed with echocardiography. Characteristic features include valve leaflet thickening, restriction of motion of one or more valve leaflets, and distortion and thickening of the subvalvar apparatus, typically of the mitral valve (Andrejak and Tribouilloy 2013). These pathologic changes contribute to regurgitant blood flow. Recently, some authors have suggested that more rigorous criteria are needed to diagnose drug-induced VHD, such as using an integrative approach combining both color Doppler and valvular morphological characteristics, and pathologic findings, if available, to arrive at a more accurate diagnosis (Cosyns and Droogmans 2015).
Assessment of Drugs for 5-HT2B Receptor Agonist Activity
Evidence implicating 5-HT2B in drug-induced VHD has come from multiple reports showing that activation of the receptor on heart valve interstitial cells is mitogenic, resulting in phosphorylation of extracellular signal–regulated kinase 1/2 (ERK 1/2) and the cytoplasmic tyrosine kinase sarcoma (Src) and activation of PKC with subsequent cellular proliferative responses and deposition of extracellular matrix material (Hutcheson et al. 2011).
Accordingly, various in vitro and animals models have been developed to determine whether drugs possess 5-HT2B receptor agonist activity. These are described as follows:
Radioligand binding assays: Performed using intact cell or cell membrane preparations expressing human 5-HT2B receptors, and results expressed as follows: Affinity (Ki
or inhibitory dissociation constant) of a compound for 5-HT2B is determined by competition displacement binding of a selective 5-HT2B radioligand by the test compound. An IC50 is determined that can then be converted to Ki
values using the Cheng–Prusoff equation (Cheng and Prusoff 1973). Kinetic binding assays are performed to calculate association (k
on) and dissociation (k
off) rates of test compounds to cell membrane preparations (Unett et al. 2013). The dissociation constant (Kd
) for the ligand is determined by the equation: Kd
= k
off/k
on.
These binding assays cannot identify agonist or antagonistic properties of the test compound. Such properties are determined only by various functional assays, including those that are cell based, as described below.
Functional cell-based assays: Drugs with significant 5-HT2B receptor binding activity are differentiated for agonistic or antagonistic properties by their ability to activate or block, respectively, in a concentration-dependent manner, cytosolic signaling pathways in various cell lines (e.g., Chinese hamster ovary, cells of simian origin [COS]-7, and human embryonic kidney cells [HEK]-293) expressing human 5-HT2B receptors. These signaling pathways include inositol phosphate (IP) accumulation, calcium release, mitogen-activated protein kinase 1/2 (MAPK 1/2) phosphorylation, nuclear factor of activated T cells (NFAT) activation, and β-arrestin recruitment/translocation (Huang et al. 2009; Unett et al. 2013; Wacker et al. 2013; Cavero and Guillon 2014). Dose–response data are fitted to a nonlinear curve-fitting program to calculate an EC50 for agonists or IC50 for antagonists in order to define the potency of the test compound. Additional parameters calculated are percentage of the maximum response (E
max%) for efficacy relative to maximum (100%) 5-HT (serotonin) activity. Animal models of drug-induced VHD: The role of 5-HT in inducing VHD in an animal model was first shown by administering 5-HT by subcutaneous injection to Sprague-Dawley rats at 50 mg/kg for the first 3 days and 20 mg/kg/day thereafter for 3 months (Gustafsson et al. 2005). Results showed morphological and echocardiographic changes in aortic and pulmonary valve insufficiency similar to those seen in human carcinoid heart disease, including shortened and thickened aortic cusps and carcinoid-like plaques characterized by myofibroblasts within an extracellular matrix of collagen ground substance. An animal model of drug-induced VHD was first reported in the Wistar rat exposed to the serotonergic drug pergolide, given intraperitoneally at 0.5 mg/kg/day for 20 weeks (Droogmans et al. 2009). At the end of treatment, pergolide-treated rats developed valvular lesions, as assessed by echocardiography and histopathology, with features very similar to those described in humans. Use of cyproheptadine, a 5-HT2B receptor antagonist, prevented the development of pergolide-induced valvular lesions in this rat model. Such animal models have shown utility in assessing the in vivo risk of drug-induced VHD during the drug development and regulatory review processes.
Regulatory Considerations for Risk Assessment of Drugs with 5-HT2B Agonist Activity
Given the well-described association of drugs with 5-HT2B agonist activity as a risk factor for VHD, safety assessments typically performed by pharmaceutical companies during early drug development include 5-HT2B binding studies for determination of any potentially significant off-target activity, and if a significant Ki or IC50 value is identified (typically <10 µM), then follow-up functional assays in cell-based systems may be performed to determine whether or not the drug possesses 5-HT2B agonist activity, a necessary characteristic for a drug to produce VHD. However, as discussed further below, there appears to be significant variability in the ability of different in vitro cellular functional assays to be able to distinguish between those 5-HT2B agonists that produce VHD from those 5-HT2B agonists that do not when compared at clinically relevant exposures and conditions of use; that is, no consistent pattern of predictivity emerges.
Based on the data collected from published sources (Huang et al. 2009; Unett et al. 2013; Wacker et al. 2013; Cavero and Guillon 2014), MAPK1/2 and NFAT assays show greater sensitivity than the other three readouts (IP accumulation, calcium release, and β-arrestin translocation) for the recognized valvulopathogens (Table 1). Further, in a study that screened nearly 2,200 U.S. Food and Drug Administration (FDA)-approved and investigational drugs and drug-like scaffolds (i.e., molecular frameworks) for potential to induce VHD using calcium-based high-throughput screening, 27 were shown to be 5-HT2B receptor agonists and included the 7 known valvulopathogens (Huang et al. 2009). This later study demonstrated that no single pattern of functional selectivity using multiple assays could distinguish known valvulopathic 5-HT2B agonists from nonvalvulopathic 5-HT2B agonists. However, the authors suggested the need to perform multiple functional assays to screen compounds for possible 5-HT2B agonist activity.
Correlation between Ki /Human fC max and EC50/Human fC max for Human 5-Hydroxytryptamine (Serotonin) Receptor 2B (5-HT2B) Agonists.
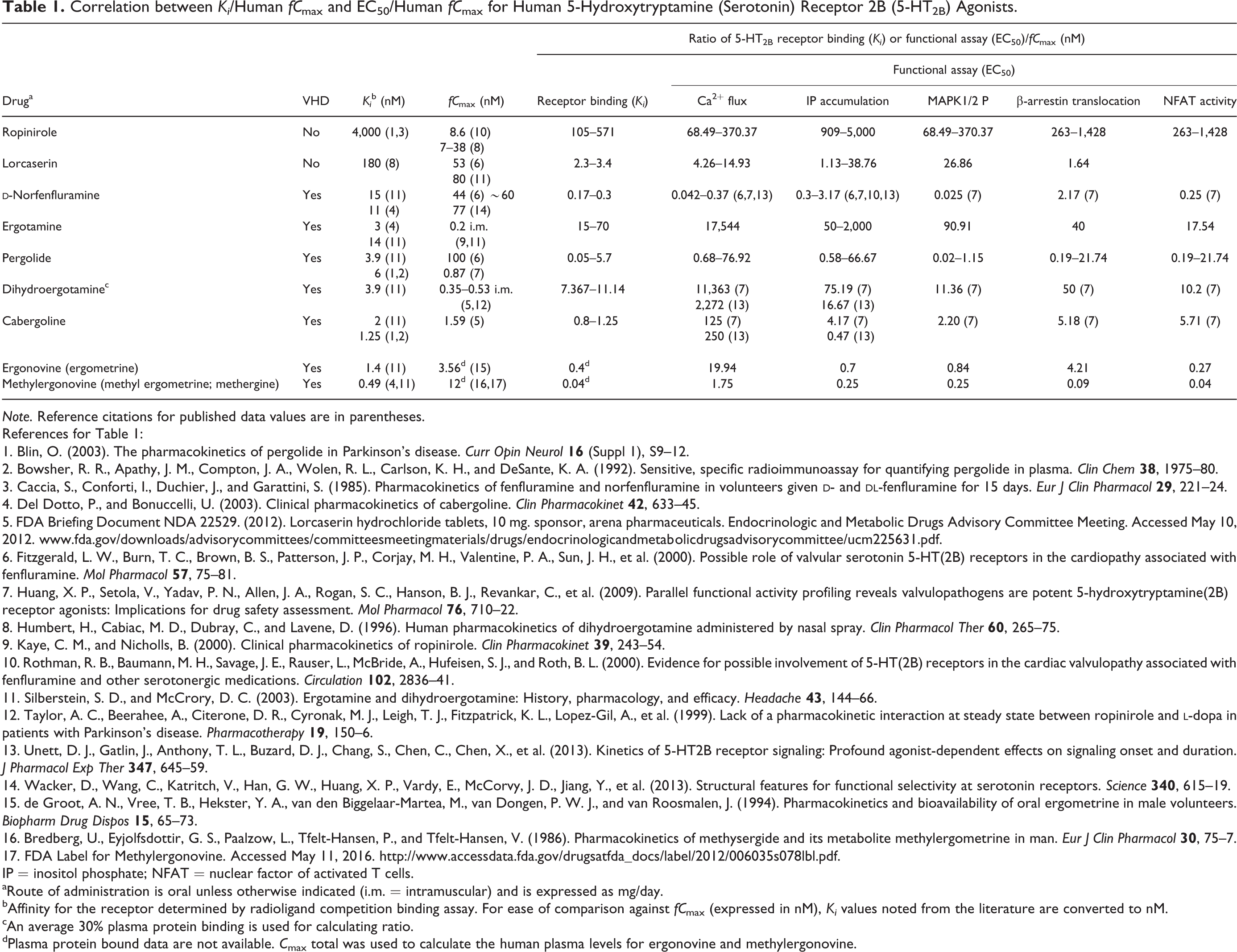
Note. Reference citations for published data values are in parentheses.
References for Table 1:
1. Blin, O. (2003). The pharmacokinetics of pergolide in Parkinson’s disease. Curr Opin Neurol
2. Bowsher, R. R., Apathy, J. M., Compton, J. A., Wolen, R. L., Carlson, K. H., and DeSante, K. A. (1992). Sensitive, specific radioimmunoassay for quantifying pergolide in plasma. Clin Chem
3. Caccia, S., Conforti, I., Duchier, J., and Garattini, S. (1985). Pharmacokinetics of fenfluramine and norfenfluramine in volunteers given
4. Del Dotto, P., and Bonuccelli, U. (2003). Clinical pharmacokinetics of cabergoline. Clin Pharmacokinet
5. FDA Briefing Document NDA 22529. (2012). Lorcaserin hydrochloride tablets, 10 mg. sponsor, arena pharmaceuticals. Endocrinologic and Metabolic Drugs Advisory Committee Meeting. Accessed May 10, 2012. www.fda.gov/downloads/advisorycommittees/committeesmeetingmaterials/drugs/endocrinologicandmetabolicdrugsadvisorycommittee/ucm225631.pdf.
6. Fitzgerald, L. W., Burn, T. C., Brown, B. S., Patterson, J. P., Corjay, M. H., Valentine, P. A., Sun, J. H., et al. (2000). Possible role of valvular serotonin 5-HT(2B) receptors in the cardiopathy associated with fenfluramine. Mol Pharmacol
7. Huang, X. P., Setola, V., Yadav, P. N., Allen, J. A., Rogan, S. C., Hanson, B. J., Revankar, C., et al. (2009). Parallel functional activity profiling reveals valvulopathogens are potent 5-hydroxytryptamine(2B) receptor agonists: Implications for drug safety assessment. Mol Pharmacol
8. Humbert, H., Cabiac, M. D., Dubray, C., and Lavene, D. (1996). Human pharmacokinetics of dihydroergotamine administered by nasal spray. Clin Pharmacol Ther
9. Kaye, C. M., and Nicholls, B. (2000). Clinical pharmacokinetics of ropinirole. Clin Pharmacokinet
10. Rothman, R. B., Baumann, M. H., Savage, J. E., Rauser, L., McBride, A., Hufeisen, S. J., and Roth, B. L. (2000). Evidence for possible involvement of 5-HT(2B) receptors in the cardiac valvulopathy associated with fenfluramine and other serotonergic medications. Circulation
11. Silberstein, S. D., and McCrory, D. C. (2003). Ergotamine and dihydroergotamine: History, pharmacology, and efficacy. Headache
12. Taylor, A. C., Beerahee, A., Citerone, D. R., Cyronak, M. J., Leigh, T. J., Fitzpatrick, K. L., Lopez-Gil, A., et al. (1999). Lack of a pharmacokinetic interaction at steady state between ropinirole and
13. Unett, D. J., Gatlin, J., Anthony, T. L., Buzard, D. J., Chang, S., Chen, C., Chen, X., et al. (2013). Kinetics of 5-HT2B receptor signaling: Profound agonist-dependent effects on signaling onset and duration. J Pharmacol Exp Ther
14. Wacker, D., Wang, C., Katritch, V., Han, G. W., Huang, X. P., Vardy, E., McCorvy, J. D., Jiang, Y., et al. (2013). Structural features for functional selectivity at serotonin receptors. Science
15. de Groot, A. N., Vree, T. B., Hekster, Y. A., van den Biggelaar-Martea, M., van Dongen, P. W. J., and van Roosmalen, J. (1994). Pharmacokinetics and bioavailability of oral ergometrine in male volunteers. Biopharm Drug Dispos
16. Bredberg, U., Eyjolfsdottir, G. S., Paalzow, L., Tfelt-Hansen, P., and Tfelt-Hansen, V. (1986). Pharmacokinetics of methysergide and its metabolite methylergometrine in man. Eur J Clin Pharmacol
17. FDA Label for Methylergonovine. Accessed May 11, 2016. http://www.accessdata.fda.gov/drugsatfda_docs/label/2012/006035s078lbl.pdf.
IP = inositol phosphate; NFAT = nuclear factor of activated T cells.
aRoute of administration is oral unless otherwise indicated (i.m. = intramuscular) and is expressed as mg/day.
bAffinity for the receptor determined by radioligand competition binding assay. For ease of comparison against fC max (expressed in nM), Ki values noted from the literature are converted to nM.
cAn average 30% plasma protein binding is used for calculating ratio.
dPlasma protein bound data are not available. C max total was used to calculate the human plasma levels for ergonovine and methylergonovine.
These findings suggest that the experimental conditions or protocols for determining 5-HT2B agonist activity in cell-based functional assays are somewhat variable, and efforts to address these issues appear to be limited due to lack of available resources within various research groups both public and private (Cavero and Guillon 2014). Importantly, current cell-based functional assays alone for derivation of EC50 values do not appear to be able to distinguish between 5-HT2B agonists that produce VHD in humans from those agonists that do not (Table 1). In light of the demonstrated variability in current cell-based functional assays, consideration of additional factors, such as inherent binding constants (Ki ), maximum plasma free drug concentrations (fC max), and duration of exposure (weeks to months), is necessary to assess whether a drug poses a significant risk for development of VHD in humans.
To explore this issue in more detail, we examined whether reported binding affinity data (Ki ) for 5-HT2B receptors, Ki relative to human fC max, and Ki relative to 5-HT (serotonin; Fitzgerald et al. 2000; Huang et al. 2009; Unett et al. 2013) would more closely correlate with the potential of a compound to be valvulopathic when compared to data from cell-based functional assays. We found that for all of the known 7 valvulopathic compounds, the Ki values for the 5-HT2B receptor are close to or less than an order of magnitude (i.e., <10×) relative to the Ki values of 5-HT (Table 2). In contrast, there is a >100-fold difference in Ki values for the nonvalvulopathic 5-HT2B agonist ropinirole relative to the Ki values of 5-HT, even though results of functional assays give a strong signal for it to be valvulopathic (Table 1). Further, there did not appear to be a good correlation between a drug’s Ki and its fC max values (Ki /fC max ratio) for predicting whether a drug has the potential to produce VHD (Table 2).
Relationship between (1) Affinity to Human 5-Hydroxytryptamine (serotonin) Receptor 2B (5HT2B) (Ki ), (2) Ki /Human fC max Ratio, and (3) Ki /5HT Ratio for 5HT2B Agonists as a Function of Risk for VHD.

Note. Reference citations for published data values are in parentheses.
References for Table 2:
1. Blin, O. (2003). The pharmacokinetics of pergolide in Parkinson’s disease. Curr Opin Neurol
2. Belmer, A., Doly, S., Setola, V., Banas, S. M., Moutkine, I., Boutourlinsky, K., Kenakin, T., and Maroteaux, L. (2014). Role of the N-terminal region in G protein-coupled receptor functions: Negative modulation revealed by 5-HT2B receptor polymorphisms. Mol Pharmacol
3. Caccia, S., Conforti, I., Duchier, J., and Garattini, S. (1985). Pharmacokinetics of fenfluramine and norfenfluramine in volunteers given
4. Del Dotto, P., and Bonuccelli, U. (2003). Clinical pharmacokinetics of cabergoline. Clin Pharmacokinet
5. FDA Briefing Document NDA 22529. (2012). Lorcaserin hydrochloride tablets, 10 mg. sponsor, arena pharmaceuticals. Endocrinologic and Metabolic Drugs Advisory Committee Meeting. Accessed May 10, 2012. www.fda.gov/downloads/advisorycommittees/committeesmeetingmaterials/drugs/endocrinologicandmetabolicdrugsadvisorycommittee/ucm225631.pdf.
6. Huang, X. P., Setola, V., Yadav, P. N., Allen, J. A., Rogan, S. C., Hanson, B. J., Revankar, C., et al. (2009). Parallel functional activity profiling reveals valvulopathogens are potent 5-hydroxytryptamine(2B) receptor agonists: Implications for drug safety assessment. Mol Pharmacol
7. Humbert, H., Cabiac, M. D., Dubray, C., and Lavene, D. (1996). Human pharmacokinetics of dihydroergotamine administered by nasal spray. Clin Pharmacol Ther
8. Kaye, C. M., and Nicholls, B. (2000). Clinical pharmacokinetics of ropinirole. Clin Pharmacokinet
9. Millan, M. J., Maiofiss, L., Cussac, D., Audinot, V., Boutin, J. A., and Newman-Tancredi, A. (2002). Differential actions of antiparkinson agents at multiple classes of monoaminergic receptor. I. A multivariate analysis of the binding profiles of 14 drugs at 21 native and cloned human receptor subtypes. J Pharmacol Exp Ther
10. Kvernmo, T., Hartter, S., and Burger, E. (2006). A review of the receptor-binding and pharmacokinetic properties of dopamine agonists. Clin Ther
11. Rothman, R. B., Baumann, M. H., Savage, J. E., Rauser, L., McBride, A., Hufeisen, S. J., and Roth, B. L. (2000). Evidence for possible involvement of 5-HT(2B) receptors in the cardiac valvulopathy associated with fenfluramine and other serotonergic medications. Circulation
12. Silberstein, S. D., and McCrory, D. C. (2003). Ergotamine and dihydroergotamine: History, pharmacology, and efficacy. Headache
13. Tfelt-Hansen, P., and Paalzow, L. (1985). Intramuscular ergotamine: Plasma levels and dynamic activity. Clin Pharmacol Ther
14. Thomsen, W. J., Grottick, A. J., Menzaghi, F., Reyes-Saldana, H., Espitia, S., Yuskin, D., Whelan, K., et al. (2008). Lorcaserin, a novel selective human 5-hydroxytryptamine2C agonist: in vitro and in vivo pharmacological characterization. J Pharmacol Exp Ther
15. Unett, D. J., Gatlin, J., Anthony, T. L., Buzard, D. J., Chang, S., Chen, C., Chen, X., et al. (2013). Kinetics of 5-HT2B receptor signaling: Profound agonist-dependent effects on signaling onset and duration. J Pharmacol Exp Ther
16. de Groot, A. N., Vree, T. B., Hekster, Y. A., van den Biggelaar-Martea, M., van Dongen, P. W., and van Roosmalen, J. (1994). Pharmacokinetics and bioavailability of oral ergometrine in male volunteers. Biopharm Drug Dispos
17. Bredberg, U., Eyjolfsdottir, G. S., Paalzow, L., Tfelt-Hansen, P., and Tfelt-Hansen, V. (1986). Pharmacokinetics of methysergide and its metabolite methylergometrine in man. Eur J Clin Pharmacol
18. FDA Label for Methylergonovine. Accessed May 11, 2016. http://www.accessdata.fda.gov/drugsatfda_docs/label/2012/006035s078lbl.pdf.
aRoute of administration is oral unless otherwise indicated (i.m. = intramuscular) and is expressed as mg/day.
bAffinity for the receptor was determined by radioligand competition binding assay. For ease of comparison against fC max (expressed in nM), Ki values noted from the literature are converted to nM.
cAn average 30% plasma protein binding is used for calculating ratio.
dPlasma protein bound data are not available. C max total was used to calculate human plasma levels for ergonovine and methylergonovine.
e Ki values used for 5-HT were 2.5 nM (15) and 3.9 nM (2,11).
For compounds whose Ki lies between 10-fold and 100-fold of 5-HT, such as lorcaserin, additional testing or clinical monitoring may still be required. Although the current label for lorcaserin contains a warning about possible valvulopathy based on in vitro binding data and early clinical trials, the postmarketing database of spontaneous MedWatch reports submitted to the FDA thus far do not add additional evidence of VHD with its use (unpublished data). The adjusted reporting ratio or data mining signal scores of the association of this drug with VHD overlap the ones from known negative controls. In addition, all the cases reported describe underlying conditions and risk factors, including cardiovascular diseases, diabetes, and obesity in elderly patients that preclude making, thus far, a causal relationship with this drug (Szarfman, Machado, and O’Neill 2002).
Taken together, and in general agreement with recent reports (Cavero and Guillon 2014; Whitebread et al. 2016), these sets of data, although limited, suggest that the inherent binding potency (i.e., Ki ) of a compound for the 5-HT2B receptor, and in relation to that of 5-HT, gives a better indication of VHD risk than does potency of functional activity (i.e., EC50 or EC50/human fC max; Table 1), provided a compound is identified as a 5-HT2B agonist in cell-based assays and is intended to be administered for a prolonged period of time (i.e., weeks to months or longer).
In summary, given the current difficulty in using functional assays to distinguish known valvulopathic drugs from those not known to produce VHD, and the inconsistency in deriving SMs based on EC50/human fC max ratios, use of 5-HT2B receptor affinity or that relative to 5-HT should be considered as an initial risk assessment for VHD. To minimize variability, such binding assays for 5-HT2B may choose to include 5-HT as an internal control to better correlate a drug’s Ki to the Ki of 5-HT. Although the Ki of a drug relative to 5-HT provides the same degree of prediction as the drug’s Ki itself, having a value relative to 5-HT may provide a useful reference point for assessing potency (Table 2).
These recommendations for assessing human risk are based on the assumptions that for a drug to be considered a possible risk for VHD under therapeutic conditions, it (1) shows an affinity to 5-HT2B that is within an order of magnitude (<10×) to that of 5-HT, (2) is positive for 5-HT2B agonist activity in more than one cell-based functional assay, and (3) is indicated for long-term clinical use (i.e., weeks to months or longer). From a regulatory perspective, additional data or analyses may be needed to confirm these correlations, and ongoing efforts to standardize experimental conditions or protocols for determining 5-HT2B agonist activity in a consistent and reproducible manner in human cell-based functional assays should continue. Relatively simple and reproducible cell-based assays can provide meaningful information on the degree of 5-HT2B agonist activity of a potential therapeutic that can then be better correlated with in vivo exposure once that data become available.
Possible Utility of in Silico Predictions
In silico methods based on chemoinformatics, shape signatures, or quantitative structure–activity relationships have the potential to offer a faster and more efficient method for predicting drugs with 5-HT2B receptor binding activity with a relatively high degree of confidence (Reid, Kumar, and Wang 2013; Garcia-Serna et al. 2015). Further, their ability to determine whether a drug is an agonist or not appears to depend on particular motifs of the molecule (Reid, Kumar, and Wang 2013).
In a regulatory setting, in silico approaches are currently being used to predict possible adverse drug events postmarketing (Duggirala et al. 2015). However, their utility for predicting off-target drug activity preclinically, or before human testing is initiated, has yet to be thoroughly evaluated. Such approaches, if feasible and validated, would allow early regulatory assessments of human risk beyond use of current in vitro secondary pharmacology studies. It could prove particularly useful for new molecular entities, first-in-class drugs, or drugs with active metabolites. In cases where binding or functional data for the 5-HT2B are not available or difficult to obtain, in silico prediction methods may prove useful for assessing this critical target in certain situations.
Conclusions
There is general consensus that in vitro secondary pharmacology data provide a wealth of information relating to a drug’s potential to produce pharmacodynamic activity unrelated to its intended mechanism of action. Such information is commonly generated by pharmaceutical companies during early drug development as a general screen for candidate drug selection and to assess potential safety risks of any selected drugs submitted to regulatory agencies for proposed investigational use in humans.
Drugs with agonist activity for the 5-HT2B possess a clear risk for producing VHD, and it is essential that appropriate and relatively simple in vitro screening for this unwanted activity be conducted early. However, there does not appear to be a clear consensus on what constitutes an appropriate SM for drugs with 5-HT2B agonist properties, in contrast to what others have proposed for drugs that block the cardiac hERG/IKr and hNav1.5 channels (Redfern et al. 2003; Harmer, Valentin, and Pollard 2011). This difference may be attributable to inconsistencies in the various methods and end points assessed in 5-HT2B-mediated cell-based functional assays among different laboratories, lack of standardized protocols, or simply due to inherent variability in cellular functional responses assessed in vitro. Although based on a limited data set of 5-HT2B agonists with actual documented clinical experience, we believe that use of SMs based on Ki values from binding assays or those relative to 5-HT Ki values appears to be a better predictor than use of EC50 or EC50/human fC max values from cellular functional assays for determining whether known 5-HT2B agonists have resulted in VHD in humans. Although no clear cutoff value for an appropriate SM can be extrapolated from available published data, and other factors (e.g., intended duration of use and real or projected in vivo fC max values) need to be considered, use of 5-HT2B receptor affinity (Ki ) or that relative to 5-HT may prove useful for initial assessments of risk until cell-based functional assays for 5-HT2B receptor agonist activity become standardized to allow more reliable use of EC50/human fC max ratio.
Footnotes
Acknowledgments
We wish to thank Drs. Haw-Jyh Chiu and Shwu-Luan Lee for constructive comments.
Authors’ Contribution
All authors (TP, JG, MS, NS, AR, BY, AL, IK, and AS) contributed to conception or design; data acquisition, analysis, or interpretation; drafting the manuscript; and critically revising the manuscript. All authors gave final approval and agreed to be accountable for all aspects of work in ensuring that questions relating to the accuracy or integrity of any part of the work are appropriately investigated and resolved.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.