
Abstract
Naphthoquine phosphate (NP) was considered as a partner drug with a promising antimalarial drug candidate. Here we report unexpected adverse clinical signs and microscopic findings in a canine pilot toxicology study with NP. Male and female dogs were dosed daily by oral gavage with NP at 2, 10, or 50 mg/kg/day for a maximum of 14 days. NP was not tolerated at ≥10 mg/kg/day; several animals were sacrificed in moribund condition and marked neurological clinical signs were noted at 50 mg/kg/day. The main microscopic observation was central nervous system vasculocentric inflammation (mainly lymphocytes and macrophages) in the white and gray matter of various regions of the brain at ≥2 mg/kg/day and at lower incidence in the spinal cord at ≥10 mg/kg/day. Vasculocentric microscopic changes predominantly centered on the centrilobular vein were also observed in the liver at ≥2 mg/kg/day. Females were more sensitive than males with comparable NP plasma exposure. In conclusion, under the conditions of this study, the administration of NP to dogs via daily oral gavage for up to 2 weeks was not tolerated causing moribundity, marked neurological clinical signs, and vasculocentric microscopic changes in the central nervous system and the liver.
Malaria is caused by a protozoan parasite of the genus Plasmodium transmitted by the bite of anopheles mosquitoes. There are 5 known species of Plasmodium parasites that affect humans, the most important being Plasmodium falciparum and Plasmodium vivax (Muller and Hyde 2010; White, Pukrittayakamee, Hien et al. 2014 ; World Health Organization [WHO] 2014). Malaria is endemic in 97 countries and territories, and it is estimated that 198 million cases of malaria and 584,000 malaria-related deaths occurred globally in 2013 (WHO 2014). Malaria is most common and lethal in Africa and particularly impacts children under 5 years of age (Muller and Hyde 2010; White, Pukrittayakamee, Hien et al. 2014; WHO 2014).
Chloroquine was discovered more than 75 years ago and has been the drug of choice to treat uncomplicated malaria for decades (Jensen and Mehlhorn 2009). Resistance to chloroquine and later to sulfadoxine–pyrimethamine has resulted in the current recommended treatment of artemisinin (AT)-based combination therapies (ACTs) for uncomplicated malaria in most areas of the world (WHO 2010). There is increasing evidence of the emergence of resistance to ACT in some areas of the world (Amaratunga et al. 2012; Breman 2012), so novel antimalarial therapies are required.
Naphthoquine phosphate (NP) is a tetra-aminoquinoline antimalarial drug registered in China in 1993 with structural similarities to chloroquine (Wang et al. 2004). It is reported to have good parasiticidal activity in various plasmodia, including those resistant to chloroquine (Wang et al. 2004). Like other quinoline antimalarial compounds, the exact mode of action is incompletely understood although many mechanisms such as inhibition of protein synthesis, hemozoin formation, hemoglobin transport vesicle trafficking, food vacuole phospholipases, and aspartic proteinases have been proposed (Roberts et al. 2008; O’Neill 2012). NP is currently marketed by ARCO (Kunming Pharmaceutical, Kunming, China) as part of a combination treatment for malaria containing 78.3 mg of NP (equivalent to 50 mg of naphthoquine) and 125 mg of AT in each tablet (Qu et al. 2010; Benjamin et al. 2012; Meremikwu et al. 2012; Liu et al. 2013). NP has a slow onset of action and a relatively long half-life of approximately 10 days (Hombhanje and Quigyun 2010), with relatively few reported adverse events (Hombhanje et al. 2009; Toure et al. 2009; Tun et al. 2009; Hombhanje and Quigyun 2010; Qu et al. 2010; Benjamin et al. 2012; Meremikwu et al. 2012; Liu et al. 2013).
Due to its reported favorable pharmacokinetic, therapeutic, and toxicology characteristics, NP was considered as a partner drug for a combination therapy with KAE609, a promising antimalarial drug candidate (White, Pukrittayakamee, Phyo et al. 2014). A 17.5-mg/kg daily oral dose of NP in combination with AT (containing 7 mg of NP) for 14 days was reported to be safe (Wang et al. 2004). In the same publication, doses of 87.5 mg/kg/day (35 mg of NP) and 140 mg/kg/day (56 mg of NP) were cited as toxic to the bone marrow (erythroid series) and the liver. In patients receiving NP alone or in combination with AT, elevated alanine aminotransferase and aspartate aminotransferase enzyme activities were also observed (Wang et al. 2004).
The Novartis Institutes for Biomedical Research, Inc. (Cambridge, MA) sponsored a repeat dose study (oral gavage) in dogs for a planned duration of 14 days to better understand (a) the toxicity profile of NP and associated dose response, (b) any toxicities specific to the dog and the associated exposure relative to the clinical efficacious exposure, and (c) the need for additional combination toxicology studies to further the development of a combination therapy with KAE609 and NP.
Here we report the unexpected adverse clinical signs and associated microscopic findings observed in this dog pilot toxicology study. NP was not tolerated at 10 and 50 mg/kg/day with marked neurological clinical signs at 50 mg/kg/day and moribundity at ≥10 mg/kg/day resulting in early euthanasia. In addition, central nervous system and hepatic vasculocentric inflammation were noted in females dosed with ≥2 mg/kg/day and males dosed with ≥10 mg/kg/day.
Material and Method
Test Article and Dose Formulations
NP (batch no. 1003103, molecular weight 605.94 g/mol [C24H28ClN3O·2H3PO4]) was obtained from Shanghai New Hualian, China. Content was confirmed by Novartis Pharma, Basel, Switzerland. NP was stored at room temperature and used as supplied.
Dose formulations were prepared as suspension of NP in 0.5% (w/v) methylcellulose (Type 400 cps; Sigma-Aldrich, St. Louis, MO) and 0.5% (v/v) polysorbate 80 (J.T. Baker, Avantor, Center Valley, PA), NF (Tween® 80) in reverse osmosis water at concentrations of 0 (vehicle control), 0.4, 2, and 10 mg/ml. NP concentrations were corrected for lot-specific drug content (93.1%) and salt content (salt/base ratio = 1.478), using a total correction factor of 1.588 on the free base weight.
Animals and Treatment
The study was conducted in accordance with the Animal Welfare Act, the Guide for the Care and Use of Laboratory Animals, and the Office of Laboratory Animal Welfare. The study was conducted at Covance Laboratories, Inc. (Madison, WI); the study protocol was reviewed and approved by both the Covance and Novartis Animal Care and Use Committees.
Animals were obtained from Marshall BioResources (North Rose, NY), and at the initiation of dosing were approximately 14 months of age and weighed 7.8 to 10.1 kg.
One male and 1 female dog were assigned to the vehicle control group (0 mg/kg/day), low-dose group (2 mg/kg/day), and mid-dose group (10 mg/kg/day), while 2 males and 2 females were assigned to the high-dose group (50 mg/kg/day). Dose groups were staggered with regard to the first day of dosing, beginning with the lowest dose. Control animals were dosed concurrently with the 10-mg/kg/day group.
All dogs were dosed daily at approximately 8:00 a.m. by oral gavage at a dose volume of 5 ml/kg. Clinical signs were recorded daily, and all animals were evaluated twice daily (a.m. and p.m.) for mortality, abnormalities, and signs of pain or distress. Body weights were recorded on all surviving dogs on days 1, 4, 8, 11, and 15. Quantitative food consumption was determined on days 1 to 4, 4 to 8, 8 to 11, and 11 to 15 (survival permitting). The 2 surviving high-dose animals had detailed observations, body weights, and food consumption collected on day 3 of the dosing phase prior to unscheduled euthanasia. Electrocardiograms (ECGs) were collected using jacketed external telemetry predose and on days 1, 4, and 13 (survival permitting).
Blood samples for toxicokinetic analysis were collected via the jugular vein on days 1, 4, and 13 approximately 0.5-, 1-, 3-, 7-, and 24-hr postdose (plasma was frozen at or below −60°C). Blood samples were collected for hematology, clinical chemistry, and coagulation assessments once during the predose phase for all animals and on day 15 (prior to scheduled euthanasia), survival permitting. Blood samples were also collected from animals euthanized early with the exception of the high-dose male euthanized on study day 2.
All animals were weighed immediately prior to necropsies. All animals (including those euthanized early) were necropsied, and tissues were collected in 10% neutral-buffered formalin (adrenal, aorta, brain, cecum, cervix, colon, duodenum, epididymis, esophagus, femur with bone marrow [articular surface of the distal end], gall bladder, heart, ileum, jejunum, kidney, lacrimal gland, larynx, liver, lung, lymph nodes, mammary gland, mesentery, skeletal muscle, ovary, pancreas, pituitary gland, prostate, rectum, salivary gland, sciatic nerve, skin/subcutis, spinal cord [cervical, thoracic, and lumbar], spleen, sternum with bone marrow, stomach, thymus, thyroid/parathyroid, tongue, trachea, ureter, urinary bladder, uterus, and vagina) or modified Davidson’s fixative (eye, optic nerve, testis). Tissues were then embedded in paraffin, processed to slides, stained with hematoxylin and eosin, and examined microscopically. The brain was trimmed according to a method adapted from that published by Bolon et al. (2013). Three coronal (transverse) levels were sampled using anatomic landmarks on the ventral aspect of the brain: level 1 was sampled between the optic chiasma and the mammillary body, level 2 was sampled rostral to the pons, and level 3 was sampled through the pons/medulla to obtain a suitable representation of the cerebellum. From each level, the left hemisphere was subsequently sampled. Due to the large size of the dog brain, the hemisphere from levels 1 and 2 was subdivided in a dorsal section (1a and 2a) and ventral section (1b and 2b) for a total of 5 brain sections per animal.
In addition, formalin-fixed, paraffin-embedded brain and liver tissue were cut at 5 μm, mounted on glass slides, and submitted for special stains or immunohistochemistry (IHC) as described below.
All slides submitted for special stains were baked at 60°C for 1 hr, deparaffinized, and hydrated prior to stain. Perl’s Prussian blue stain for detection of iron was performed by immersing slides in potassium ferrocyanide/hydrochloric solution for 30 min, rinsing in deionized (DI) water, and counterstaining with nuclear fast red. The long Ziehl–Neelsen stain for detection of lipofuscin was performed by incubating slides at 56°C for 3 hr in carbol fuchsin, rinsing in DI water, differentiating in acid alcohol, and counterstaining with light hematoxylin. Hall’s bilirubin stain was performed by immersing slides in trichloroacetic acid/ferric chloride solution (Fouchet’s reagent) for 15 min, rinsing in running water, and counterstaining with nuclear fast red. Antibodies used for IHC are detailed in Table 1. All IHC was performed with the Ventana XT Discovery Platform (Ventana Medical System, Tucson, AZ). Slides were baked at 60°C for 1 hr prior to loading on the Ventana XT. Slides were deparaffinized using EZ Prep (Ventana Medical System, Tucson, AZ; cat#950-100) followed by heat-induced epitope retrieval at varying times with Cell Conditioning 1 (CC1; cat#950-124; pH 8 at 95–100°C). Primary antibodies were incubated for 60 min, followed by secondary incubation with Omnimap Multimer (Ventana Medical System, Tucson, AZ; cat#760-4310, OmniMap anti-MS HRP; cat#760-4311, OmniMap anti-Rb HRP) and detected with ChromoMap 3,3-diaminobenzidine kit (Ventana Medical System, Tucson, AZ; cat#760-159). Hematoxylin and bluing (cat#760-2021, cat#760-2037) were used as counterstains.
Immunohistochemical Methods for Characterization of Brain Microscopic Changes.
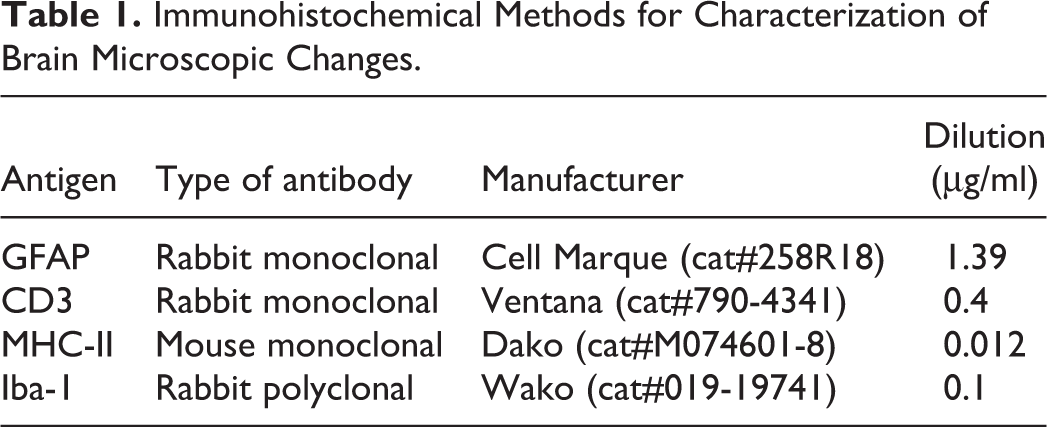
In all instances, microscopic findings were attributed a severity grade with regard to the intensity and extent of the change in their respective organ as follows: 1 = minimal, 2 = slight, 3 = moderate, 4 = marked, and 5 = severe.
Plasma Concentration Analysis of Naphthoquine
All samples were maintained frozen at or below −60°C prior to analysis. Plasma samples were processed using protein precipitation with an acetonitrile/methanol mixture. Samples were centrifuged and analysis was performed by liquid chromatography tandem-mass spectrometry (LC-MS/MS), using positive atmospheric pressure chemical ionization as an interface. The calibration curve was valid from 0.500 to 10,000 ng/ml using 0.0400 ml of plasma. Calibration standards were within the range of ±30% at all concentration levels. Quality control (QC) samples were within the range of ±30% for at least 2 (of 3) of the individual values. At least 1 value at each QC level fulfilled the acceptance criteria.
The C max (highest plasma concentration) was reported for the male and female dog in each group individually or as the mean of the 2 animals for the high-dose group. Similarly, the time the C max value was observed was reported as the T max value individually or as the mean values for the high-dose group. Total exposure was calculated for each animal as the area under the plasma curve from time 0- to 24-hr postdose (AUC0–24hr) using the trapezoidal rule and reported individually or as the mean of the 2 males and females in the high-dose group.
Results
Clinical Observations
Animals dosed with 10 or 50 mg/kg/day were euthanized before the end of the study based on clinical observations. Following the second daily dose of 50 mg/kg/day, 1 male and 1 female had convulsions with a combination of ataxia, recumbency, excessive salivation, and mucoid feces, resulting in early termination that same day. The 2 remaining animals given 50 mg/kg/day were euthanized on day 3, as further dosing was not considered appropriate based on the adverse NP-related clinical observations seen at this dose level and the presence of notably elevated body temperature which may have been secondary to convulsions that were not observed clinically. Both dogs dosed with 10 mg/kg/day lost substantial body weight by day 11 (0.6 or 0.7 kg from day 1), and food consumption during the period of day 8 through 11 was considerably reduced compared with earlier intervals and with the concurrent vehicle control group. Clinical observations of vomiting and warm to touch began to appear on day 10. Based on this pattern of effects, animals were not dosed on day 11 and were euthanized. Both animals dosed with 2 mg/kg/day survived to scheduled euthanasia on day 15. The only clinical observations noted for this dose were reduced food consumption and slight body weight loss (0.5 kg) in the female from day 11 to 15. As the control animals were dosed on the same schedule as animals dosed with 10 mg/kg/day, they were also euthanized on day 11.
No NP-related changes were observed in ECG parameters (PR interval, QRS duration, QT interval, corrected QT interval, or heart rate), and no rhythm abnormalities or qualitative time- or dose-dependent changes attributed to NP were observed during qualitative assessment of the ECGs.
Clinical Pathology
The only hematology or coagulation changes considered NP related were observed in the 50 mg/kg/day female animal euthanized on day 2. These findings included mildly increased red cell mass (hematocrit was +9% relative to baseline) and absolute neutrophil count (+52% relative to baseline) and mildly decreased absolute lymphocyte and eosinophil counts (−34% and −71% relative to baseline, respectively). Increased red cell mass was consistent with splenic contraction, and the leukocyte changes were consistent with a stress response from endogenous catecholamine and glucocorticoid release. These effects were likely secondary to the convulsions observed prior to euthanasia.
The only NP-related clinical chemistry findings in this study were noted in the same 50 mg/kg/day female animal euthanized on day 2 and were also likely secondary to convulsions. These findings included moderately decreased glucose concentration (−67% relative to baseline), moderately increased aspartate aminotransferase and creatine kinase activities (5.0- and 8.4-fold above baseline, respectively), and moderately decreased inorganic phosphorus and potassium concentrations (−28% and −31% relative to baseline, respectively). The decreased glucose concentration and increased muscle enzyme activities were consistent with glucose consumption and muscle injury associated with convulsive activity. Specific mechanisms for the changes in inorganic phosphorus and potassium concentrations were not apparent, but none of the other NP-dosed animals exhibited similar changes.
Necropsy
There were no NP-related macroscopic changes noted in this study.
Histopathology
The main NP-related microscopic change consisted of minimal to marked vasculocentric inflammation in the central nervous system and adjacent meninges in females dosed with ≥2 mg/kg/day and males dosed with ≥10 mg/kg/day with occasional hemorrhages (Table 2). Central nervous system inflammation was characterized by lymphocytes and macrophages with rare neutrophils filling and expanding the Virchow–Robin space and frequently obscuring the vessel wall with multifocal, generally perivascular, areas of increased cellularity (infiltrative inflammatory cells and gliosis; Figure 1). These changes were observed in both the white and gray matter of various brain regions as presented in Table 3, but they were most commonly found in the midbrain and brain stem. In 2 of 4 females dosed with NP, similar vasculocentric inflammatory changes and/or focal areas of gliosis were noted in the white and gray matter of the spinal cord (cervical, thoracic, and lumbar sections; Figure 1), and similar focal, sparsely distributed, vasculocentric areas of inflammation were observed in the meninges in 3 female animals (Table 3). Although microscopic changes were consistently centered on the vasculature, no morphological evidence of direct vascular damage, such as fibrinoid necrosis or pyknotic debris, was observed. Microscopic findings in the brain of the 2 male animals administered 50 mg/kg/day were generally of lesser severity and more acute in nature (with an increased proportion of neutrophils in the inflammatory infiltrate) when compared with animals euthanized on days 11 to 15 presumably due to the short duration (2–3 days) of treatment of these animals. However, 1 female administered 50 mg/kg/day and euthanized on study day 3 exhibited marked inflammation of a similar nature and distribution to those euthanized later. The cellular composition of the inflammatory infiltrate was confirmed by immunohistochemical labeling with cluster of differentiation 3 (CD3; lymphocytes) and major histocompatibility complex class II (Figure 2). In addition, a microglial reaction was demonstrated by ionizing calcium-binding adaptor molecule 1 (Iba-1) immunohistochemical staining (Figure 2). No differences in the nature, intensity, or pattern of glial fibrillary acidic protein (GFAP) labeling (astrocytes) were noted in affected animals as compared with controls (Figure 2).
Group Incidence and Severity of Naphthoquine Phosphate–related Microscopic Findings in Central Nervous System and Liver.
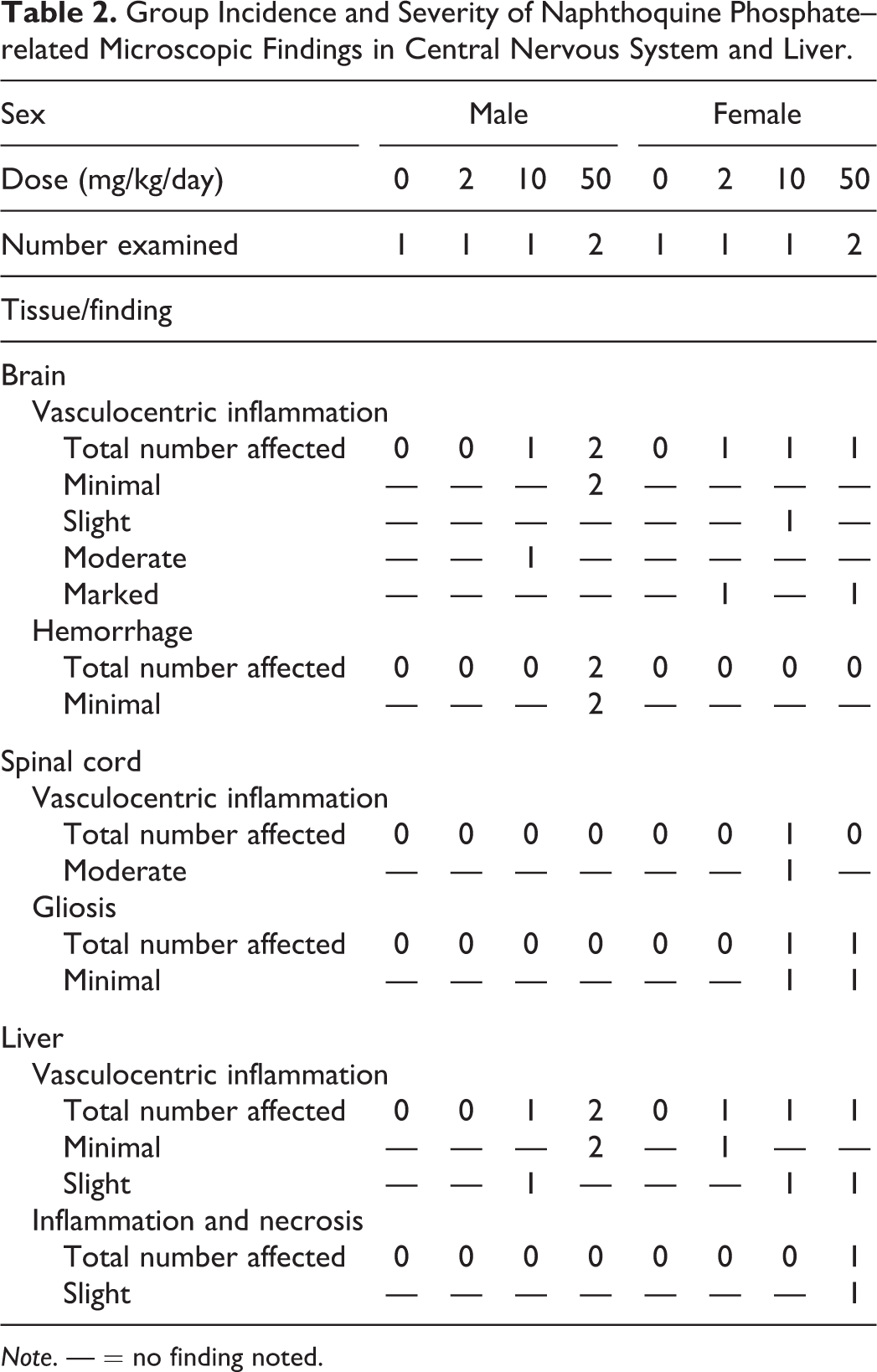
Note. — = no finding noted.
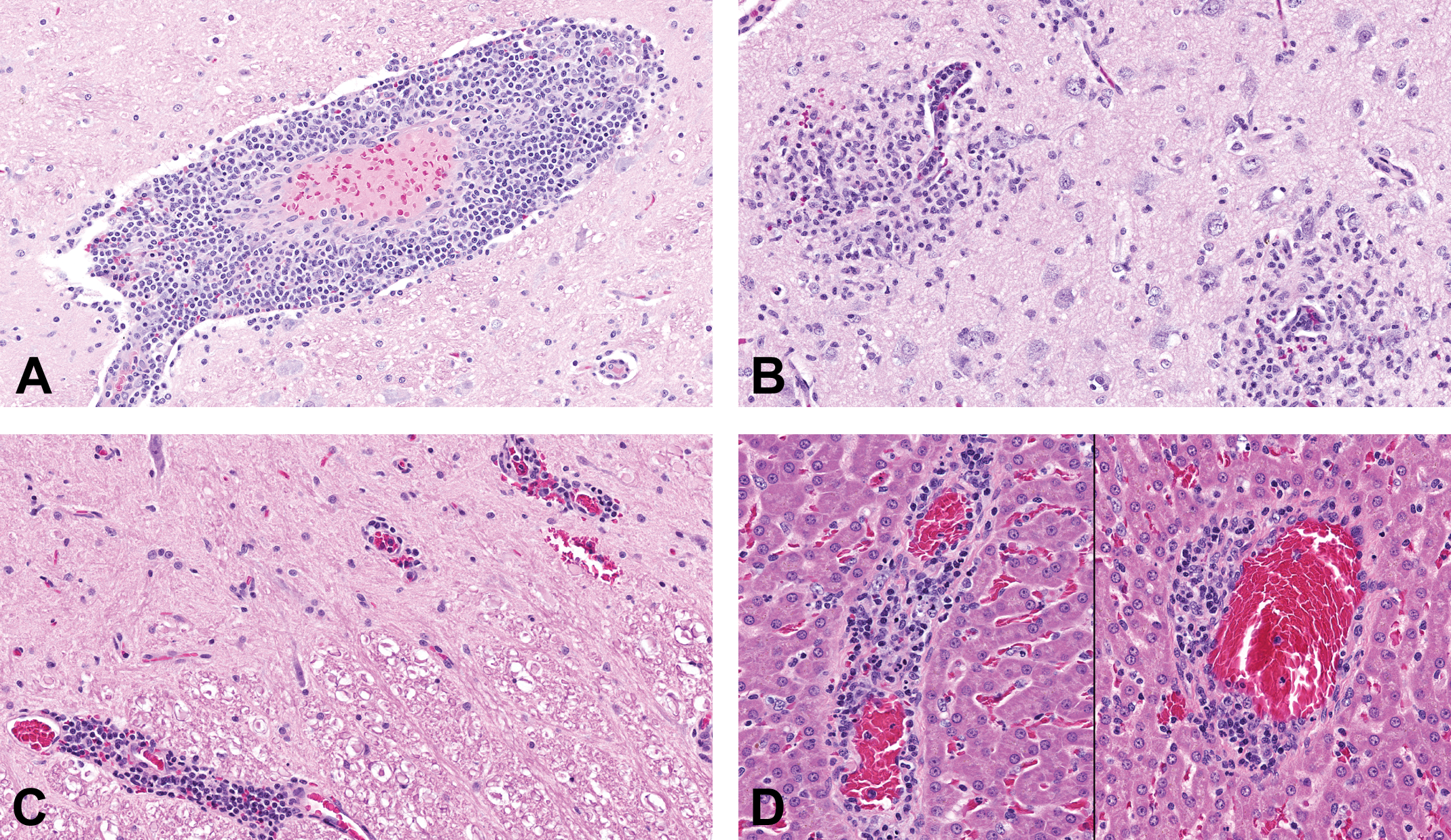
Representative hematoxylin and eosin–stained section of vasculocentric inflammation in the brain, spinal cord and, liver. (A) Thalamus of a female dog administered naphthoquine phosphate (NP) at 2 mg/kg/day for 14 days. (B) Pyriform lobe of a female dog administered NP at 10 mg/kg/day for 11 days (preterminally euthanized) with significant inflammation and gliosis in the neighboring neuropil. (C) Spinal cord of a female dog administered NP at 10 mg/kg/day for 11 days (preterminally euthanized). (D) Centrilobular veins (liver) of a female dog administered NP at 50 mg/kg/day for 2 days.
Distribution and Severity of Naphthoquine Phosphate–related Microscopic Findings in the Brain.
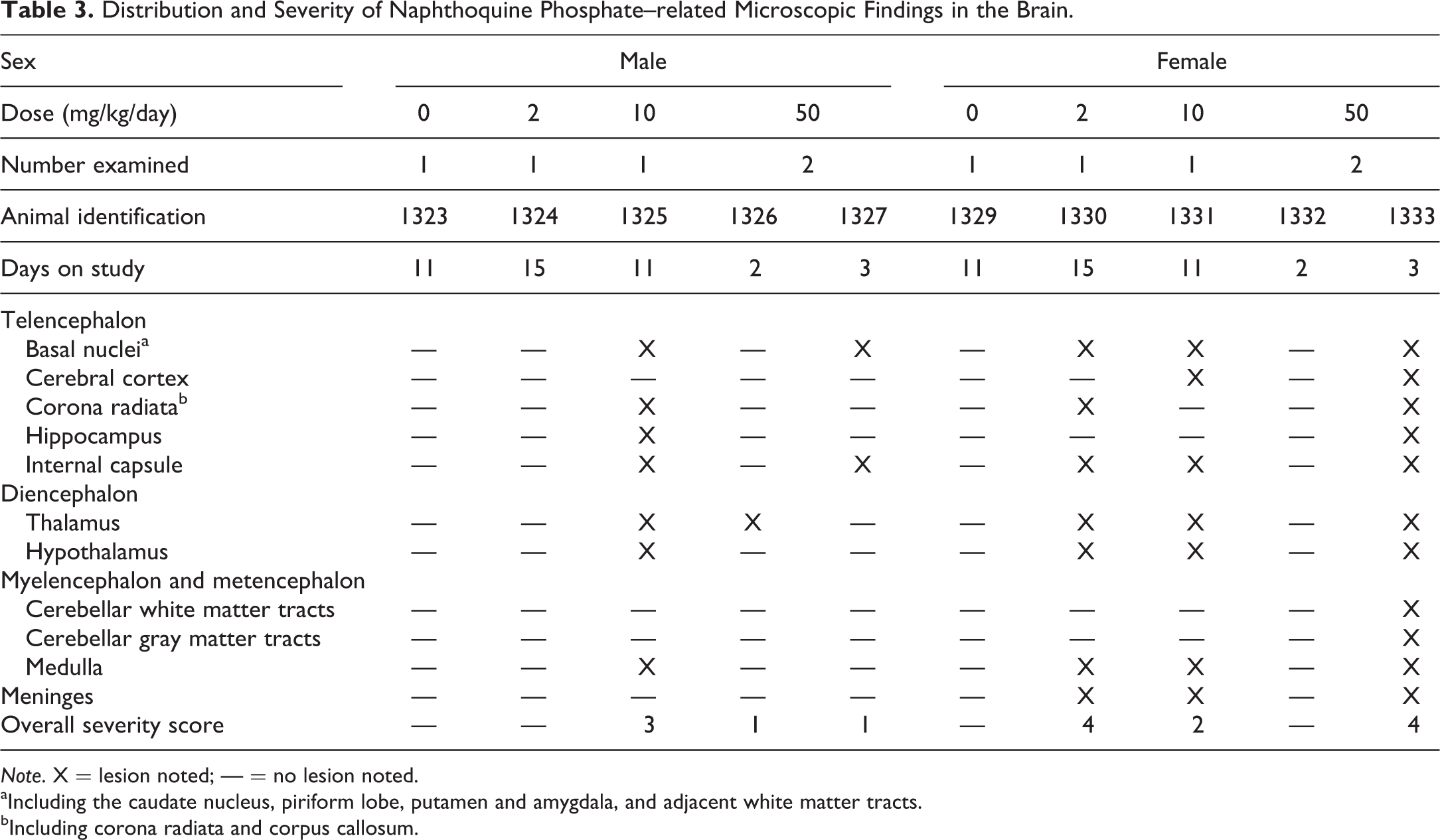
Note. X = lesion noted; — = no lesion noted.
aIncluding the caudate nucleus, piriform lobe, putamen and amygdala, and adjacent white matter tracts.
bIncluding corona radiata and corpus callosum.
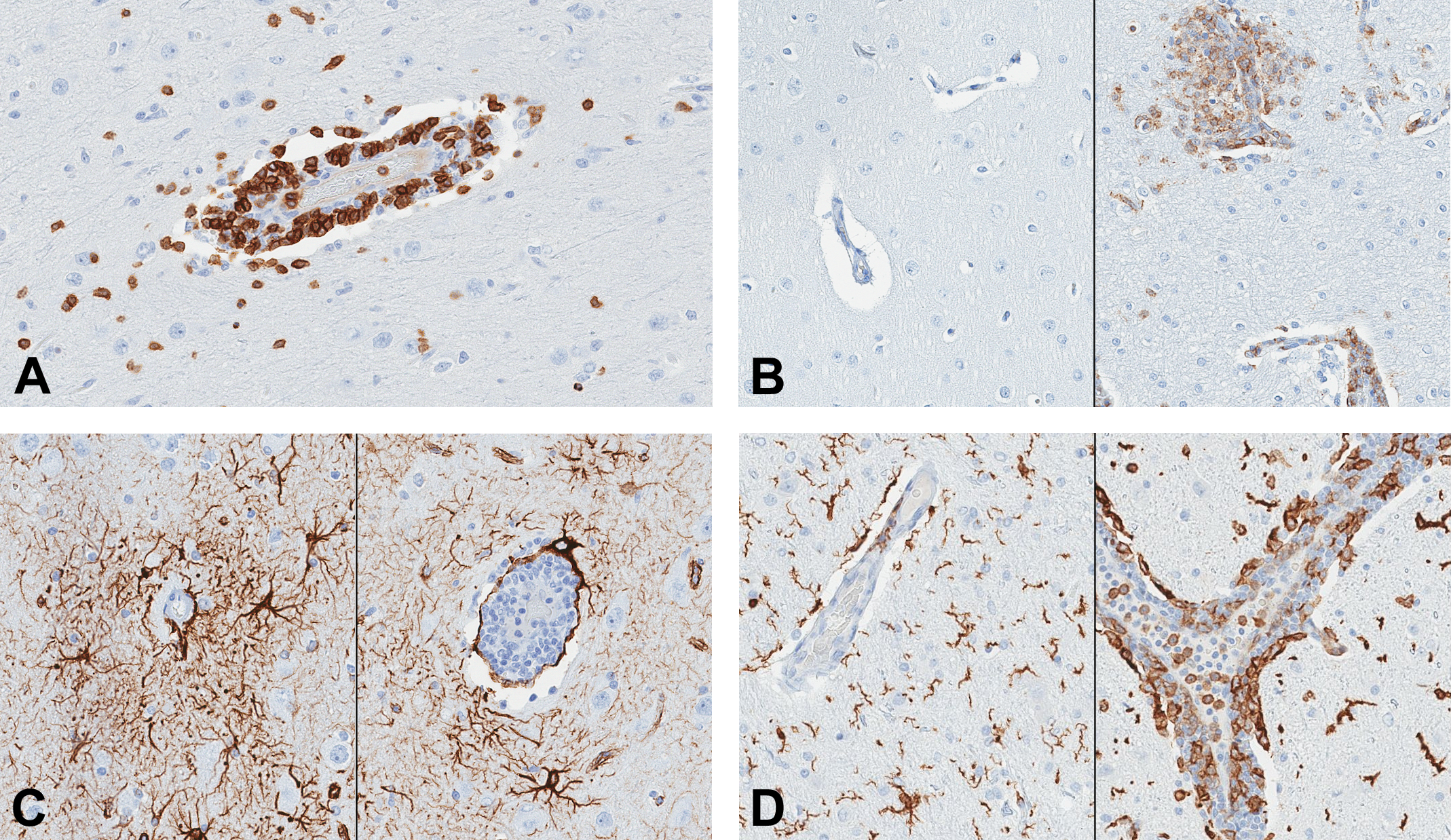
Immunohistochemical labeling of brain sections. (A) Vasculocentric infiltration of CD3-positive lymphocytes (pyriform lobe, female dog, naphthoquine phosphate [NP] at 2 mg/kg/day for 14 days). (B) Immunohistochemical labeling of major histocompatibility complex (MHC) class II. Left: control; right: vasculocentric infiltration of MHC class II positive mononuclear phagocytic cells (pyriform lobe, female dog, NP at 2 mg/kg/day for 14 days). (C) Immunohistochemical labeling of GFAP positive astrocytes in the pyriform lobe of a control (left) and NP-treated (right, NP at 2 mg/kg/day) female dogs. No differences were noted in the nature, intensity, or pattern of GFAP labeling between control and treated animals. (D) Immunohistochemical labeling of Iba-1-positive microglial cells and mononuclear phagocytic cells in the thalamus of a control (left) and NP-treated (right, NP at 50 mg/kg/day) female dogs. Note the presence of reactive microglial cells with large cell body and short processes in the neuropil of the NP-treated dog.
NP-related vasculocentric microscopic changes were also noted in the liver (Figure 1) and were characterized by minimal to slight vascular and perivascular inflammation centered predominantly on the centrilobular vein in females dosed with ≥2 mg/kg/day and males dosed with ≥10 mg/kg/day. In addition, slight hepatocellular single-cell necrosis accompanied by parenchymal inflammation was noted in 1 female dosed with 50 mg/kg/day and iron pigment deposits (positive with Perl’s Prussian blue) were noted in the Kupffer cells of females dosed with ≥10 mg/kg/day.
A constellation of minor NP-related microscopic changes were noted in various other tissues and organs with a predilection for the high-dose group. These changes included vacuolated pulmonary alveolar macrophages infiltrates, vacuolation of the adrenal cortex, vacuolation of the pancreatic acinar cells with occasional single-cell necrosis, neutrophilic infiltrate in the gastrointestinal tract mucosa, single-cell necrosis in the cecum and colon (mucosal epithelial cells), hematopoietic hypocellularity of the bone marrow, and lymphoid depletion/necrosis in the gastrointestinal-associated lymphoid tissue, spleen, retropharyngeal lymph node, and mesenteric lymph node.
Toxicokinetic Assessment
No measurable concentration of the test article was observed in any samples from vehicle control animals. Plasma NP exposure generally increased overproportionally to the increase in dose between 2 and 10 mg/kg/day, and there was accumulation. No consistent sex-related exposure differences were noted. Mean toxicokinetic parameters are presented in Table 4.
Toxicokinetic Parameters.
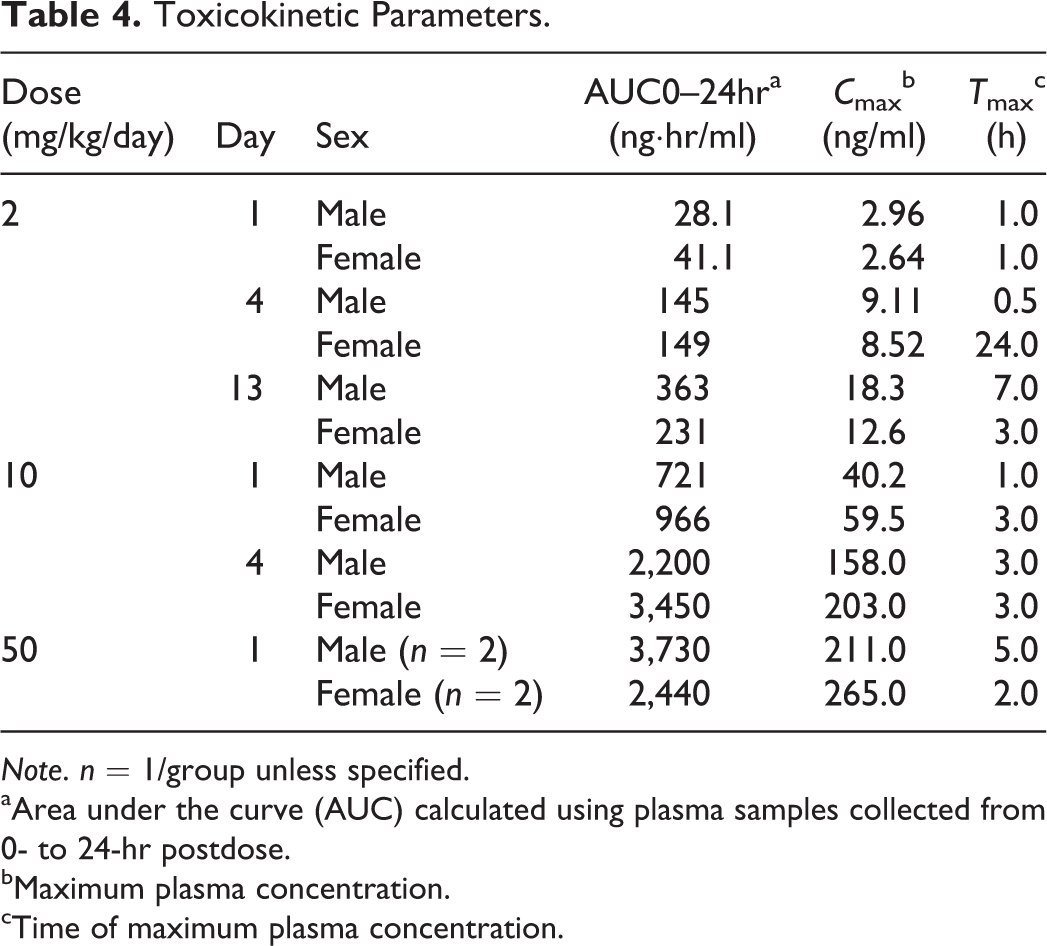
Note. n = 1/group unless specified.
aArea under the curve (AUC) calculated using plasma samples collected from 0- to 24-hr postdose.
bMaximum plasma concentration.
cTime of maximum plasma concentration.
Discussion
NP was considered as a potential combination partner for KAE609, a promising antimalarial drug candidate. While clinical safety data reported for combination therapy using NP and AT show good tolerability (Hombhanje et al. 2009; Toure et al. 2009; Tun et al. 2009; Hombhanje and Quigyun 2010; Qu et al. 2010; Benjamin et al. 2012; Meremikwu et al. 2012; Liu et al. 2013), there are limited published preclinical safety data available for this drug combination (Wang et al. 2004). Furthermore, to our knowledge, there are no preclinical or clinical safety data published for NP as a monotherapy. Wang et al. (2004) reported that 14-day oral gavage toxicology studies conducted in rats and dogs with NP/AT identified bone marrow (erythroid series) and liver to be target organs of toxicity. Due to the paucity of available preclinical data and no preclinical exposure data, it was decided to profile NP in a pilot 14-day toxicology study in dogs to enable development of a potential combination therapy with NP and KAE609.
NP doses of 2 to 50 mg/kg/day, based on free base, were selected to cover the range of NP doses reported from safe (7 mg/kg) to toxic (56 mg/kg; Wang et al. 2004). Administration was via oral gavage, using a formulation frequently used for oral dosing in dogs and a standard dose volume of 5 ml/kg. Unexpectedly, dogs dosed with 10 or 50 mg/kg/day had to be preterminally euthanized. One male and 1 female dog dosed with 50 mg/kg/day presented symptoms of significant neurological toxicity by day 2, including convulsions, ataxia, and recumbency. Minimal to marked vasculocentric central nervous system inflammation was observed in all males dosed with ≥10 mg/kg/day and all females dosed with ≥2 mg/kg/day, with the exception of 1 female in the 50-mg/kg/day dose group. The lack of microscopic findings in the central nervous system in this 1 female was most likely due to the decision to euthanize this animal early (day 2) due to clonic convulsions and recumbency, as it is not unusual for clinical signs to precede the appearance of light microscopic morphological changes in the brain (Summers 1995). It is, however, interesting to note that marked inflammation of the central nervous system characterized by the presence of lymphocytes and macrophages was noted in the other female dosed with 50 mg/kg/day and euthanized on day 3, without neurological clinical signs of convulsions, ataxia, or recumbency.
The mechanisms and associated potential biomarkers of drug-induced vascular injury (DIVI) in preclinical species, including the dog, have been recently reviewed and include hemodynamic alterations, altered endothelial nitric oxide synthase signaling, immune complex deposition, modulation of the immune system, and direct toxicity to endothelial cells (Mikaelian et al. 2014). In this study, vasculocentric inflammation in the liver and the central nervous system without degenerative or necrotic changes in the adjacent neuropil or the hepatic parenchyma suggests a primary systemic vascular process. However, no specific morphological evidence of vascular damage such as medial hemorrhage or fibrinoid necrosis was noted, other than inconsistent and rare adventitial microhemorrhages in the brain, suggestive of increased permeability.
In contrast to the acute onset of findings in this study with administration of NP for a duration ranging from 1 to 8 days, systemic DIVI in humans is often due to an immune-mediated mechanism secondary to prolonged treatment with a drug such as propylthiouracil or a monoclonal antibody that induces antineutrophil cytoplasmic antibody–associated vasculitis (Radic, Martinovic Kaliterna, and Radic 2012). While activation of the immune system can result in DIVI, a classical type III or IV hypersensitivity reaction would not be expected to occur so early in a repeat-dose study; a type III hypersensitivity reaction would be expected to present with a greater prominence of neutrophils accompanied by vascular necrosis, and a type IV hypersensitivity reaction would not be dose related, nor would it be expected to occur at such a high incidence (Clemo et al. 2003; Radic, Martinovic Kaliterna, and Radic 2012).
Dogs, as a preclinical species, are considered exquisitely sensitive to drug-induced hemodynamic toxicity, and changes are most often observed in the coronary arteries (Louden and Morgan 2001; Clemo et al. 2003). However, this mechanism of toxicity appears unlikely to explain the observations from the study described here, owing to the nature of the findings including inflammatory cells obscuring the vessels walls but with no evidence of vessel wall necrosis or arterial wall thickening, and the anatomic distribution of the vasculocentric changes favoring the small diameter vessels in the liver and central nervous system.
Finally, while primary toxic vasculitis cannot be excluded, such a finding would be expected to present histological features of segmental fibrinoid necrosis with more prominent neutrophilic inflammation (Clemo et al. 2003).
In summary, the characteristics of the vasculocentric inflammatory cells, being primarily composed of lymphocytes and macrophages, with little reaction in the adjacent neuropil or hepatic parenchyma and no discernable necrosis or other morphologic alterations of the vessel walls, is considered unusual, if not unique, and does not fit neatly into the morphologic criteria of any of the classically described mechanisms of DIVI. The authors were not able to identify a specific cause for the distribution of the changes (limited to the brain, spinal cord, and liver) nor a specific mechanism of toxicity for the inflammatory vascular changes described.
The clinical and histopathological findings appeared to be associated with relatively high plasma NP concentrations. The day 1 peak levels in the 50-mg/kg/day group averaged 238 ng/ml, while the AUC0–24hr value was 3,090 ng·hr/ml. Similar levels were achieved in the 10-mg/kg/day animals on day 4, while the levels in the 2-mg/kg/day animals reached only ∼10% of these values, indicating an overproportional increase in exposure with increasing dose as well as accumulation following repeat dosing. There was no difference between males and females in plasma exposure that would explain the increased sensitivity of female dogs to NP in this study.
Based on the clinical and histopathological findings in this study, further development of NP as a combination therapy with KAE609 was discontinued.
The lack of concordance with literature reports of the toxicological profile of NP in preclinical species is not immediately apparent. Published data on NP and other 4-aminoquinolines give little insight into the pathogenesis of the vasculocentric inflammation noted in the present study. No serious neurological or hepatic adverse events have been reported in clinical trials involving NP/AT (Hombhanje et al. 2009; Toure et al. 2009; Tun et al. 2009; Hombhanje and Quigyun 2010; Qu et al. 2010; Benjamin et al. 2012; Meremikwu et al. 2012; Liu et al. 2013). In one of these trials, a single dose of AT (1,000 mg) and NP (400 mg) in healthy volunteers produced an average NP C max of 27.4 ng/ml with an AUC0–216 of 955 ng·hr/ml, which is closest to the pharmacokinetic data associated with a single 10-mg/kg dose of NP in the present study (Qu et al. 2010). The 0- to 24-hr sampling in the present study would yield a lower estimate of AUC compared to the reported 0- to 216-hr AUC value. Chloroquine, amodiaquine, mefloquine, piperaquine, and primaquine are all aminoquinolines with structures similar to that of NP yet none of these drugs are reported to cause serious clinical neurological adverse events, although mefloquine has been associated with neuropsychiatric adverse reactions (Chattopadhyay, Mahajan, and Kumar 2007; Jensen and Mehlhorn 2009; O’Neill 2012). Of these drugs, only amodiaquine is reported to cause serious hepatic toxicity, although the nature of the amodiaquine-associated microscopic changes is different (mostly cholestatic or necrotic changes; Neftel et al. 1986) to what was observed for NP in the present study. Explaining the mechanism of toxicity via the mode of action of NP is also challenging since the exact mode of action of 4-aminoquinolines has not been completely elucidated, although most are thought to act by interfering with the sequestration/detoxification of ferriprotoporphyrin IX, a toxic by-product of hemoglobin degradation essential for plasmodium growth and division (Jensen and Mehlhorn 2009; O’Neill 2012).
In summary, the administration of NP to beagle dogs via oral gavage for 2 weeks was not tolerated at doses of 10 and 50 mg/kg/day, with marked neurological clinical signs observed at 50 mg/kg/day, and moribundity at ≥10 mg/kg/day resulting in early euthanasia. In addition, central nervous system and hepatic vasculocentric inflammation were noted in females dosed with ≥2 mg/kg/day and males dosed with ≥10 mg/kg/day.
Footnotes
Acknowledgments
The authors thank Peter Smith, Keith Mansfield, Karen Killary, Jennifer Marlowe, and Vito Sasseville for editorial assistance with this article and Clare Thomas for preparing the IHC-labeled sections.
Authors’ Contribution
Authors contributed to the conception or design (JG, PH, WL); data acquisition, analysis, or interpretation (JG, EM, PH, WL); drafting the manuscript (JG); and critically revising the manuscript (EM, PH, WL). All authors gave final approval and agreed to be accountable for all aspects of work in ensuring that questions relating to the accuracy or integrity of any part of the work are appropriately investigated and resolved.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This study was funded by the Novartis Institutes for BioMedical Research (USA).