
Abstract
Ferroptosis, an iron-dependent form of programmed cell death, is characterized by iron overload, increased reactive oxygen species (ROS) generation, and depletion of glutathione (GSH) and lipid peroxidation. Lipophilic antioxidants and iron chelators can prevent ferroptosis. GSH-dependent glutathione peroxidase 4 (GPX4) prevents lipid ROS accumulation. Ferroptosis is thought to be initiated through GPX4 inactivation. Moreover, mitochondrial iron overload derived from the degradation of ferritin is involved in increasing ROS generation. Ferroptosis has been suggested to explain the mechanism of action of organ toxicity induced by several drugs and chemicals. Inhibition of ferroptosis may provide novel therapeutic opportunities for treatment and even prevention of such organ toxicities.
Introduction
Human exposure to several drugs and chemicals from different sources has been linked to specific target organ toxicity.1–3 Such exposure can result in different mechanisms of cell death such as apoptosis, ferroptosis, autophagy, and necrosis. 4
Ferroptosis in organ toxicity.

COX2: cyclooxygenase 2; CytOx: cytochrome oxidase; DMT1: divalent metal transporter 1; FTH1: ferritin heavy chain 1; GLS2: glutaminase 2; GPX4: glutathione peroxidase4; GSH: glutathione; HEPH: hephaestin; HK-2 cells: human kidney-2 cells; 4-HNE: 4-hydroxynonenal; HO-1: heme oxygenase 1; JNK: c-Jun NH2-terminal kinases; LC3B: the microtubule-associated protein 1 light chain 3 beta; LDHA: lactate dehydrogenase; MDA: malondialdehyde; MMP: mitochondrial membrane potential; NADPH: nicotinamide adenine dinucleotide phosphate; NCOA4: nuclear receptor coactivator 4 mediates this process; Nrf2: nuclear factor-erythroid 2 related factor 2; NRVC: neonatal rat ventricular cardiomyocytes; PDH: pyruvate dehydrogenase; PDK1: pyruvate dehydrogenase kinase 1; PKM2: pyruvate kinase M2; PTGS2: prostaglandin-endoperoxide synthase 2; ROS: reactive oxygen species; SLC1A5: solute carrier family 1 member 5; SLC38A1: solute carrier family 38 member 1; TFR: transferrin receptor; TR-1: transferrin receptor-1; VDAC3: voltage-dependent anion channel 3.
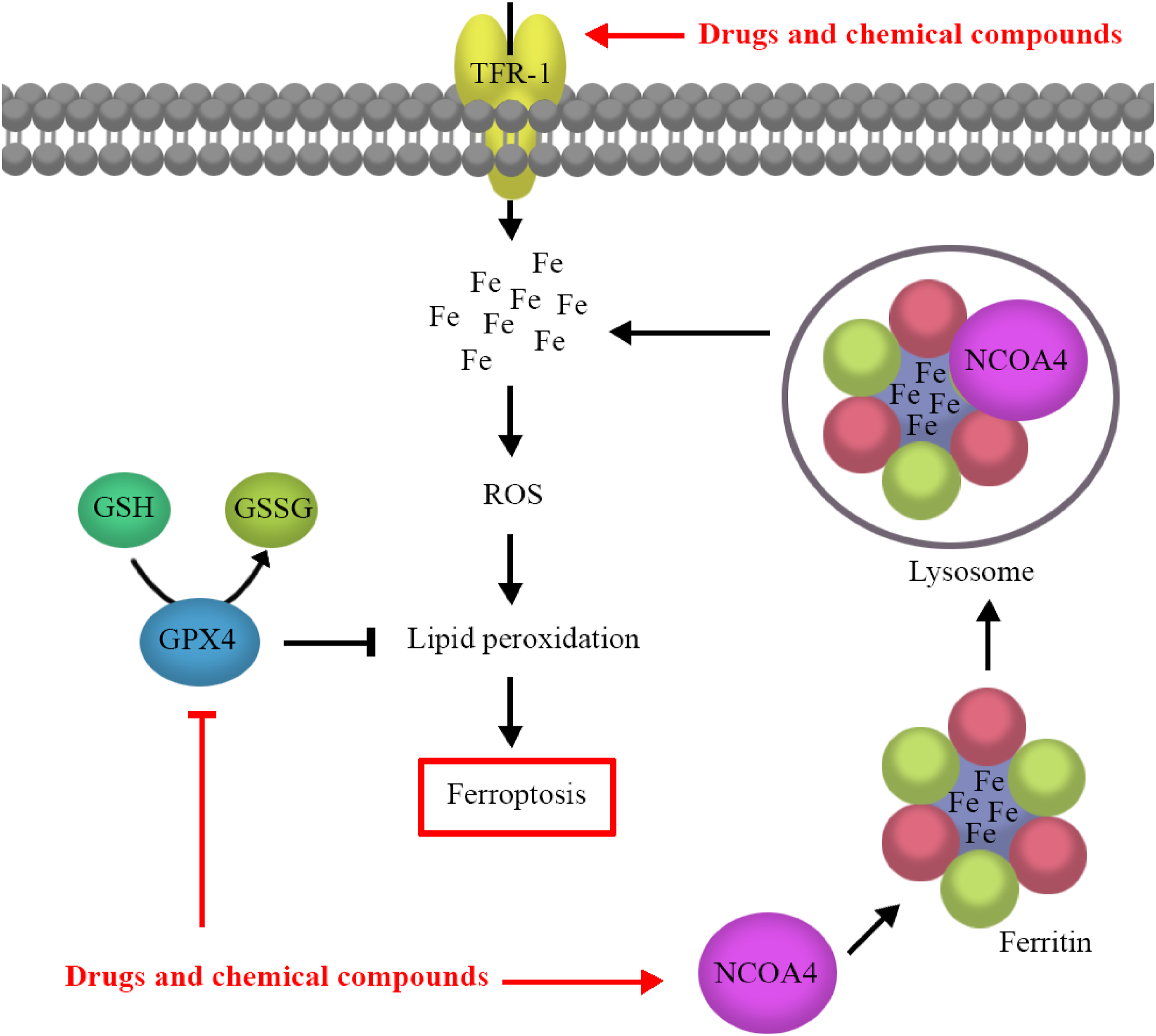
A schematic drawing suggesting potential drug and chemical compound toxicity-induced ferroptosis. TFR-1 mediates the cellular iron overload, which is an important factor for ROS production. NCOA4 induces ferritin degradation and increases cellular iron content by binding to ferritin and delivering it to the lysosome. Iron overload–dependent excessive ROS accumulation causes lipid peroxidation and ferroptosis. GPX4 modulates ROS levels and lipid peroxidation. Ferroptosis is initiated by drugs and chemical compounds through decreasing GPX4 activity and increasing THR-1 and NCOA4 activities. GPX4, glutathione peroxidase 4; GSH, glutathione; GSSG, glutathione disulfide; NCOA4, nuclear receptor coactivator 4; ROS, reactive oxygen species; TFR-1, transferrin receptor-1.
An overview of the ferroptosis
Iron is an essential component of enzymes involved in several physiological processes, including oxygen transport and energy production. 13 Ferritin is a blood iron storage protein composed of both heavy chain (FTH1) and light chain (FTL) ferritin subunits. 14 Transferrin (TF), a marker of iron homeostasis, is secreted from hepatocytes into the blood and transported to various tissues. 13 Transferrin receptor-1 (TFR-1), a transmembrane glycoprotein, mediates the cellular uptake of TF and ferritin. 14 These proteins play a major role in maintaining iron homeostasis. 14 Dysregulation of iron homeostasis is linked to cell death by the generation of reactive oxygen species (ROS). 15 Iron overload is considered an important factor for ROS generation through the Fenton reaction, where hydrogen peroxide breaks down to yield hydroxyl radicals.16,17
Ferritinophagy is an autophagic process that induces ferritin degradation, resulting in increased cellular iron content.6,18 Nuclear receptor coactivator 4 (NCOA4) mediates this process by binding to ferritin and delivering it to the lysosome. 19 Lysozyme-catalyzed ferritin degradation increases the availability of intracellular iron, resulting in the production of ROS. 19 The overproduction of ROS potentially contributes to cellular oxidative injury. 20 ROS levels are controlled by enzymatic and non-enzymatic antioxidants, including glutathione (GSH), superoxide dismutase (SOD), and glutathione peroxidase 4 (GPX4).20,21 An imbalance between ROS generation and its detoxification can lead to DNA damage and the peroxidation of proteins and lipids. 21
Lipid peroxides (LPOs) are an important class of ROS that exert their toxic effects through altering membrane properties and producing DNA-protein crosslinks. 7 The role of LPOs as key mediators of cellular death has been confirmed. 22 Ferroptosis involves lipid peroxidation-induced cell death following iron overload, ROS generation, depletion of GSH, and reduction of GPX4 activity, a GSH-dependent anti-oxidant defense pathway.5,23
Ferroptosis and neurotoxicity
The role of ferroptosis in arsenic-induced neurotoxicity
The peripheral and central nervous systems are targets of arsenic over-exposure. 24 Free radical formation, lipid peroxidation, GSH depletion, and mitochondrial impairment are involved in arsenite (most common toxic form of arsenic)-induced neurotoxicity. 25 Arsenite has been shown to trigger ferroptosis by ferritinophagy activation in neuronal cells. 26 Arsenite decreased both ferritin and protein NCOA4 expressions in the hippocampus and PC-12 cells. 26 NCOA4 mediates ferritinophagy by promoting ferritin transport to autophagosomes. 26 Arsenite also has been shown to enhance the level of the microtubule-associated protein 1 light chain 3 beta (LC3B). 26 Inhibition of ferroptosis by ferrostatin-1 (Fer-1) attenuated neuronal cell death induced by arsenite. 26 Co-treatment of cells with arsenite and the autophagy inhibitors 3-methyladenine (3-MA) and bafilomycin A1 (BafA1) decreased the cytotoxic effects of arsenite 26 suggesting that ferroptosis is involved in its neurotoxicity. 26
The role of ferroptosis in atrazine-induced neurotoxicity
Atrazine is a synthetic herbicide that is used to protect crops against grassy weeds. 1 It has been shown that atrazine exposure threatens human health and the environment. 27 Atrazine is well recognized as a neurotoxic agent that can cause neurodegenerative disorders. 28 The effect of long-term exposure to atrazine on the midbrain of rats has been investigated. 1 Atrazine increased midbrain iron content in a dose-dependent manner. 1 Both mRNA and protein expression of iron homeostasis proteins, such as TFR-1, divalent metal transporter 1, ferroportin 1, and hephaestin in the midbrain were affected by atrazine. 1 Iron overload and ferroptosis, a consequence of long-term exposure to atrazine, may enhance the neurotoxicity of atrazine. 1
The role of ferroptosis in cobalt nanoparticles–induced neurotoxicity
Occupational exposure to cobalt (Co) is associated with various adverse health effects, including lung cancer, cardiomyopathy, and neurotoxicity.12,29 It has been reported that Co nanoparticles (NPs) induced dose-dependent ferroptosis in neuronal cells through increasing cytosolic calcium, enhancing lipid production, and depletion of GSH. 12 Co NPs also suppressed mRNA and protein expression of GPX4 in neuronal cells. 12 The protective effect of N-acetylcysteine, as a precursor to GSH synthesis, also has been shown in Co NPs–exposed neuronal cells. 12 Liproxstatin-1 attenuated Co NPs–induced ferroptosis through inhibition of lipid peroxidation in neuronal cells. 12 Overall, these data suggest that neuronal cell death induced by Co NPs may occur through ferroptosis. 12
The role of ferroptosis in formaldehyde-induced neurotoxicity
Formaldehyde (FA) is an industrial contaminant found in the workplace and numerous products including adhesives and cosmetics. 30 Neurotoxic effects have been reported following occupational exposure to FA. 30 FA-induced neuronal ferroptosis has been demonstrated in HT22 cells. 31 FA reduced GSH levels and increased MDA, 4-HNE, and ROS levels, and the iron content in neuronal cells. 31 FA also up-regulated the ferroptosis-related genes, including solute carrier family 1 member 5 (SLC1A5)/SLC38A1, glutaminase 2 (GLS2) and prostaglandin-endoperoxide synthase 2 (PTGS2) in HT22 cells. 31 FA also decreased the mitochondria cross-sectional area and increased the ROS level and iron content in hippocampal tissue. 31
The Warburg effect (aerobic glycolysis) is the process of energy metabolism in the human brain that converts glucose to lactate in the absence of oxygen. 32 FA has been reported to induce the Warburg effect in both HT22 cells and hippocampal tissue by up-regulating pyruvate kinase M2, lactate dehydrogenase, and pyruvate dehydrogenase kinase 1 proteins and down-regulating pyruvate dehydrogenase. 31 Dichloroacetate reversed FA-induced ferroptosis via inhibition of the Warburg effect. 31 FA may induce ferroptosis in neuronal cells by up-regulating aerobic glycolysis. 31
The role of ferroptosis in isoflurane-induced neurotoxicity
Prolonged exposure to isoflurane, an inhaled anesthetic, can induce neurodegeneration changes. 33 Ferroptosis may contribute to isoflurane-induced neurotoxicity.34,35 Isoflurane has been reported to provoke ferroptosis in a dose- and time-dependent manner in the hippocampus. 34 In vitro exposure of neurons to isoflurane resulted in increased ROS generation, lipid peroxidation, mitochondrial membrane potential disruption, and suppression of GPX4 expression, which was attenuated by the selective ferroptosis inhibitor Fer-1. 35 Isoflurane also increased the cytochrome oxidase activity in the mitochondrial electron transport chain, which also may contribute to ferroptosis. 34 Dimethylfumarate, a mitochondria activator, has been reported to inhibit isoflurane-induced ferroptosis. 34 Mitochondria-dependent ferroptosis may play a pivotal role in isoflurane neurotoxicity. 34
The role of ferroptosis in zinc oxide nanoparticles–induced neurotoxicity
Zinc oxide nanoparticles (ZnONPs) are functional inorganic materials that are used in cosmetics and medicine. 36 Exposure to ZnONPs has exhibited toxicological effects such as respiratory toxicity, cardiotoxicity, and neurotoxicity.36,37 Pulmonary exposure to ZnONPs has been reported to trigger ferroptosis in cerebral cortex tissue and the PC-12 cell line, which is alleviated by Fer-1. 37 ZnONPs-induced ferroptosis is characterized by mitochondrial dysfunction, reduction of ATP, enhanced iron levels, increased amounts of lipid peroxidation, decreased SCL7A11 and GPX4 protein expression, and increased voltage-dependent anion channel 3 (VDAC3) protein expression. 37 GPX4 and SCL7A11 are key regulators in the reduction of ferroptosis. 38 VDAC3 protein, located at the mitochondrial outer membrane, has been shown to induce ferroptosis. 39
Activation of the c-Jun NH2-terminal kinases (JNK) signaling pathway has been reported to contribute to ferroptosis cell death. 37 The protein expression of p-JNK was enhanced in ZnONPs-treated mice and PC-12 cells. 37 The inhibition of the JNK pathway by SP600125 and a reduction of lipid peroxidation and ferroptosis in ZnONPs-treated PC-12 cells have been demonstrated. 37 Qin and his colleagues (2020) 37 have suggested that ZnONPs exposure may induce JNK-involved neuronal ferroptosis.
The role of ferroptosis in paraquat and maneb–induced neurotoxicity
Environmental exposures to the neurotoxic pesticides paraquat and maneb have been associated with an increased risk of Parkinson’s disease 40 by triggering ferroptosis in dopaminergic cells. 41 Lipid peroxidation and GPX4 expression and decreased GSH levels occurred in SHSY5Y cells treated with paraquat and maneb. 41 Paraquat and maneb–induced ferroptosis was inhibited by the ferroptosis inhibitors, Fer-1 and liproxstatin-1 and the iron chelator deferoxamine (DFP). 41 Paraquat and maneb also have been reported to stimulate nicotinamide adenine dinucleotide phosphate (NADPH) oxidase activation in dopaminergic cells. 41 NADPH oxidase inhibition by apocynin suppressed ferroptosis in these cells. 41 NADPH oxidase activation by paraquat and maneb may contribute to neurodegeneration through ferroptosis. 41
The role of ferroptosis in tetrachlorobenzoquinone-induced neurotoxicity
Tetrachlorobenzoquinone (TCBQ) is a metabolite of the fungicide hexachlorobenzene. 42 TCBQ exposure triggers both hepatotoxicity and neurotoxicity.42,43 Ferroptosis is a novel programmed cell death driven by TCBQ. 43 TCBQ decreased GPX4 levels and induced iron accumulation, up-regulated the protein and mRNA level of PTGS2 in PC12 cells, which was alleviated by Fer-1 or DFP. 43 Moreover, a deficiency in the nuclear factor-erythroid 2 related factor 2 (Nrf2) enhanced the susceptibility of PC12 cells to ferroptosis. 43 Nrf2 activation improved the iron storage capacity through up-regulating FTH1 expression. 43 Nrf2 also increased GPX4 activity by elevating the levels of GSH. 43 Nrf2 deficiency appears to modulate the outcome of TCBQ-induced ferroptosis, contributing to its neurotoxicity. 43
The role of ferroptosis in cardiotoxicity
The role of ferroptosis in doxorubicin-induced cardiotoxicity
Anthracyclines comprise a class of chemotherapeutic drugs, including doxorubicin (DOX), daunorubicin, idarubicin, and epirubicin, which are used to treat various malignancies. 44 Irreversible cardiomyopathy, however, limits the clinical use of these anthracyclines.44,45 DOX has been reported to induce various forms of regulated cell death, including necroptosis, apoptosis, ferroptosis, and pyroptosis in cardiomyocytes. 9 Potential mechanisms underlying DOX-induced cardiotoxicity include excess ROS generation, disrupted iron metabolism, mitochondrial iron accumulation, and mitochondrial dysfunction.2,9,45 Iron overload–mediated generation of ROS has been shown to induce lipid peroxidation and eventually accelerate cardiac damage. 2 Moreover, heme oxygenase 1 (HO-1) up-regulation has been suggested as the primary mechanism of DOX-induced ferroptosis in cardiac tissue. 44 HO-1 releases free iron in cardiac cells by degradation of heme, leading to oxidized lipid generation in the mitochondria membrane. 44 This mitochondrial-mediated cell death is a major cause of DOX-induced cardiac injury. 2 DOX has been shown to down-regulate GPX4 followed by the accumulation of lipid peroxides through the DOX-iron complex in the mitochondria. 2 DOX-induced cardiac impairment ameliorated the overexpression of GPX4 in GPX4 hetero-deletion mice. 2 The overexpression of GPX4 or iron chelation in the mitochondria inhibited DOX-induced ferroptosis, suggesting that DOX triggered ferroptosis in the mitochondria of cardiomyocytes. 2 Inhibition of ferroptosis with Fer-1 suppressed the DOX-induced cardiomyocyte death. 2 Mitochondria-dependent ferroptosis may be a primary cause of the cardiotoxicity progression in DOX treatment. 2 Liu and colleagues 9 have shown the anti-ferroptosis effect of acyl-CoA thioesterase 1 (Acot1) against DOX-induced cardiac cell death through inhibition of lipid peroxidation.
The role of ferroptosis in hepatotoxicity
The role of ferroptosis in acetaminophen-induced hepatotoxicity
Acetaminophen (APAP) or paracetamol is a common pain medication. 46 APAP can be metabolized to a reactive metabolite, N-acetyl-p-benzoquinone imine (NAPQI), following excessive APAP exposure which can form a mitochondrial protein adduct, resulting in mitochondrial oxidative/nitrosative stress.46,47 It has been shown that oxidative/nitrosative stress–induced mitochondrial dysfunction plays a key role in APAP-induced liver injury. 46 NAPQI is detoxified by intracellular GSH 47 ; however, in APAP overdose the GSH level is decreased and the highly reactive NAPQI can cause hepatotoxicity. 46 GSH depletion may induce hepatic lipid peroxidation and ferroptosis. 23 It has been shown that ferritinophagy plays a critical role in the delivery of ferritin to the lysosomes and its degradation. 48 APAP overdose triggers ferritinophagy and mitochondrial iron uptake, leading to the mitochondrial permeability transition pore opening in hepatic mitochondria and eventually cell death. 19
The effect of Fer-1, a ferroptosis-specific inhibitor, in a mice model of APAP-induced hepatotoxicity has been investigated.10,47 Fer-1 pretreatment reduced the elevated serum levels of ALT and AST in mice treated with APAP. 47 Moreover, Fer-1 improved centrilobular necrosis in the liver. 47 This study also assessed the expression of PTGS2, a ferroptosis marker, and 4-hydroxynonenal (4-HNE), a product of lipid peroxidation. Increased PTGS2 and 4-HNE expression was reduced by Fer-1 in the liver of APAP-injected mice. 47 In addition, malondialdehyde (MDA) levels and GSH depletion were restored by Fer-1. 47
The protective effect of clausenamide (CLA) against APAP-induced liver injury through inhibiting hepatocyte ferroptosis has been reported. 49 APAP induced hepatic ferroptosis by the accumulation of ROS, inhibition of GPX4 enzymatic activity, depletion of GSH, and inducing lipid peroxidation. 49 CLA inhibited APAP-induced ferroptosis thereby reducing liver injury. 49 Jaeschke et al.,50,51 however, have reported that there is no quantitative evidence for sufficient iron-dependent LPO to cause ferroptosis-induced APAP liver injury.
The role of ferroptosis in alcohol-induced hepatotoxicity
Chronic and excessive alcohol (i.e., ethanol) consumption has been shown to produce a spectrum of liver lesions, including hepatitis, steatosis, and fibrosis/cirrhosis. 52 Alcohol is metabolized to acetaldehyde and acetate primarily in hepatocytes. 53 Alcohol metabolism generates excessive amounts of ROS in hepatic cells.53,54 The ROS react with proteins and unsaturated lipids, exacerbating oxidative stress and lipid peroxides. 52 The accumulation of acetaldehyde also can result in pathological levels of iron deposition in hepatocytes and depletion of mitochondrial GSH. 55
Ferroptosis is involved in alcohol-induced cell death, and the inhibition of ferroptosis by Fer-1 has been reported to prevent or at least reduce liver injury. 53 Alcohol-induced lipid peroxidation and GSH depletion in alcohol-treated hepatic cells (L-02 cells) are accompanied by the down-regulation of GPX4 and SLC7A11 protein expression and the up-regulation of the ferroptosis marker PTGS2 mRNA levels. 53 SLC7A11 has been reported to promote cystine uptake and GSH biosynthesis, resulting in protection against ferroptosis cell death. 56 Alcoholic liver damage and lipid peroxidation are markedly suppressed by Fer-1. 53
The role of ferroptosis in nephrotoxicity
The role of ferroptosis in cisplatin-induced nephrotoxicity
Cisplatin is a major anti-cancer drug used for the treatment of solid tumors such as testicular cancer. 57 Its therapeutic potential, however, is limited by its nephrotoxicity. 58 Ferroptosis has been investigated as a potential mechanism for cisplatin-induced renal injury. 3 Cisplatin treatment augmented the concentration of renal ferrous iron by increasing iron content and expressions of ferritin and transferrin receptor-1 in the kidney of mice. 3 On the other hand, increased level of hydroxyl radicals and expressions of cyclooxygenase 2 (COX2) and 4-HNE attenuated in kidney tissue by Fer-1, an inhibitor of ferroptosis. 3 Moreover, cisplatin-induced inflammation, apoptosis, and necroptosis were inhibited by Fer-1. Also, the expression of COX-2 and 4-HNE was decreased by DFP, an iron chelator. 3 Generally, these findings have suggested a role for ferroptosis in cisplatin-induced nephrotoxicity. 3
The role of ferroptosis in aristolactam I–induced nephrotoxicity
Chinese herbal medicine has been used for centuries as an alternative therapy in the treatment of chronic disorders, including chronic kidney disease (CKD) and cancer.59,60 Some Chinese medicines contain potentially toxic compounds such as aristolactam I (ALI), which is found in herbs of the Asarum species. 61 It has been reported that prolonged administration of a herbal medicine containing ALI can result in nephrotoxicity. 61 Gökmen and Lord 62 have suggested that oxidative stress, apoptosis, ferroptosis, and inflammation are involved in the renal toxicity induced by ALI.
The role of mitochondrial iron overload–mediated ferroptosis in ALI-induced nephrotoxicity has been investigated. 59 ALI decreased GSH levels, increased the protein levels of 4-HNE, and induced mitochondrial iron overload and ROS levels in human kidney-2 cells (HK-2 cells), which was particularly or completely reversed by deferoxamine mesylate, an iron-chelating agent. 59 Fer-1 alleviated or at least reduced the ALI-induced nephrotoxicity by decreasing the expression of GPX-4 in a dose-dependent manner. 59 Fer-1 also elevated GPX4 expression by scavenging lipid peroxides in HK-2 cells. 59 Ferroptosis-regulated cell death may be involved in ALI-induced nephrotoxicity. 59
The role of ferroptosis in testicular toxicity
The role of ferroptosis in arsenic-induced testicular toxicity
Arsenic is a heavy metal toxicant abundantly found in the environment. 63 Exposure to arsenic is a major public health problem. 63 Male reproductive toxicity of arsenite, the most toxic species, has been well-documented. 63 Mitochondrial injury, accumulation of iron, production of ROS, GSH depletion, and lipid peroxidation in the testes of mice treated with arsenite have been reported. 11 The protein and RNA expressions of GPX4, SLC7A11, and iron-responsive element-binding protein 2 (IREB2) decreased in the testes of arsenite-treated mice, while voltage-dependent anion-selective channel protein 3 (VDAC3) expression was elevated. 11
Anti-ferroptosis therapeutic strategies
Iron has an important role in the progress of ferroptosis. Regulating iron homeostasis by diminishing iron uptake and improving iron storage can contribute to the inhibition of ferroptosis. 64 It has been suggested that cytochrome P450 substrate drugs have anti-ferroptosis properties by preventing lipid peroxidation. 65 Moreover, the potential antioxidant therapeutic agents can reduce ferroptosis through increasing GSH levels and GPX4 activation.64,66 In addition, NCOA4 is important in the ferritinophagy turnover of the ferritin in ferroptosis. NCOA4 inhibition can be considered as a therapeutic target against ferroptosis. 67
Conclusion
The potential role of ferroptosis in drug and chemical–induced organ toxicity has been reported in a growing number of experimental studies. It has been suggested that several ferroptosis inhibitors, including Fer-1 and liproxstatin-1, suppress programmed-cell-death events induced in the brain, heart, liver, kidney, and testicular tissue. Activation of drug and chemical–induced ferritinophagy has been shown to promote ferroptosis by degradation of ferritin, resulting in an iron-overloaded condition and excessive generation of ROS in different organs. An understanding of the mechanism(s) responsible for inducing ferroptosis in different organs will be important to control their toxicity. Iron chelation therapy with DFO has been reported to abolish or at least reduce ferroptosis. Repression of ferroptosis might provide a novel therapeutic strategy for treating these toxicities.
Footnotes
Acknowledgments
The authors thank Mashhad University of Medical Sciences for financial support.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.