
Abstract
The emerging field of translational safety genetics is providing new opportunities to enhance drug discovery and development. Genetic variation in therapeutic drug targets, off-target interactors and relevant drug metabolism/disposition pathways can contribute to diverse drug pharmacologic and toxicologic responses between different animal species, strains and geographic origins. Recent advances in the sequencing of rodent, canine, nonhuman primate, and minipig genomes have dramatically improved the ability to select the most appropriate animal species for preclinical drug toxicity studies based on genotypic characterization of drug targets/pathways and drug metabolism and/or disposition, thus avoiding inconclusive or misleading animal studies, consistent with the principles of the 3Rs (replacement, reduction and refinement). The genetic background of individual animals should also be taken into consideration when interpreting phenotypic outcomes from toxicity studies and susceptibilities to spontaneous safety-relevant background findings.
Safety genetics addresses the impact of genetic variations in drug target biology, metabolism, and disposition of drug-induced phenotypes. With the latest advances in the field of genomics and newest sequencing technologies, genomes of different toxicology animal species have been sequenced or their quality dramatically improved. Genetic variations in therapeutic targets, off-target interactors, and drug metabolism and disposition pathways may cause differences in both pharmacologic and toxicologic responses across species, strains, and/or animals from diverse geographic origins.
New molecular entities and biological products are required to be tested in a rodent or nonrodent species prior to human clinical studies, following the fundamental principles of the 3Rs: replacement, reduction, and refinement (Törnqvist et al. 2014). Improved preclinical study design, coordination, and proper methodology can lead to a reduction in the number of animals needed in a study. Important sources of guidance for selecting an appropriate species for preclinical toxicity studies and safety assessment include the International Conference on Harmonization (ICH), U.S. Food and Drug Administration, and European Medicines Agency (EMA; ICH guideline 2012; ICH 1997; EMA 2008; Table 1).
Recommendations for Drug Target Characterization within Regulatory Guidelines for Preclinical Safety Testing.

Note. ICH = International Conference on Harmonization; EMA = European Medicines Agency; FDA = Food and Drug Administration; CHMP = Committee for Medicinal Products for Human Use; FIH = first-in-human.
This review discusses the importance of characterizing genetic variation within preclinical animal species and opportunities for enhancing drug safety through translational safety genetics.
Species Selection for Toxicology
Several animal species are currently used in toxicology including mice, rats, dogs, minipigs, and monkeys. These species are subject to well-characterized spontaneous pathology findings that can vary across species, strains, and geographic origins (Bradley, Mukaratirwa, and Petersen-Jones 2012; Chamanza et al. 2010; Jeppesen and Skydsgaard 2014; Mahler et al. 2016; Sato et al. 2012). We outline below the most commonly used toxicology species and how their genetic characterization can enhance the design and interpretation of toxicology studies.
Nonrodents
Nonhuman primates (NHPs) have an important role in basic and translational research, and especially in toxicology studies of biologics or advanced therapeutics due to their similarity with humans in evolutionary ancestry. Frequently, NHPs are the only nonhuman species capable of pharmacological responses to novel biotherapeutics (Capitanio and Emborg 2008). Some of the most conclusive human-relevant data for safety assessment has been obtained from monkey toxicology studies (Capitanio and Emborg 2008).
Cynomolgus monkeys are the most commonly used nonrodent species for biologics (Brennan et al. 2010), while marmosets (Marmoset Genome Sequencing and Analysis Consortium 2014), African green monkeys (Warren et al. 2015), and Rhesus monkeys are only rarely used for preclinical toxicology and safety testing.
Dogs are frequently used in the pharmaceutical industry when selecting nonrodent species for toxicological studies, mostly for low molecular weight compounds (Smith et al. 2002; Robertson, Breider, and Milad 2001). However, dogs are sensitive to sympathomimetic and hypotensive drugs and are susceptible to cardiotoxicity (Dogterom, Zbinden, and Reznik 1992; Lehmann 1998), making them a less relevant species for certain compound classes that are likely to affect those specific pathways.
Minipigs are another nonrodent species used for comparative biology at molecular, biochemical, cellular, and tissue levels (Vamathevan et al. 2013) as well as toxicity assessment. Since their skin structure is similar to humans, the minipig is important for characterizing dermal compounds as well as investigation of immunopathological mechanisms and skin-related safety signals (Eaglstein and Mertz 1978; Swindle et al. 2012; Vana and Meingassner 2000). Given the similarity in anatomy, physiology, and biochemistry of minipigs to humans, it is also a particularly attractive species in reproductive toxicology testing, where early onset of sexual maturity and large size of litter is an advantage (Mitchell and Creasy 2007).
Other less common nonrodent species used in specific toxicology studies include rabbits for reproductive toxicology because of their high rate of reproduction and ferrets for safety pharmacology (Foote and Carney 2000; Gad 2000).
Rodents
Both mouse and rat are used for rodent toxicity and efficacy studies (Pritchard et al. 2003; Tirmenstein et al. 2015). Even though they are phenotypically similar to each other, significant evolutionary differences need to be considered for optimal animal species selection (Iannaccone and Jacob 2009). For example, while the mouse has been one of the most commonly used animal species in fundamental research, its value as a model for the majority of human diseases is limited (Seok et al. 2013). Mouse models often serve to replicate specific processes within a disease but not the whole spectrum of physiological changes that occur in humans in the disease setting (Fingleton 2007; Mak, Evaniew, and Ghert 2014).
Additional rodent models that are less frequently used for toxicity studies include hamsters (Brinkrolf et al. 2013; Dipaolo, Nelson, and Donovan 1972; Erexson, Periago, and Spicer 2001) and Guinea pigs (Rocca and Wehner 2009).
Paradigm Shift in Genetic Characterization of Toxicological Species
Given the challenges associated with identifying reliable animal species for toxicity studies of a diverse range of innovative therapeutic modalities, it is important to optimize species selection using genetic data to ensure the use of a pharmacologically relevant species.
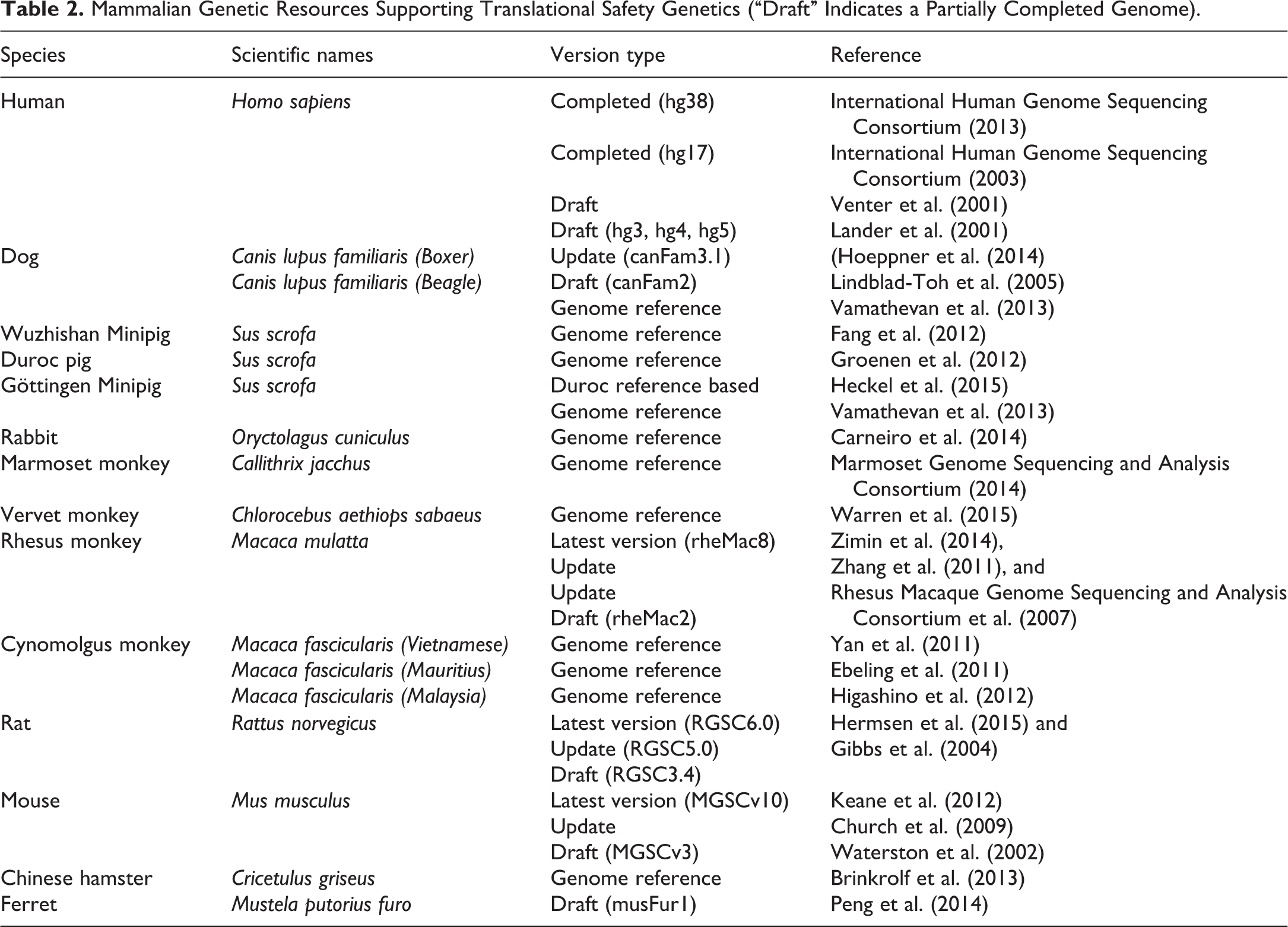
Despite access to comprehensive human and mouse genome sequences for more than a decade, progress in preclinical safety genetics has been constrained by a lack of comprehensive genome data in animals commonly used for preclinical toxicology testing such as NHPs, dogs, minipigs, rats, and mice. Furthermore, breed or strain variations in these animal species add to the complexity of establishing genotype–phenotype relationships. Nevertheless, recently there has been tremendous progress in the whole genome sequencing of additional mammalian species that can be leveraged for toxicity studies and safety assessment (Table 2).
Mammalian Genetic Resources Supporting Translational Safety Genetics (“Draft” Indicates a Partially Completed Genome).
The understanding of the baseline genetic variation in monkey is particularly important, as NHP studies are usually composed of a small number of animals due to the ethical and animal welfare concerns inherent to NHP work. Furthermore, only relatively limited access to monkeys from specific geographic origins is feasible. This is exemplified by a shift toward the usage of Chinese-origin rhesus monkeys for HIV research due to export limitations even though Indian-origin rhesus monkeys are a natural model for HIV (Marthas et al. 2001). Little attention was previously given to the geographic origins of the monkeys enrolled in toxicology programs. Differences in spontaneous pathological findings have been observed in cynomolgus monkeys from diverse geographic origins (Drevon-Gaillot et al. 2006; Vidal et al. 2010), as well as diverse major histocompatibility complex genetics (Campbell et al. 2009; Kita et al. 2009), and even geographic origin-dependent genetic variation within captive cynomolgus monkey stocks (Stevison and Kohn 2009).
The first genome draft of the rhesus monkey, Macaca mulatta, was a major advance in biomedical sciences (Rhesus Macaque Genome Sequencing and Analysis Consortium et al. 2007). This genome draft enabled the identification of macaque-specific genes and also the assessment of population diversity at single nucleotide resolution. The first draft genome of the cynomolgus monkey (long-tailed macaque), Macaca fascicularis, was assembled using a whole-genome shotgun sequencing approach employing two independent deep sequencing technologies (Yan et al. 2011; Ebeling et al. 2011). These studies have been reinforced by numerous additional NHP genome sequencing efforts that are enabling the detailed characterization of both coding and noncoding parts of the macaque genome.
We have been able to detect evidence at the exome level that geographic origin strongly influences the genetic background of cynomolgus monkeys. Whole genome sequencing initiatives provide an opportunity for enhanced early target characterization and toxicology species selection spanning all relevant rodent and nonrodent species. The identified genetic variants can help define the molecular basis for interindividual, intrastrain, and interspecies variation at the molecular, cellular, and physiologic levels.
In particular, the systematic genotyping of therapeutic antibody targets/pathways within nonhuman primates combined with an assessment of human target genetic variation in the intended patient population can support optimal toxicology species selection and ensure optimal design of pharmacokinetics/pharmacodynamics assays (Kronenberg et al. 2013). Beyond genetic variation in the intended therapeutic target, additional confounding factors include the potential for species and strain differences in Fc receptor (FcR) binding and distinct Immunoglobulin G (IgG)-FcR interactions observed between cynomolgus monkeys and humans (Warncke et al. 2012). Species differences have also been observed for IgG-neonatal Fc receptor (FcRn) interactions (Abdiche et al. 2015). For example, human IgG1 binds to cynomolgus monkey FcRn with a 2-fold higher affinity than human FcRn, whereas it binds rat FcRn with a 10-fold higher affinity than human FcRn. An overarching strategy for incorporating knowledge on genetic variation in drug targets, metabolism, and disposition into translational safety genetics is outlined in Figure 1.
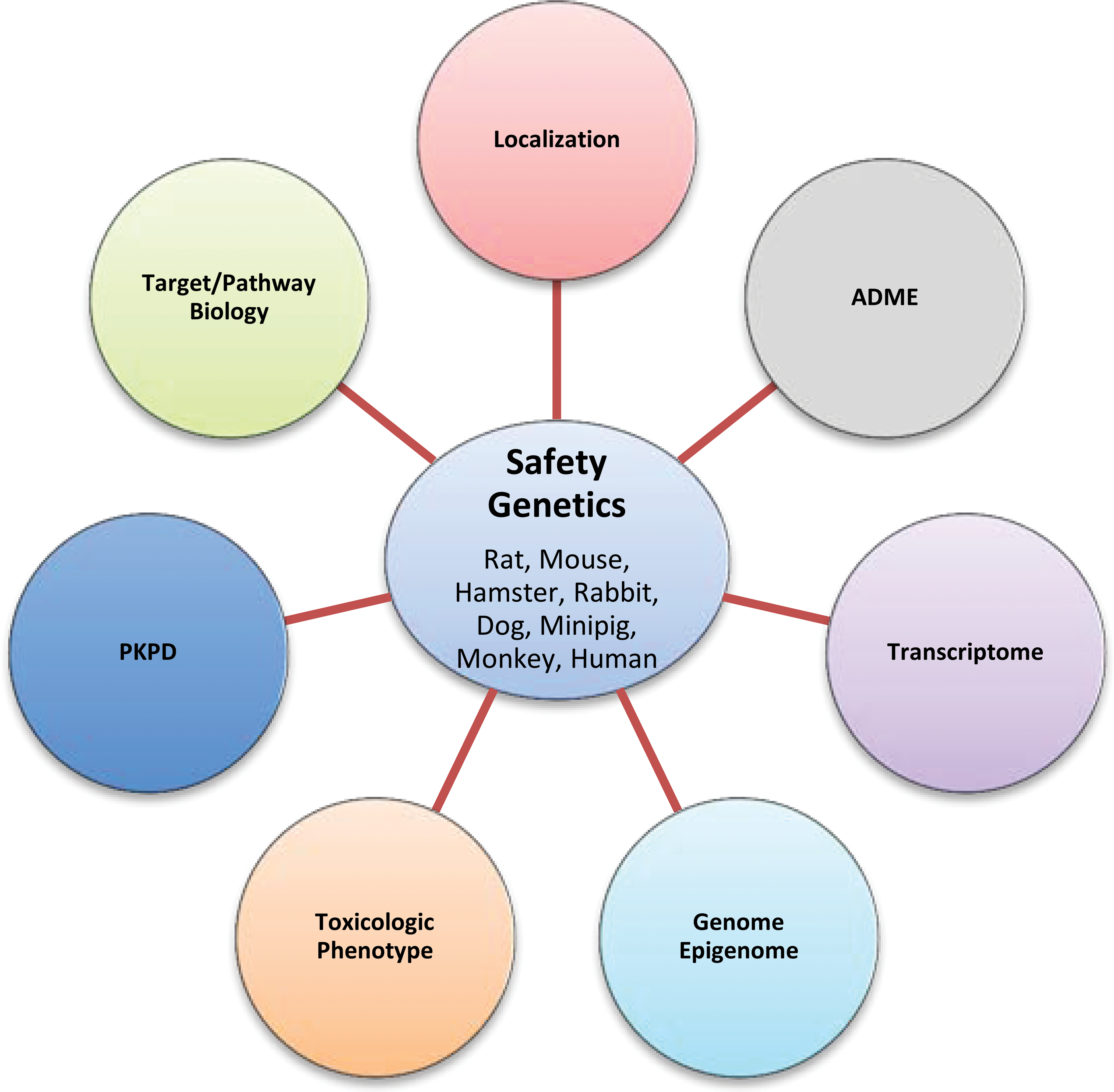
Integrating genetic variation in drug targets, metabolism, and disposition into translational safety genetics.
Clinical Relevance of Genetic Variations
The use of animal species for advancing medical science is widely accepted and recognized (Matthews 2008). However, the safety and efficacy profiles of therapeutics in animal species do not always correlate with human clinical trial outcomes (Mak, Evaniew, and Ghert 2014). This was exemplified by the TGN1412 clinical trial (Attarwala 2010) in which an anticluster of differentiation 28 (anti-CD28) therapeutic antibody led to organ failure in humans despite being deemed safe based on animal studies due to species differences in CD28 expression on CD4+ effector memory T-cells (Eastwood et al. 2010). Hence, it is critical to identify an appropriate animal species for human-relevant toxicity studies and safety assessment.
It is also common to observe differences in drug responses between individual patients, which in some cases may be attributed to genetic variations. There is an increasing repertoire of examples where genetic factors play a role in the drug metabolism and/or toxicity phenotypes (Table 3). In the case of Lumiracoxib, the elucidation of a specific human leucocyte antigen (HLA) genotype association with drug-induced liver injury provides the potential to improve the drug’s safety profile by identifying individuals at elevated risk for liver injury and excluding them from treatment (Singer et al. 2010). Carbamazepine, a drug prescribed for seizures, was observed to cause skin blisters (Steven–Johnson syndrome) in some patients carrying the HLA-B*1502 marker, a strong indicator of a genetic factor influencing drug response (Chung et al. 2004). It was subsequently found that the T-cell receptor (TCR) clonotype plays a key role in the pathogenic mechanism of Steven–Johnson syndrome (Ko et al. 2011). Thus, it appears that specific combinations of TCR and HLA genotypes with therapeutic drug exposure underlay this drug-induced hypersensitivity reaction. These findings are not restricted to immune-driven responses. Ischemic stroke patients treated with Alteplase and Tenecteplase and carrying polymorphisms in alpha-2-macroglobulin and coagulation factor 12 were more prone to suffer hemorrhagic transformations and inhospital death (Del Río-Espínola et al. 2012).
Examples of Studies Where Genetic Factors Play a Role in the Drug Metabolism and/or Toxicity.

Note. AOX1 = aldehyde oxidase-1; A2M = alpha-2-macroglobulin; BN = Brown Norway (Rat); CYP2D15 = cytochrome P450 2D; F12 = coagulation factor XII; GK = Goto-Kakizaki (Rat); HLA = human leucocyte antigen; MAP kinase = mitogen-activated protein kinase; PolR2A = polymerase (RNA) II subunit A; TCR = T-cell receptor; TYR = tyrosinase; Ugt2b = UDP glucuronosyltransferase 2b; WKY = Wistar-Kyoto (Rat).
Genetic Differences within Preclinical Models
Similar to humans, genetic variations underlie differences in drug response within animals from the same species as well as between different species. Celecoxib is a cyclooxygenase-2 inhibitor that readily metabolizes in dogs. A study on Beagle dogs identified 3 new variants in cytochrome P450 2D (CYP2D15; homologue to human CYP2D6) and a novel variant of CYP3A12, which were associated with different rates of metabolism of celecoxib in poor metabolizer (PM) and extensive metabolizer dog populations (Paulson et al. 1999). This was one of the earliest studies in dog that described a genetic polymorphism in a drug metabolizing enzyme. Another example in a rat study showed linkage of quantitative trait loci to a gut microbial metabolite (benzoate), which can be explained by deletion of the UDP glucuronosyltransferase 2b (Ugt2b) gene in several rat strains including Goto-Kakizaki (Rat), Brown Norway (Rat), and Wistar-Kyoto (Rat) (Dumas et al. 2007).
Furthermore, Pol2RA mutations are associated with resistance to α-amanitin in mice (Bartolomei and Corden 1995), a natural toxin whose use in chemotherapy has been recently reinvigorated (Liu et al. 2015). Finally, the genetic variations can have an overt phenotypic impact such as the altered skin color caused by tyrosinase in rats (Bartolomei and Corden 1995; Blaszczyk et al. 2005).
Translatability of Preclinical Findings
The preclinical evaluation of a novel p38 kinase inhibitor across inbred mice strains, dog, rats, and monkeys exemplifies the potential for species differences in pharmacokinetic profiles compared to humans, where rapid clearance of the drug was observed (Zhang et al. 2011). Differences in animal versus human metabolism were attributed to differences in enzyme activity of aldehyde oxidase-1 (AOX1) and the pattern of genetic variation in AOX1 from inbred mouse strains correlated with the rate of drug biotransformation (Zhang et al. 2011).
The recommendation to assess the safety of therapeutic monoclonal antibodies in a preclinical species that is pharmacologically responsive to the treatment (Brennan et al. 2010) can be challenging. This is exemplified by a fully humanized C3b therapeutic antibody candidate (S77) that did not bind rhesus C3b due to a H897F substitution (Katschke et al. 2009). Such observations emphasize the need to assess functional equivalence of biotherapeutic drug epitopes in the animal species versus humans in order to optimize toxicology species selection.
An important future aim for preclinical findings would be to predict the differences between animal phenotypes and clinical responses for various adverse events. This might be achieved in part through the characterization of genetic variants associated with drug metabolism and targeted pathways, combined with in vitro and in vivo assessments of functional consequences (Roden and George 2002).
Leveraging Animal Species Genome Sequencing Initiatives for Enhanced Safety Assessment
In order to better understand the genetic variation between cynomolgus monkey populations, we have recently generated an exome sequencing-derived genome resource for rapid in silico assessment of genetic variation in targets and pathways. 10 individual cynomolgus monkeys from each of the 3 distinct geographic origins (Vietnam, Mauritius, and Philippines) were selected and exome sequencing was performed (Del Rio-Espinola, manuscript in preparation). The resultant database containing genetic data is being used to support early target characterization and toxicology species selection; define interindividual, intrastrain, and interspecies genetic variants; and assess potential functional impact on gene/protein expression/function and translation to human genetic variation in intended patient populations. Similar applications are envisaged for rodent, canine, and minipig genome variation sequencing initiatives.
Conclusion
Characterizing genetic variation in commonly used animal species can enhance toxicology species selection for translational safety assessment. Species-specific genotypes, strain-specific genotypes, intrastrain genetic variation, influence of geographic and breeder origins, and their impact on drug absorption, distribution, metabolism, excretion, pharmacology, and toxicology are some of the factors that need to be considered for enhanced safety genetics. While identifying genetic variation is relatively straightforward, assessing potential functional consequences is much more challenging and requires extensive in vitro– and in vivo–based characterization of candidate genetic variants.
Some key future directions in translational safety genetics are (1) advanced genome resources for cross-species assessment of target/pathway genetic variation (enabling in silico assessment, hypothesis generation, and assessment of human relevance), (2) defining the importance of genetic variants by functionalization, (3) systematic sampling in toxicology studies for retrospective assessment of preclinical genotype–phenotype relationships, (4) proactive rather than reactive preclinical genotyping for specific drug targets where functional variants have been identified, and (5) selection of appropriate preclinical species for new drug modalities (e.g., bispecific monoclocal antibodies; cell and gene therapies).
Footnotes
Acknowledgments
We would like to thank Leslie Pond and the Education Office in Novartis for their encouragement and support. We would also like to thank Dominique Brees and Page Bouchard (PreClinical Safety, Novartis) for reviewing the manuscript.
Authors’ Contribution
Authors contributed to conception or design (PB, AD, FH, JM, OG); data acquisition, analysis, or interpretation (PB); drafting the manuscript (PB); and critically revising the manuscript (AD, FH, JM, OG). All authors gave final approval and agreed to be accountable for all aspects of work in ensuring that questions relating to the accuracy or integrity of any part of the work are appropriately investigated and resolved.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.