
Abstract
The skin has a relatively limited range of responses to injury regardless of the specific mechanism underlying the insult. When the skin’s barrier function is disrupted, it mounts an inflammatory and proliferative response in an effort to restore this essential function. The epidermal keratinocyte is central to the initiation of the skin’s response, triggering an immunologic cascade that leads to the stereotypic morphologic responses that we encounter as pathologists. Drug-induced immune-mediated cutaneous injuries or “drug eruptions” are relatively common, sometimes with overlapping mechanisms, and it is often possible to classify these based on the classical hypersensitivity-type reactions. A specific type of immune-mediated skin injury is psoriasis. The pathogenesis of psoriasis is multifactorial but involves the interaction of environmental factors with a genetic predisposition. The initial stimulus triggering the development of psoriatic lesions involves activation of epidermal keratinocytes, with subsequent amplification driven by cross talk between the adaptive and innate immune systems. Several cytokines produced by Th17 T helper cells have recently been shown to be important in the pathogenesis of psoriasis, namely, interleukin-23 (IL-23) and IL-17, due to demonstrated clinical efficacy of cytokine blockade; and IL-22, based on its effects in both in vitro and in vivo models.
Immune-mediated Skin Injury
The skin, like other organ systems, has a relatively limited range of responses to injury regardless of the specific mechanism underlying the insult. Despite this, there is still a great deal that can be ascertained from the different morphologic, physiologic, and molecular alterations that arise in response to injury. One of the skin’s primary functions is to serve as a physical and physiologic protective barrier against injury from the external environment and from loss of water and solutes from the body. When the skin is exposed to irritants, such as xenobiotics, infectious agents, or ultraviolet (UV) radiation that may damage or disrupt this barrier, it mounts an inflammatory and proliferative response in order to prevent further damage and to restore a morphologically and physiologically functioning barrier (Nestle et al. 2009).
A core premise underlying the skin’s response to injurious stimuli is that the epidermis, and particularly epidermal keratinocytes and dendritic cells, including Langerhans cells, are central to the initiation of the skin’s response to injury (Nickoloff 2006; Nestle et al. 2009; Di Meglio, Perera, and Nestle 2011).
The hypothesis that the epidermal keratinocyte is a primary initiator of the skin’s response to noxious stimuli has gained widespread acceptance (Pittelkow 2005; Nickoloff 2006; Nestle et al. 2009; Di Meglio, Perera, and Nestle 2011). Epidermal keratinocytes can recognize pathogen-associated molecular patterns (PAMPs) of microbial origin and danger-associated molecular patterns (DAMPs), such as xenobiotics and other irritants through Toll-like receptors (TLRs), including TLR-1, TLR-2, TLR-4, TLR-5, and TLR-6 on their surface and TLR-3 and TLR-9 in their endosome that trigger an inflammatory cascade leading to the generation of antimicrobial peptides such as β-defensins, cathelicidins, and S100 family proteins; proinflammatory chemokines such as interleukin-8 (IL-8), CXCL9, CXCL10, CXCL11, CCL27, and CCL20; and proinflammatory cytokines such as IL-1β, tumor necrosis factor (TNF), IL-6, and IL-18 (Nestle et al. 2009; Di Meglio, Perera, and Nestle 2011).
These chemokines and cytokines recruit and activate leukocytes and convert the initial innate immune response into an adaptive immune response. In addition, keratinocytes express nucleotide-binding domain, leucine-rich repeat-containing (NLR) proteins that recognize cytoplasmic PAMPs, DAMPs, and UV radiation. When engaged, NLRs trigger a proinflammatory signaling pathway through a large multiprotein complex termed an inflammasome that is formed by an NLR, an adaptor protein termed apoptosis-associated speck-like protein containing a caspase recruitment domain and procaspase 1 (Nestle et al. 2009; Di Meglio, Perera, and Nestle 2011). Inflammasome assembly activates caspase-1, which in turn cleaves pro-IL-1β to active IL-1β (Figure 1).
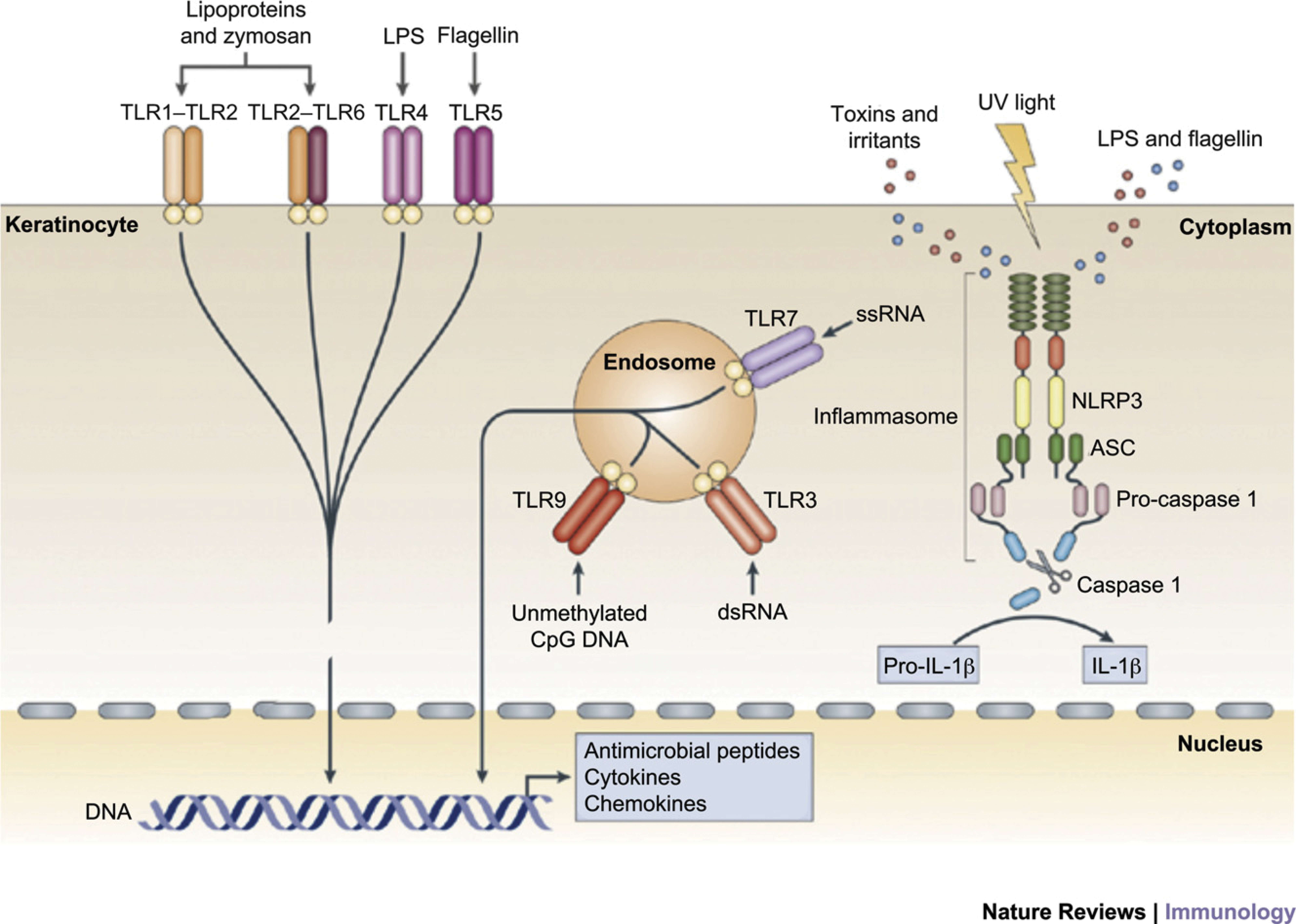
Keratinocytes as sensors of danger: keratinocytes are central skin sentinels and can recognize foreign and dangerous agents such as pathogen-associated molecular patterns (PAMPs) of microbial origin and danger-associated molecular pattern (DAMPs), such as irritants and toxins, through Toll-like receptors (TLRs) and the inflammasome complex. Keratinocytes also express nucleotide-binding domain, leucine rich repeat-containing (NLR) family, pyrin domain containing 3 (NLRP3), which belongs to a class of proteins encoded by the NLR gene family. These proteins can recognize PAMPs that are in the cytoplasm (such as lipopolysaccharide and flagellin), DAMPs, and ultraviolet (UV) light and in turn activate the inflammasome complex. This multimeric complex is formed by an NLR, an adaptor protein termed apoptosis-associated speck-like protein containing a caspase recruitment domain (ASC) and procaspase 1, and its assembly leads to the activation of caspase 1, which processes prointerleukin-1b into biologically active IL-1b. Reprinted from Nestle et al. (2009), with permission from Macmillan Publishers Limited.
The immune response triggered by keratinocyte activation is believed to be central to the skin’s response to a wide range of stimuli (Nickoloff 2006; Nestle et al. 2009; Di Meglio, Perera, and Nestle 2011) and leads to the stereotypic morphologic response(s) that we encounter as pathologists. Even the immune-mediated/autoimmune disease psoriasis is now believed to be at least partially caused by inappropriate or poorly regulated activation of epidermal keratinocytes, which in turn leads to inflammation and the hallmark morphologic changes associated with this condition (Nestle et al. 2009; Di Meglio, Perera, and Nestle 2011; Lowes, Suárez-Fariñas, and Krueger 2014). The ability of cytokines such as IL-22 and oncostatin M to induce morphologic and differentiation features in keratinocytes that mimic those found in psoriatic epidermis in 3-dimensional reconstituted human epidermal (RHE) models in the absence of blood vessels and leukocytes is remarkable and reinforces the hypothesis that epidermal keratinocytes are central to the initiation of cutaneous inflammation as well as the initiation of cutaneous response to injury, infection, and toxicity (Boniface et al. 2007; Sa et al. 2007).
Thus, the current model for the cutaneous response to injury, regardless of the specific type or etiology of the injury, is that the response is initiated by epidermal keratinocyte recognition of PAMPs or DAMPs via engagement of TLRs and/or NLRs, thus triggering proinflammatory signaling pathways and an inflammatory cascade that leads to the generation of antimicrobial peptides such as β-defensins, cathelicidins, and S100 proteins; proinflammatory chemokines such as IL-8, CXCL1, CXCL9, CXCL10, and CXCL11; and cytokines such as IL-1β, TNF, IL-6, and IL-18. These keratinocyte-derived chemokines and cytokines further recruit and activate dendritic cells (DCs) and other leukocytes to elaborate additional cytokines and chemokines, such as interferon (IFN)-α from plasmacytoid DCs (pDCs) and IL-12 and IL-23 from dermal DCs, which further recruit and activate T lymphocytes of both the Th1 and particularly the Th17/Th22 lineage to release proinflammatory cytokines such as IFNγ, IL-17, and IL-22, thus converting the initial innate immune response to an adaptive immune response and providing cross talk between the 2 arms of the immune system (Figure 2; Nestle et al. 2009; Di Meglio, Perera, and Nestle 2011). This immune response is believed to be central to the skin’s response to a wide range of injurious stimuli, regardless of the exact nature of the stimulus (Nestle et al. 2009; Di Meglio, Perera, and Nestle 2011).
Skin-resident immune sentinels. Ultraviolet (UV) light, trauma, irritants, or infection (essentially any type of barrier disruption) triggers a coordinated immune response to maintain skin homeostasis. Skin-resident immune cells are key sentinels for restoring homeostasis but can also be effector cells during cutaneous injury. Keratinocytes sense and react to noxious stimuli by producing proinflammatory cytokines such as interleukin-1b (IL-1b), IL-6, IL-18, and tumor necrosis factor, which in turn activate dermal dendritic cells (DCs). Innate immune cells, such as plasmacytoid DCs, activated by stress signals derived from keratinocytes, can also contribute to dermal DC activation by releasing interferon-α. Dermal DCs activate and promote the clonal expansion of skin-resident memory CD4+ or CD8+ T cells. Proinflammatory cytokines and chemokines from T cells can then in turn further stimulate keratinocytes, thus forming an amplification feedback loop for the inflammatory reaction. Finally, skin-resident T cells can migrate into the epidermis, engaging in cross talk between immune cells and keratinocytes. Reprinted from Nestle et al. (2009), with permission from Macmillan Publishers Limited.
Drug-induced Cutaneous Hypersensitivity
Cutaneous toxicity that occurs as the result of immune-mediated mechanisms falls into the same categories as systemic immune-mediated diseases (Lee and Thompson 2006; Segal et al. 2007; Ramdial and Naidoo 2009). Type I hypersensitivity is acute and classically manifests cutaneously as urticaria mediated by immunoglobulin (IgE) antibodies bound to the surface of mast cells and basophils (Friedmann et al. 2010). Type II hypersensitivity is characterized by cytotoxicity induced by IgG or IgM antibodies with complement activation. The various forms of pemphigus are an example of cutaneous type II hypersensitivity, with autoantibodies directed against keratinocyte antigens and inducing acantholysis or loss of cell–cell adhesion. While the pathogenesis of pemphigus has been understood to involve autoantibodies against the adhesion molecules desmogleins 1 and 3 for some time (Maruani et al. 2008), more recently it has been suggested that engagement of autoantibodies against keratinocyte acetylcholine receptors is also involved in the initial pathogenesis (Ruocco et al. 2013).
Type III hypersensitivity is mediated by IgG or IgM antigen–antibody immune complex deposition. Cutaneous toxicities due to type III hypersensitivity reactions are drug-induced cutaneous vasculitis and cutaneous lupus erythematosus (LE). Approximately 20% to 30% of cutaneous vasculitides are drug-induced, generally arising 7 to 10 days following administration of the inducing drug substance (Lee and Thompson 2006; Segal et al. 2007; Ramdial and Naidoo 2009). Vasculitis is characterized by inflammation of small cutaneous vessels with or without vascular necrosis and/or thrombosis. Vasculitis is also a relatively common drug-induced finding in the nonclinical setting; one particular manifestation is vasculitis that is induced by antidrug antibodies (ADAs) in an animal model directed against human or humanized therapeutic proteins. While the most common presentation for ADA-induced vasculitis is systemic with occurrence in multiple internal organs (Leach et al. 2014), ADA-induced cutaneous vasculitis can also occur (Figure 3). Drug-induced cutaneous lupus can have a variety of histologic presentations but is often characterized by an interface dermatitis consisting of a lichenoid inflammatory cellular infiltrate at the dermal–epidermal interface. Lichenoid drug eruptions have a very similar histopathologic appearance to cutaneous forms of lupus (Lee and Thompson 2006; Segal et al. 2007; Ramdial and Naidoo 2009).
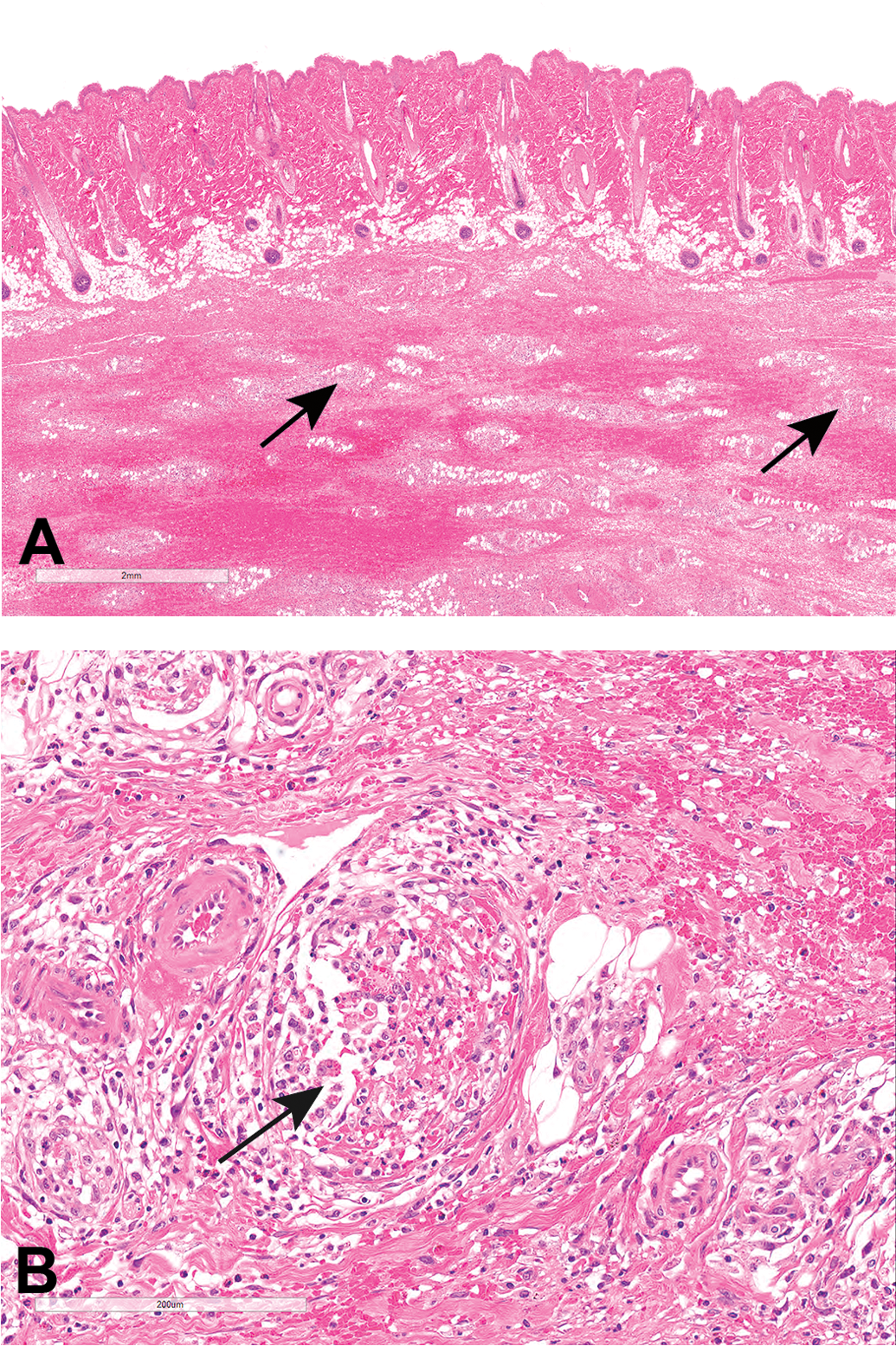
Skin and subcutis from a cynomolgus monkey administered a humanized therapeutic monoclonal antibody via repeated subcutaneous injections. The injection site subcutis contains relatively marked mixed inflammatory cellular infiltration, often surrounding and infiltrating into subcutaneous vessels that sometimes exhibit necrosis and/or thrombosis (perivasculitis and vasculitis). This animal had a very high antidrug antibody (ADA) titer of >log5, strongly suggesting that this subcutaneous vascular inflammation was the result of ADAs directed against the foreign protein.
Drugs most commonly implicated in drug-induced LE are carbamazepine, chlorpromazine, hydralazine, isoniazid, methyldopa, minocycline, penicillamine, procainamide, quinidine, terbinafine, omeprazole, and the TNF-α inhibitors infliximab and etanercept, both of which are biologics. The exact pathogenesis of drug-induced LE is unknown, but reactive drug metabolites are implicated for those induced by xenobiotics (Lee and Thompson 2006; Segal et al. 2007; Ramdial and Naidoo 2009).
It has been hypothesized that cutaneous autoimmune inflammation as exemplified in cutaneous lupus is dependent on pDC activation by nucleic acid antigens via the innate immune receptors TLR7 and TLR9 (Guiducci et al. 2010; Harr and French 2010). In the specific case of cutaneous lupus induced by the TNF-α inhibitor biologics, nucleic acid antigens termed nucleosomes become detectable in the plasma of rheumatoid arthritis (RA) patients after the start of TNF-α inhibitor therapy, suggesting that this rise in plasma nucleosome levels might contribute to a break of tolerance and thereby induce autoantibodies in susceptible individuals. This hypothesis is supported by the finding that antinucleosome antibodies correlated strongly with the presence of antinuclear antibodies, a hallmark of systemic LE, in anti-TNF-α treated RA patients (Guiducci et al. 2010).
In contrast to type I, II, and III hypersensitivities, which are generally systemic, type IV hypersensitivity is often a local reaction, and in the skin is referred to as allergic contact dermatitis (Lee and Thompson 2006; Segal et al. 2007; Ramdial and Naidoo 2009). Different toxic agents can induce more than one immune-mediated mechanism. As an example, penicillin, which is often cited as the classic example of a drug acting as a hapten, can cause both an IgE-mediated type I hypersensitivity reaction manifesting as urticaria (Friedmann et al. 2010) and non-IgE-mediated reactions that manifest as variable degrees of epidermal keratinocyte necrosis, ranging from erythema multiforme to toxic epidermal necrolysis and Stevens–Johnson Syndrome, severe conditions with widespread full-thickness epidermal necrosis and detachment from the underlying dermis (Figure 4; Lee and Thompson 2006; Segal et al. 2007; Ramdial and Naidoo 2009).
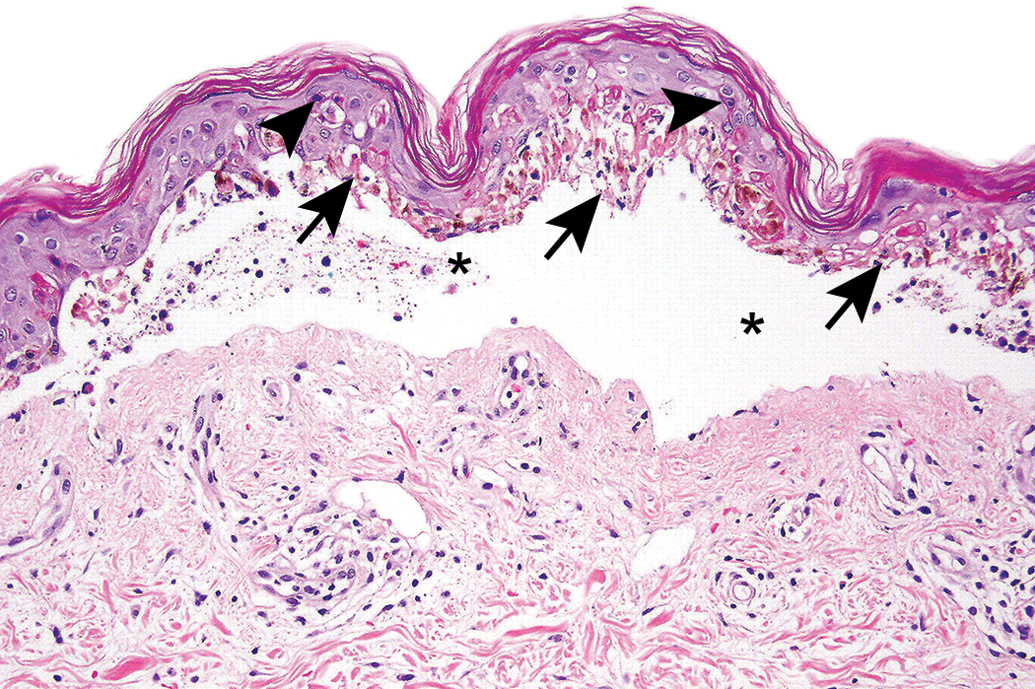
Skin from a human with toxic epidermal necrolysis illustrating widespread necrosis of the entire basal layer of the epidermis (arrows) with scattered necrosis of suprabasal keratinocytes (arrowheads) and epidermal–dermal separation and cleft formation (asterisks). The cleft is filled with a small amount of cellular debris and amorphous material. There is little or no associated inflammatory reaction in either the epidermis or dermis. Image courtesy of Dr. Phillip McKee.
The pathogenesis of immune-mediated cutaneous toxicity is not completely understood, although the current understanding suggests involvement of both the adaptive and innate immune system (Nestle et al. 2009; Di Meglio, Perera, and Nestle 2011). It had long been surmised that drugs and other xenobiotics acted as haptens, as mentioned earlier for penicillin, and when conjugated to proteins, presented to the immune system and elicited an immune response (Lee and Thompson 2006; Segal et al. 2007; Ramdial and Naidoo 2009). However, there is still much that is not fully understood. As mentioned previously, there is quite a bit of overlap in the mechanisms and lesions that can be induced by any 1 drug, such as penicillin, which can induce both type I and type IV hypersensitivity reactions. In addition, a single morphologic and diagnostic entity, such as cutaneous LE, can have more than one underlying pathogenesis. As mentioned earlier, the cutaneous LE that is related to therapy with anti-TNF biologics is likely distinct from that induced by nonbiologic drugs (Williams, Gadola, and Edwards 2009). With cutaneous LE induced by biologics, the underlying mechanism is believed to be due to the pharmacologic effect of lowered TNF-α levels leading to pDC activation by nucleic acid antigens via the innate immune receptors TLR7 and TLR9 (Guiducci et al. 2010; Harr and French 2010). In contrast, LE induced by xenobiotics is believed to be caused by reactive drug metabolites inducing haptens or by direct stimulation of the innate immune system (Lee and Thompson 2006; Segal et al. 2007; Ramdial and Naidoo 2009).
The innate immune system and, particularly, the epidermal keratinocyte have recently come to the forefront in the pathogenesis of drug-induced immune-mediated skin injury. As described earlier, keratinocytes sense DAMPSs through their TLRs, which trigger the release of antimicrobial peptides such as S100A8/S100A9 complexes, cytokines, and chemokines that in turn recruit and activate leukocytes (Nestle et al. 2009; Di Meglio, Perera, and Nestle 2011; Pasparakis, Haase, and Nestle 2014). Completing the loop, leukocytes that are recruited to the epidermis then release cytotoxic factors, such as perforin and granulysin from CD8+ cytotoxic cells and NK cells, as well as TWEAK, TRAIL, Fas ligand, and other TNF family members from macrophages and DCs that in turn induce the keratinocyte death that underlies drug-induced blistering syndromes such as erythema multiforme and toxic epidermal necrolysis (Chung et al. 2008; Guiducci et al. 2010; Harr and French 2010).
Gene expression analysis of cells from the blister fluid of patients with toxic epidermal necrolysis (TEN) and Stevens–Johnson Syndrome recently identified secretory granulysin, a cationic cytolytic protein secreted by cytotoxic T cells, NK cells, and NKT cells as a key molecule responsible for the induction of keratinocyte death (Chung et al. 2008). Blister fluid cells express high levels of granulysin messenger RNA, the protein is found in increased concentrations in blister fluid, and when injected intradermally into mice, granulysin induces keratinocyte necrosis and histopathologic lesions similar to those of TEN and Stevens–Johnson syndrome. Evidence suggests that granulysin alone is insufficient to induce cytotoxic T cell–mediated cell death, and that the simultaneous release of perforin is also required to fully induce apoptosis (Chung et al. 2008).
Psoriasis
A specific example of immune-mediated skin injury is psoriasis. Psoriasis is a common inflammatory condition of human skin characterized by focal to coalescing raised cutaneous plaques with consistent scaling and variable erythema (Krueger and Bowcock 2005). There are both generalized and localized forms of psoriasis, with the generalized form manifesting as broad areas of cutaneous coalescing plaques with scaling and variable erythema and the localized form having similar features but being limited to areas of friction/irritation (the Koebner phenomenon). Typical histologic features of psoriasis include epidermal hyperplasia with elongated rete ridges, a less discrete epidermal granular layer (hypogranulosis), parakeratosis, and leukocytic infiltration of the dermis and epidermis (Krueger and Bowcock 2005; Lowes, Bowcock, and Krueger 2007; Nickoloff and Nestle 2004). The cellular composition of the psoriatic leukocytic infiltrate is variable but very consistently contains both CD4+ and CD8+ T lymphocytes, with CD4+ T lymphocytes predominating in the dermis, and CD8+ T lymphocytes, particularly, those expressing the αEβ7 integrin, preferentially infiltrating the epidermis (Krueger and Bowcock 2005; Pasparakis et al. 2002). Psoriasis is the most common autoimmune disease in man, with a prevalence of between 2% and 4% worldwide (Kruger and Bowcock 2005; Lowes, Bowcock, and Krueger 2007). While the specific etiology of psoriasis is unknown, a genetic basis has been suspected for some time, and a number of different psoriasis susceptibility gene clusters, designated PSORS1, PSORS2, …, PSORS6, as well as over 30 single nucleotide polymorphisms (SNPs), particularly in the IL-23/IL-17 axis (more details on this axis are given below) have been identified, underscoring the heterogeneous nature of psoriasis (Krueger and Bowcock 2005; Nickoloff and Nestle 2004; Lowes, Suárez-Fariñas, and Krueger 2014).
The specific pathogenesis of psoriasis is still not fully understood, but there has been much recent progress in understanding many of its complex underlying mechanisms, which involve an interplay between epidermal keratinocytes, leukocytes (including dendritic cells and other antigen presenting cells), and vascular endothelium (Krueger and Bowcock 2005; Mease et al. 2000). Originally, psoriasis was considered to be primarily a disorder of dysregulated epidermal proliferation and differentiation, and indeed, agents such as retinoids and vitamin D analogs that treat epidermal differentiation defects have shown some efficacy in the treatment of psoriasis (Barker 1991; Kopp et al. 2001; Nickoloff 2006). With the discovery that immunosuppressive agents such as cyclosporin A and corticosteroids are often effective in treating psoriasis, the pendulum then swung toward psoriasis being considered primarily a disorder of the immune system, with T cells and specifically CD4+ T cells felt to play a central role (Nickoloff and Wrone-Smith 1999; Voskas et al. 2005). Initially, psoriasis was believed to primarily be a Th1 T helper cell–mediated process, driven by IFNγ and related cytokines (Krueger and Bowcock 2005; Lowes, Bowcock, and Krueger 2007). More recently, it has been shown that IL-23, a cytokine involved in the polarization of T helper cells into the Th17 subset (Park et al. 2005), plays a major role in psoriasis, as demonstrated by increased expression of the p19 and p40 subunits of IL-23 in psoriatic skin (Lee et al. 2004) and particularly by the convincingly demonstrated clinical efficacy shown by an anti-IL-23 p40 monoclonal antibody, ustekinumab, in moderate to severe plaque psoriasis (Blauvelt 2007; Kauffman et al. 2004; Kimball et al. 2012, Lee et al. 2004; Nickoloff 2007). Additional even more recent compelling evidence for the involvement of the Th17 T helper subset comes from convincing clinical efficacy demonstrated by an anti-IL-17A monoclonal antibody, secukinumab, in moderate to severe plaque psoriasis (Langley et al. 2014). Finally, as mentioned earlier, a strong association between psoriasis and several SNPs in the IL-23/IL-17 axis has been identified (Lowes, Suárez-Fariñas, and Krueger 2014).
In addition to this recent work establishing the important roles of both IL-23 and IL-17 in the immunopathogenesis of psoriasis, relatively recent data from several genetically engineered mouse models targeting epidermal keratinocytes have thrust the epidermal keratinocyte back into playing a central role in the pathogenesis of psoriasis and suggest that psoriasis is due to interactions and cross talk between epidermal keratinocytes and the immune system (Pasparakis et al. 2002; Nickoloff 2006; Pittelkow 2005; Sano et al. 2005; Stratis et al. 2006; Zenz et al. 2005). One possible candidate for linking the immune system and epidermal keratinocytes is IL-22, another cytokine that is produced by Th17 polarized T cells that are stimulated by IL-23 (Zheng et al. 2007). Unlike IL-23 or IL-17, however, IL-22 acts on epidermal keratinocytes and has been shown to induce epidermal hyperplasia with hypogranulosis, and upregulation/induction of many of the features found in psoriatic epidermis, such as upregulation of cytokeratin 16 and S100A7, and activation/phosphorylation of Stat3 in epidermal keratinocytes (Boniface et al. 2005; Nickoloff 2007; Sa et al. 2007; Sano et al. 2005). Additional evidence for the importance of IL-22 in psoriasis comes from the demonstration that an anti-IL-22 antibody greatly ameliorates skin lesions in the CD45RBHi SCID immune transfer mouse model of psoriasis (Ma et al. 2008). Therefore, Th17 cells and the cytokines that both drive their polarization (IL-23) and that are produced by these cells (IL-17 and IL-22) appear to play important roles in the immunopathogenesis of psoriasis (Blauvelt 2007; Zheng et al. 2007; Nograles et al. 2008).
Regardless of its specific underlying pathogenesis, psoriasis is felt to be initiated by an environmental trigger or injury, with subsequent amplification and immune cross talk between keratinocytes and leukocytes via an inflammatory cascade, much as is seen in generalized immune-mediated skin injury as described earlier (Figure 5; Nestle et al. 2009; Di Meglio, Perera, and Nestle 2011; Lowes, Suárez-Fariñas, and Krueger 2014). Many of the distinct epidermal features in psoriatic lesions, which are characterized by dysregulated epidermal hyperplasia with hypogranulosis and parakeratosis, upregulated expression of the hyperproliferative cytokeratins 6 and 16 and antimicrobial peptides such as the S100 proteins, and activation of the transcription factor Stat3 (Bowcock and Krueger 2005; Sano et al. 2005), are also features that are induced by IL-22 in RHE; Boniface et al. 2005; Sa et al. 2007; illustrated in figure 5 in Danilenko, Lewis Phillips, and Diaz (in this issue). In addition, psoriatic lesions contain dermal and epidermal leukocytic infiltrates with dilation of dermal blood vessels—all lesions that are maintained by the complex interplay between T cells and their cytokines, other leukocytes, vascular endothelium, and epidermal keratinocytes (Bowcock and Krueger 2005; Lowes, Bowcock, and Krueger 2007) via secretion of cytokines and growth factors, and the upregulation of signaling and adhesion molecules on their surfaces (Krueger and Bowcock 2005; Lowes, Bowcock, and Krueger 2007; Lowes, Suárez-Fariñas, and Krueger 2014).
The immunopathogenesis of psoriasis. In the psoriasis initiation phase, environmental factors coupled with psoriasis-susceptibility genes trigger an orchestrated cascade of pathogenic events leading to disease initiation and eventually plaque formation. Initially, injured or stressed keratinocytes (KCs) release self-nucleic acids and LL-37. These recruit plasmacytoid dendritic cells (pDCs) to produce interferon (IFN)-α, activate dermal DCs (DDCs), and inflammatory DDCs (iDDCs), which in turn produce IL-23, tumor necrosis factor (TNF), and nitric oxide radicals that promote the activation of skin-resident and newly recruited T cells that lead to plaque formation. Plaque progression occurs when IL-23 stimulates T helper 17 (Th17) and T cytotoxic 17 (Tc17) cells to express cutaneous leukocyte antigen (CLA), CCR6, and CCR4, plus very late antigen-1 (VLA-1) in the epidermis and to release IL-17A, IL-17F, IL-22, and IFN-γ. IFN-γ further activates DDCs while IL-17A and IL-17F act on KCs to promote production of T cells and neutrophil (Neut)-attracting chemokines (CXCL1, 3, 8, 11; CCL17–20) and antimicrobial peptides (AMPs), such as S100 proteins and LL-37. IL-22, which is also produced by Th1 cells as well as Th22 and Tc22 cells, induces epidermal hyperplasia by delaying KC terminal differentiation. Recruited unconventional Vg9Vd2T cells, which express CLA and CCR6, are activated by pDC-derived IFN-α and release further proinflammatory cytokines (IL-17A, IFN-γ, and TNF) as well as chemokines (CCL3-5) that attract Neuts and Th1 cells infiltrating Neut, mast cells, and macrophages, all contribute to the proinflammatory environment by producing cytokines (IL-17A, TNF), AMPs (S100 proteins, LL-37), and chemokines. Cross talk between KCs producing IL-1, TNF and transforming growth factor beta (TGF-β), and fibroblasts, which in turn release KCGF, epidermal GF, and TGF-β, and possibly Th22 cells releasing fibroblast GF, all contribute to tissue reorganization. Reprinted from Di Meglio, Perera, and Nestle (2011), with permission from Cell Press.
Summary and Conclusions
In summary, the skin has a fairly limited range of responses to injury, regardless of cause; thus, it is often quite difficult to determine the specific mechanism that underlies a particular skin lesion. However, regardless of the specific lesion and initiating factor(s), the immune response is felt to be central to the skin’s response to a wide variety of injuries, and the epidermis, and particularly the keratinocyte, is central to the initiation and manifestation of immune-mediated skin injury. Drug-induced immune-mediated cutaneous injuries or “drug eruptions” are relatively common, sometimes with overlapping mechanisms. And despite the skin’s fairly limited range of responses to injury, it is still often possible to classify immune-mediated cutaneous lesions based on the classical hypersensitivity type reactions, I through IV.
Psoriasis is the most common immune-mediated skin disease, with a prevalence between 2% and 4% worldwide. The pathogenesis of psoriasis is multifactorial and complex but involves the interaction of environmental factors with a genetic predisposition. The initial stimulus triggering the development of psoriatic lesions involves activation of epidermal keratinocytes, with subsequent amplification and persistence of lesions driven by the adaptive immune system. Several cytokines produced by Th17 T helper cells have recently been shown to be important in the pathogenesis of psoriasis, namely, IL-23 and IL-17, due to demonstrated clinical efficacy of cytokine blockade, and IL-22, based on its effects in both in vitro and in vivo models.
Footnotes
Acknowledgments
Author Contribution
The author (DD) contributed to conception or design; data acquisition, analysis, or interpretation; drafting the manuscript; and critically revising the manuscript. The author gave final approval and agreed to be accountable for all aspects of work in ensuring that questions relating to the accuracy or integrity of any part of the work are appropriately investigated and resolved.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.