
Abstract
The Society of Toxicologic Pathology formed a working group in collaboration with the American Society for Veterinary Clinical Pathology to provide recommendations for the appropriate inclusion of clinical pathology evaluation in recovery arms of nonclinical toxicity studies but not on when to perform recovery studies. Evaluation of the recovery of clinical pathology findings is not required routinely but provides useful information on risk assessment in nonclinical toxicity studies and is recommended when the ability of the organ to recover is uncertain. The study design generally requires inclusion of concurrent controls to separate procedure-related changes from test article–related changes, but return of clinical pathology values toward baseline may be sufficient in some cases. Evaluation of either a select or full panel of standard hematology, coagulation, and serum and urine chemistry biomarkers can be scientifically justified. It is also acceptable to redesignate dosing phase animals to the recovery phase or vice versa to optimize data interpretation. Assessment of delayed toxicity during the recovery phase is not required but may be appropriate in development programs with unique concerns. Evaluation of the recovery of clinical pathology data for vaccine development is required and, for efficacy markers, is recommended if it furthers pharmacologic understanding.
Keywords
Introduction and Background
Previous literature has addressed best practices in study design and evaluation of the recovery phases for nonclinical toxicity studies (Perry et al. 2013; Pandher, Leach, and Burns-Naas 2012; Sewell et al. 2014). While clinical pathology was mentioned in these articles, specific considerations for the appropriate inclusion of clinical pathology evaluation were not explored in detail. Standard clinical pathology biomarker (alternatively known in general as “variable” and as “analyte” for a chemistry biomarker) evaluation is critical for the monitoring of potential toxicity in the clinic following characterization in nonclinical studies, and it is also crucial to the understanding of recovery from toxicity. Since regulations for inclusion of clinical pathology in recovery arms of toxicity studies are neither detailed nor stringent, an opportunity exists for a review of the common practices in the pharmaceutical industry and to make recommendations based on sound scientific practices, with input from pathologists having diverse backgrounds. The Scientific and Regulatory Policy Committee of the Society of Toxicologic Pathology (STP) formed a working group in collaboration with the Clinical Pathology Interest Group of STP and the Regulatory Affairs Committee of the American Society for Veterinary Clinical Pathology (ASVCP) to provide insight on the following points regarding inclusion of clinical pathology evaluation in recovery studies: (1) appropriate comparisons for assessment of recovery of clinical pathology, (2) considerations of tissue recovery, (3) selection of recovery biomarkers based on dosing phase data, (4) considerations for exploratory or newly approved biomarkers, (5) evaluation of delayed toxicity, (6) biomarker recovery in vaccine studies, and (6) recovery of efficacy markers.
Current Regulatory Guidelines
Regulatory guidelines from organizations and agencies responsible for the investigation of the safety and/or efficacy of chemicals or human health products, such as the International Conference of Harmonization (ICH), U.S. Food and Drug Administration (FDA), Environmental Protection Agency (EPA), European Medicines Agency (EMA), Organisation for Economic Cooperation and Development (OECD), and the Japanese Ministry of Health, Labor, and Welfare, provide specific recommendations for the inclusion of body weight, food/water consumption, hematologic and clinical biochemistry measurements, and morphologic pathology investigations in nonclinical toxicity studies. Guidelines for recovery evaluation have been recently reviewed (Pandher, Leach, and Burns-Naas 2012; Perry et al. 2013).
Few guidelines for pharmaceutical development provide detailed recommendations regarding clinical pathology evaluation in the recovery phase of nonclinical regulatory toxicology studies. Recommendations for the inclusion of a recovery phase state that at least one nonclinical study over the course of the development of a compound should incorporate a recovery phase at the end of the study to determine the reversibility or potential worsening of test article–related effects; guidelines specify that recovery groups should be monitored until reversibility is demonstrated but full recovery is not necessary (FDA 2003; OECD 2008, 2009; ICH 2011; EPA 2001). However, an alternative to including a recovery phase is using scientific justification as to whether a toxicologic effect with potential adverse clinical impact is reversible. A nondosing period is warranted in a chronic study if there is severe toxicity at relevant clinical exposures or if toxicity is only detectable at an advanced stage of the pathophysiology in humans.
A few of the EMA guidelines indicate that in repeat-dose studies, clinical pathology should be evaluated during the dosing phase as recommended by Weingand et al. (1996) and then monitored at a frequency that allows assessment of changes over time (EMA Committee of Human Medicinal Products/Safety Working Party 2010). If there is a need for single-dose toxicity studies, hematology and clinical chemistry data should be evaluated ∼24 hr after a single administration, with further evaluations conducted 2 weeks later to assess for delayed toxicity and/or recovery (EMA Committee of Human Medicinal Products 2010).
The World Health Organization (WHO) guidelines on vaccines (Guidelines on Nonclinical Evaluation of Vaccines; WHO 2005) are an exception as they provide detailed recommendations for the evaluation of findings during the recovery phase. WHO recommends that the pivotal supporting nonclinical study should include additional treatment groups, which will be evaluated at later time points after vaccination to allow evaluation of the reversibility of adverse effects observed during the dosing phase and to screen for potential delayed adverse effects. WHO indicates that hematology and serum chemistry evaluation should be considered within 1 to 3 days following the first and last dose administration and at the end of the recovery phase (2 weeks or more following the last dose; WHO 2005). At a minimum, biomarkers to be evaluated include an evaluation of relative and absolute differential white blood cell counts, albumin/globulin ratio, enzyme activities (specific enzymes not detailed in the guideline), and electrolyte concentrations. In some cases, assessment of coagulation times and fibrinogen concentration, urinalysis, and serum immunoglobulin classes are recommended.
Current Pharmaceutical Practices
Common practices for the evaluation of clinical pathology during the recovery phase of nonclinical toxicity studies were compiled based on evaluation of the literature and the experience of the working group and are presented in Table 1. The number of recovery groups may be similar to the number of dosing phase groups or limited to control and high-dose groups. The study design relative to inclusion of recovery groups may be influenced by many factors including the availability of data from previous studies, company preference, or an attempt to minimize animal usage. The number of animals designated for the recovery phase is usually smaller than that used during the dosing phase (e.g., one-third to two-thirds of that in the dosing phase), although this could vary depending on the anticipated incidence of findings. Recommendations in this article are not based on a specific study design because the number of recovery groups and the number of animals evaluated per recovery group is unique to each test article. However, it is strongly recommended that the clinical pathology recovery assessment strategy be determined in consultation with a clinical pathologist and ideally with the one who will be interpreting the data.
Common Study Designs for Clinical Pathology Recovery Evaluation.a
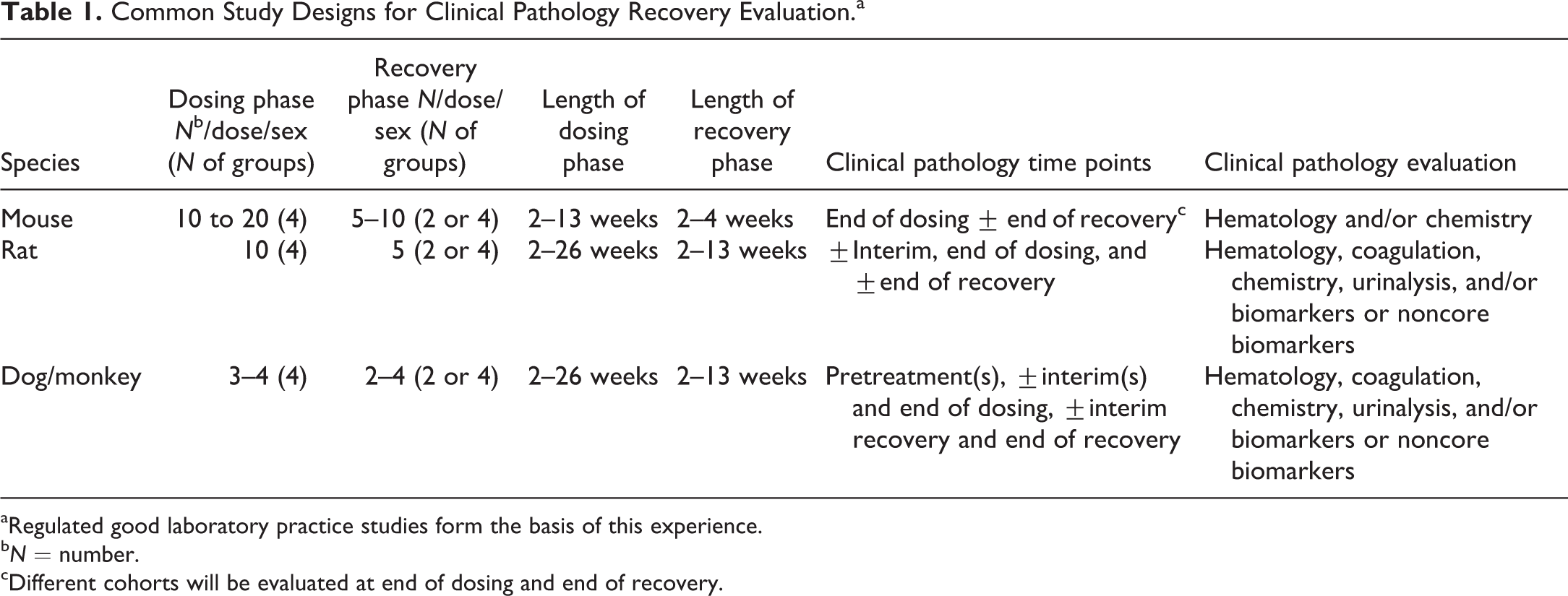
aRegulated good laboratory practice studies form the basis of this experience.
b N = number.
cDifferent cohorts will be evaluated at end of dosing and end of recovery.
Standard clinical pathology biomarkers (Tomlinson et al. 2013) are generally evaluated from individual recovery animals at the end of the dosing phase in most laboratory species (e.g., rats, dogs, and monkeys) other than mice as this helps with the assessment of reversibility in individual animals. In some studies, changes in individual clinical pathology biomarkers may help to determine which animals are to be retained for evaluation during the recovery. Particularly in rodents, evaluation of different cohorts at the end of the dosing and recovery phases may be required due to blood and sample volume limitations, thus preventing evaluation of recovery in the same individual animals. Repeated collections are rarely possible in mice as blood collection is usually a terminal procedure in this species. The length of the recovery phase is generally determined by the half-life of the test article or the nature of the test article–related findings. In most cases, all clinical pathology biomarkers that were assessed during the dosing phase are evaluated during the recovery phase. However, in the recovery phase, some investigators may opt to evaluate only the subset of biomarkers that were altered by target organ toxicity and/or test article pharmacology during the dosing phase. This decision can be determined on a case-by-case basis as long as there is scientific justification for it. For appropriate data comparisons to be made, blood sampling and analysis conditions (site of collection, fasting, anesthesia, and type of tube and analysis) should be the same for the dosing and recovery phases.
A large global retrospective analysis detailing general practices for use of recovery groups (Sewell et al. 2014) provided recommendations for minimizing recovery animals where scientifically justified, including avoiding recovery phases in first in human (FIH)-enabling studies. A broad review of the literature suggested that clinical pathology is most often incorporated in the recovery groups to evaluate reversibility of organ dysfunction (such as renal, hematopoietic, immune, and endocrine systems), with fewer reports incorporating clinical pathology to monitor tissue injury (Abraham, Gottschalk, and Ungemach 2005; Boorman et al. 1982; Henzen et al. 2000; Derelanko et al. 1985; Lefebvre et al. 1984; Rouse et al. 2011; Streck and Lockwood 1979; Terse et al. 2011).
A retrospective analysis of studies performed over the past 6 years at a large U.S.-based contract research organization indicated that recovery groups were included in ∼25% of studies. In most cases, the full clinical pathology profile was routinely included in the recovery phase. In a few instances, selected clinical pathology biomarkers were included if a test article–related effect was observed in these test(s) during the dosing phase.
Appropriate Comparisons for Assessment of Recovery in Clinical Pathology
Nonclinical study design recommendations by the FDA to support clinical trials do not specify which comparisons should be made for evaluation of reversibility. Reference values or potential comparators in toxicology studies are often obtained from 1 or more of 3 sources: a concurrent control group not exposed to the test article, baseline (alternatively known as “pretest” or “prestudy”) values compiled from all animals prior to dosing, or historical reference intervals collected from defined control and/or predose animals. Importantly, utilizing historical control values or “reference ranges/intervals” can result in misinterpretation of data, unless there is strict partitioning for animals of the same strain; age; sex; provider; comparable routes of administration; and vehicles and exclusion of animals with individual conditions, histories, and/or treatments (Hall 1997; National Committee for Clinical Laboratory Standards 1995; James 1993). The practice of strict partitioning prevents highly variable historical data which would be derived if control groups from nonclinical studies were given a variety of vehicles (Figure 1). The ASVCP has also developed guidelines for establishing reference intervals (http://www.asvcp.org/pubs/pdf/RI%20Guidelines%20For%20ASVCP%20website.pdf).

Variable alanine aminotransferase values in control male Han-Wistar rats given a variety of vehicles.
In rodent studies, a concurrent control group is considered to be necessary for the interpretation of data (Weingand et al. 1996; Leissing, Izzo, and Sargent 1985; James 1993). Using concurrent controls is recommended because there is less interanimal variation than with the use of historical data, it eliminates concerns with collecting baseline data from the same animals, and the animals are the same age at the end of the study. In accordance with suggested animal numbers (ICH 2009), rodent studies commonly incorporate larger numbers of genetically similar animals per group than nonrodent studies, resulting in lower biological variance. Due to the small differences between individual animals, a control group of ≥5 animals/gender is sufficient to assess recovery for the standard biomarkers. Because of low blood volumes, particularly in young rodents, baseline blood collection can negatively impact results and interfere with study interpretation or overall animal health. Satellite groups for other purposes such as toxicokinetics are often included to avoid impact of blood collection procedures on recovery animals. Due to the shorter life span of rodents, the periadolescent age that rodent studies typically start, and potential age-related effects on certain analytes (e.g., age-related decreases in serum phosphorus; Wolford et al. 1987), inclusion of a concurrent control group ensures comparison to appropriate age-matched controls (Pandher, Leach, and Burns-Naas 2012).
Studies in nonrodent species (dogs and nonhuman primates) generally use smaller numbers of more genetically diverse animals with higher interanimal variation but lower intraanimal variation; therefore, evaluation of baseline values is recommended to better understand the biological variance and aid in the interpretation of results during the dosing and recovery phases (Leissing, Izzo, and Sargent 1985; Boone et al. 2005; James 1993). However, the study purpose and/or complexity of the study design may dictate the need for concurrent controls in the recovery phase. For short-term studies with low animal numbers and limited blood collection or other evaluations during the dosing phase, low intra-animal variation may make it preferable to monitor the return toward baseline of potential test article–related changes in individual animals. However, as the study design becomes more complicated with additional pharmacokinetic, pharmacodynamic, antidrug antibody, immunophenotyping or other evaluations, separation of procedure-related changes from test article–related changes may not be possible without a concurrent control group. Nonhuman primates are particularly susceptible to impact red cell mass because of the blood volume requirements for additional biomarkers that can be more easily translated to the clinic than in other nonrodent species.
Considerations of Tissue Recovery
A crucial component of evaluating the reversibility of toxicity is the assessment of biomarkers indicative of structural organ damage and/or alteration of organ function. The standard recommended evaluations during the dosing phase and/or recovery phase include hematology, coagulation, clinical chemistry, and urinalysis panels to assess alterations in major organ systems (Tomlinson et al. 2013).
For the majority of test article–related changes in specific biomarkers, it is useful to monitor the return of those results back toward baseline levels, particularly when the recovery of clinical pathology findings is not well documented for a given test article. However, with some test article–related changes, there is no realistic potential that a biomarker will return to baseline, and thus measurement of the relevant biomarkers would not be warranted. An example is a permanent change in an organ such as the endocrine pancreas where the utility of evaluation of functional markers (e.g., insulin and glucagon) during the recovery phase is questionable in the presence of advanced nonrecoverable damage to the islets. Perry et al. (2013) provided examples of anatomic pathology findings that are not expected to recover; one example is injury to permanent or terminally differentiated tissues, such as cardiac muscle. As cardiomyocytes are considered postmitotic, there is no reasonable expectation that regeneration will occur. Thus, Perry et al. (2013) contend that recovery of the microscopic lesions should not be evaluated in such cases. With cardiac muscle injury, common analytes of assessment may include aspartate aminotransferase (AST) and creatine kinase (CK) activities and cardiac troponin I (cTnI). These analytes may be used to monitor cessation of active degeneration, but they are not indicative of restored functional capacity of the heart since irreversible cardiac injury resolves with fibrosis. Similarly, evaluating analytes should be carefully considered when stable tissues (quiescent with the ability to regenerate with appropriate stimuli, such as renal tubular epithelial cells or hepatocytes) have severe enough structural injury that tissue architecture is not expected to be restored. For example, with severe liver injury resulting in hepatic fibrosis that bridges and dissects liver lobules, recovery of hepatic injury marker changes (e.g., alanine aminotransferase and AST activities) would not reflect a full recovery of liver structure or function.
When tissue injury is expected to fully recover and can be easily monitored in a clinical setting, whether associated with microscopic changes or not, assessment of the recovery of biomarkers associated with that tissue injury may not be required. This decision should be based on the information available for the test article and/or pharmacologic mechanism. If the time course of damage and recovery is well understood for a test article and its related toxicity, then monitoring associated biomarkers after the recovery phase adds no additional information and is not required. For example, altered biomarkers from labile tissues (those that are continuously dividing), such as hematopoietic cells in the bone marrow and surface epithelium of the gastrointestinal tract, are expected to return to baseline/control levels. In these cases, the timing of the return toward baseline/control levels may be more critical than demonstration of full recovery. In agreement with Perry et al. (2013), when loss of architecture occurs in a labile tissue (such as bone marrow) or there is injury to a stable tissue (such as liver) but normal architecture is retained, there is more uncertainty about the ability of the tissue to recover. In those cases, morphologic recovery should be evaluated along with appropriate associated biomarkers, with consideration of the half-life of the test article and biomarkers and length of the recovery phase. For example, monitoring liver enzyme activity can provide information regarding ongoing liver injury, as well as timing of recovery. This could be particularly helpful when evaluating either small molecules or biologics with long terminal half-lives (several days to weeks).
The concurrent recovery of clinical pathology changes and clinical observations may help to distinguish changes that are secondary to poor clinical condition (e.g., decreased body weight or stress-related signs) from primary test article–related effects. For example, the return toward baseline/control levels of biomarkers associated with dehydration (e.g., urea and creatinine) and decreased food consumption (e.g., ALP, albumin, triglycerides, and effects on hematopoietic biomarkers such as reticulocytes) in the recovery phase should be correlated to the clinical signs of the recovering animals. If clinical pathology changes in the dosing phase are clearly identified as secondary or have been previously characterized in the program as secondary, evaluation at the end of the recovery phase is not required.
Selection of Recovery Biomarkers Based on Dosing Phase Data
In general, options for clinical pathology assessment following a recovery phase can include evaluation of the full panel, portions of the panel such as hematology but not clinical chemistry, or only affected biomarkers. A prescriptive recommendation cannot be made because there are aspects, such as biomarker storage stability, that affect the ability to delay assessment and thus, decision making on the assessment. An abbreviated panel can be considered if there are only changes in a portion of the clinical pathology panel, such as fibrinogen. If a decrease in fibrinogen is the only change at the end of the dosing phase, the decision could be to evaluate the coagulation panel (e.g., prothrombin time [PT], activated partial thromboplastin time [APTT], and fibrinogen) or fibrinogen alone during the recovery phase. In the latter case, the individual laboratory or sponsor should consider logistical considerations such as computer software templates to determine if there is an appropriate cost–benefit scenario to run one test instead of a typical panel of biomarkers. However, each laboratory has to weigh these considerations against the risk of creating unnecessary data versus the risk of producing a limited data set that cannot be interpreted properly. For example, if there are increases in fibrinogen alone at the end of the dosing phase, evaluation of related inflammatory parameters in hematology and serum chemistry panels should also be considered. Consultation with a clinical pathologist is recommended during this decision-making process.
There is limited utility in evaluating biomarkers after a recovery phase when there were no test article–related alterations at the end of the dosing phase. However, if clinical pathology results from the dosing phase are not available in time for interpretation and if waiting for evaluation compromises sample quality, then it may be preferable to routinely collect clinical pathology samples and/or evaluate them at the end of a recovery phase. Also, if there are specific concerns of potential alterations due to test article exposure during the recovery phase because of a long half-life, clinical pathology evaluation may be warranted.
Assessment of clinical pathology during the recovery phase should also be considered when test article–related clinical pathology changes indicate a functional effect that is not expected to have morphologic correlates or when clinical pathology changes are more sensitive predictors of toxicity than morphologic ones. For example, a decreased reticulocyte count may indicate injury to the bone marrow that is not yet apparent histologically or has not yet impacted red blood cell (RBC) mass because of the long life span of RBCs. If damage to the marrow is severe enough in a short-term study, the reticulocyte count may not improve or may decrease further during the recovery phase, and RBC mass may be decreased at the end of the recovery phase even though it was unchanged at the end of the dosing phase.
Interpretation of data can be challenging if appropriate time points and animals are not evaluated. In rodent studies, sufficient blood volume may not be available to sample every animal at every desired time point. Special attention must be paid to how animals are bled during the dosing phase and during the recovery phase to minimize preanalytic variation and optimize interpretation. For example, multiple collections during a study (interim, end of dosing, and/or end of recovery) would enable assessment at multiple time points in a single rat and correlation with histopathology data for animals terminated at the end of dosing or recovery phases. Bridging of doses between studies can facilitate comparisons of clinical pathology and histopathology data across studies; by utilizing data from shorter term studies, it is possible to minimize blood collection during the dosing phase of longer term studies, allowing collection of appropriate time points to interpret the recovery phase. For example, when only two time points can be evaluated per animal, it is most beneficial to collect data from end of dosing and end of recovery.
Animals from the control and selected dose group(s) are usually assigned to a cohort recovery group at the onset of the study. However, under certain circumstances, the assignment of animals may be altered to include animals that have been identified as having test article–related clinical pathology findings to ensure that representative changes are present in the recovery group and subsequently analyzed during the recovery phase. For example, if 5 of the 10 animals in the high-dose group exhibit marked hematologic changes, but none are assigned to the recovery group, then reassignment may be considered so that recovery of the finding can be assessed. However, if animals assigned to the recovery phase have critical levels of a biomarker (e.g., severe life-threatening thrombocytopenia or neutropenia), it might be advisable to terminate them at the end of the dosing phase. Similarly, if all animals with an increase in an injury biomarker are in the recovery group, it may be impossible to determine the histologic nature of the lesion present at the end of dosing without reassigning one or more animals to the end of dosing phase. Such reassignments are only recommended if they will make the data set more interpretable and are more likely to be utilized in studies with large animals rather than in rodents due to the smaller group size. A reasonable justification of any reassignment should be well documented in a study amendment. Reassignment is not recommended for animals with minor changes and may not be necessary for animals with changes that are expected to fully recover. Ensuring and documenting that appropriate animals are part of the recovery group to allow adequate assessment of recovery is the responsibility of the study director.
Considerations for Exploratory or Newly Approved Biomarkers
Standard clinical pathology biomarkers have been used for decades and their performance features (sensitivity, specificity, kinetics), assay formats, and caveats are well understood. This is not necessarily the case with novel safety biomarkers. Novel exploratory safety or nontraditional biomarkers should be used within a clearly defined context. The goals, anticipated outcomes, and reference standard or comparators such as the associated histopathology or functional change, should be identified prior to use in a study. The interpretation of the novel biomarkers should be based on these predefined endpoints. In some instances, very little is known about the biological role, sensitivity, kinetics, and species differences of novel biomarkers. The assays themselves are often at the exploratory stage of development and characterized as “fit for purpose” (Lee et al. 2006; Wagner 2008). Gathering data during the recovery phase will enable a progressive understanding of the candidate biomarker, as interpretation of changes in standard clinical pathology biomarkers and other study data can be used to put information on the novel marker biology in health and disease into context.
In 2011, the FDA accepted the qualification of cTnI as an indicator of the presence and extent of cardiac structural damage in dogs and rats based on data from studies of drugs with known cardiotoxicity or drugs that have shown evidence of cardiac damage when previously tested (http://www.fda.gov/downloads/Drugs/DevelopmentApprovalProcess/DrugDevelopmentToolsQualificationProgram/UCM294644.pdf). The intent of using cTnI as a biomarker of cardiac toxicity is to better define the lowest cardiotoxic dose in nonclinical studies of test articles known to induce structural cardiac damage and to help choose starting doses in clinical studies of those test articles; there is no intent to use cTnI as a general screening marker to detect unexpected cardiac toxicity. CTnI often peaks 2 to 6 hr after cardiac injury in nonclinical studies, depending on the duration and extent of cardiac injury (Clements et al. 2010; O’Brien 2008). Therefore, cTnI should be evaluated taking the short circulating half-life and the timing of the peak concentration of the drug into account, usually within 24 hr after drug administration. An increase in cTnI values, however, should be carefully evaluated. As in people, obtaining serial samples might help to evaluate the relevance of these changes in nonclinical studies. Given the kinetics and short circulating half-life of cTnI, measurement during a recovery phase may not be warranted, but the half-life of the test article must also be considered. A negative cTnI result does not indicate an absence of structural cardiac damage.
Beginning in 2010, FDA accepted several qualification submissions for 9 novel urinary biomarkers including kidney injury molecule-1 (Vaidya et al. 2010), clusterin, cystatin C, and renal papillary antigen-1 (Dieterle et al. 2010) that demonstrated improvement in the detection of acute drug-induced kidney injury in rats (Harpur et al. 2011). Subsequently, rat urinary osteopontin and neutrophil gelatinase-associated lipocalin (NGAL) have received regulatory endorsement in the form of a Letter of Support signed by Janet Woodcock on August 20, 2014. Most of the data supporting the use of these biomarkers were generated in 4- to 15-day studies, and recovery was not evaluated. Even though these renal biomarkers are accepted, there is still limited information about the kinetics, sensitivity, specificity, recovery, or assay interferences for them. The widespread use of these novel urinary biomarkers in rodent nonclinical studies, including their use to evaluate recovery of the target organ toxicity, will enable progressive qualification and further understanding of the biology unique to these markers.
Immunotoxicity findings (excluding immune-related findings secondary to stress) may be observed as intended pharmacological effects or as unexpected (off-target) effects of test articles. It is recommended that immunotoxicity findings undergo further evaluation either in follow-up studies, as described in the FDA guidance on immunotoxicology evaluation of investigational new drug (FDA Center for Devices and Radiological Health: U.S. Department of Health and Human Services 1999), or during the recovery phase of toxicity studies. For example, in the case of a lymphocyte-depleting antibody, thorough immunophenotypic evaluation of lymphocyte subsets and the kinetics of repopulation is essential for characterization of the pharmacologic activity of the test article, identification of potential adverse events, and determination of the most appropriate clinical administration regimen. However, the addition of recovery phases in order to assess immunogenicity is not required as per ICH harmonized tripartite guideline, preclinical safety evaluation of biotechnology-derived pharmaceuticals S6 (R1).
Evaluation of Delayed Toxicity
With the exception of nonclinical studies supporting microdosing in the clinic, there is no regulatory requirement for the evaluation of delayed toxicity in nonclinical studies. However, in nonclinical drug development, the addition of recovery groups should be considered when there is a specific concern for test article–related findings that could develop after the dosing phase. For example, for test articles with known class liabilities caused by effects on early hematopoietic or spermatogenic progenitors, a recovery phase may be used to monitor changes that may not manifest within the course of an FIH-enabling study. Data from recovery groups may allow decisions on the design of clinical dosing as well as subsequent nonclinical studies. In the case of biopharmaceuticals, recovery groups are commonly used to monitor for the potential of a test article to produce delayed toxicity during the off-dose phase due to either prolonged exposure or extended pharmacodynamic effects of the test article, although this is not required per ICH S6 (R1).
Although most of the concerns for delayed toxicity relate to morphologic changes, clinical pathology data can aid in the evaluation of target organ toxicities through identification of effects on organ function that might not be evident morphologically. For example, a compound affecting lymphoid stem cells in bone marrow could have delayed effects on peripheral lymphoid subpopulations due to the progressive depletion of circulating lymphocytes with a long half-life. Similarly, as T-cell maturation occurs in the thymus, a test article causing concurrent hematopoietic and thymic toxicity might exhibit a delayed or more severe effect on peripheral T-cell counts than a compound causing thymic injury alone due to the combined effects of senescence of mature T cells and decreased bone marrow lymphopoiesis. Finally, in the case of a nephrotoxicant with proximal tubular injury, concurrent urinalysis biomarkers, including evaluation of protein or albumin excretion with histopathology, could support demonstration of reversibility, particularly if there is histological evidence of tubular basophilia in recovery animals. As tubular basophilia can reflect either a regenerative response or a toxic change (Seely and Frazier 2015), recovery of urinalysis biomarkers can aid in the discrimination between regenerative and adverse tubular changes.
Biomarker Recovery in Vaccine Studies
As described earlier, regulatory guidelines for the recovery of clinical pathology biomarkers in the evaluation of vaccines are more detailed than for other areas of drug development. Vaccine programs are unique since a dose–response evaluation is not required as part of the basic toxicity assessment, and maximum tolerable doses do not need to be determined. As vaccines are given episodically rather than daily, the dosing intervals may be based on the kinetics of the primary and secondary antibody responses observed in the animal studies.
Vaccine administration causes acute phase protein increases that will usually be at their peak 24 to 48 hr postinjection. Therefore, only a mild, if any, change in acute phase proteins should be observed during or at the end of the recovery phase. While residual chronic inflammation is often described after the 2-week off-dose phase and is reflective of an expected tissue response, some recovery of the inflammation is expected during that time. Unless the immunological and inflammatory response to the vaccine is severe and prolonged, it is not considered to be adverse.
Extensive recommendations for vaccine evaluation have recently been published and are being readily adopted by companies developing vaccines (Green and Al-Humadi 2013). These recommendations provide a larger and updated list of biomarkers to consider during vaccine studies. The clinical pathology biomarkers recommended for evaluation at all time points include a complete blood cell count, a serum chemistry panel, a coagulation panel including fibrinogen concentration, and the measurement of acute phase proteins. The recommended acute phase proteins are C-reactive protein (CRP, which is the primary acute phase protein in rabbits, monkeys, and humans) and α 2-macroglobulin and α 1-acidic glycoprotein in rodents (Green and Al-Humadi 2013) or more specifically in rats (Cray, Zaias, and Altman 2009). This STP/ASVCP working group concurs with the recommendations but also recommends CRP in dogs and serum amyloid A and haptoglobin in mice (Cray, Zaias, and Altman 2009). When specific acute phase proteins are measured, serum electrophoresis analysis is not required. The measurement of CK activity, an analyte with a short half-life, is useful for the evaluation of the recovery from muscle injury, particularly in dogs (Tomlinson et al. 2013; Vassallo et al. 2009). Since vaccine administration may cause local muscular degeneration, the evaluation of CK activity or other biomarkers of skeletal muscle injury such as skeletal troponin I (Vassallo et al. 2009), dependent on the species being evaluated, is also recommended during the off-dose phase of nonclinical vaccine studies.
Recovery of Efficacy Markers
Efficacy markers enable monitoring of desired pharmacologic effects in both nonclinical studies and clinical trials and may be useful to evaluate in a recovery phase to understand persistence of these pharmacologic effects, particularly for biologic or large-molecule test articles that have long half-lives. For small molecules with short half-lives, efficacy is likely to decrease quickly following cessation of dosing, and evaluation of recovery for efficacy markers induced by these test articles is generally unnecessary.
A primary objective for the use of efficacy markers in nonclinical toxicity studies is to identify the relationship between intended pharmacologic effects and off-target or adverse findings. For example, comparison of the peak exposure for desired pharmacology with the exposure level where adverse findings occur can be used to define the relationship between on- and off-target effects. Subsequently, efficacy markers or clinical pathology biomarkers related to either desired pharmacology or target organ toxicity may be evaluated during a recovery phase in conjunction with histopathology to define the relationship between various findings or to inform the risk assessment of both intended and unintended findings. Utilizing the desired pharmacology, half-life, mode of action, and associated on- or off-target effects will optimize the design for evaluation of efficacy markers during the recovery phase.
The biological time course of a pharmacologic effect often determines duration of recovery. For example, increased glucose production due to treatment with a glucagon analog quickly returns to normal levels after test article exposure diminishes, whereas excessive production of RBCs caused by erythropoiesis-stimulating agents or an excessive number of leukocytes produced as a result of bone marrow stimulation by granulocyte-colony stimulating factor reverse slowly because the target cell populations may decrease slowly over time. In cases where cell populations are expected to change slowly or where long-lasting effects are expected, demonstration of complete recovery to baseline may not be necessary. However, frequent monitoring of efficacy markers can help to define the length of recovery required in some cases, such as for biologic test articles with longer half-lives. It may not be necessary or beneficial to assess a full panel of clinical pathology biomarkers if only intended pharmacology is being monitored.
Besides persistence of a pharmacologic effect, the need for recovery assessment may depend on the organ involved and the mechanism of the effect. With some immunomodulators, prolonged decreases in B- or T-lymphocyte populations can persist without clinical or other hematologic manifestations. In rare cases, adverse events can occur during the recovery phase due to immunosuppression or unintended immunostimulation. If such events are anticipated, close monitoring of efficacy markers, traditional clinical pathology biomarkers, and clinical observations may be warranted throughout the dosing and recovery phases. Also, with certain classes of test articles that are known to have long-lasting effects (e.g., some bisphosphonates or endocrine modulators), slowly progressive or delayed pharmacologic effects can occur and monitoring may be desired during the recovery phase.
Finally, it is recommended that for the evaluation of the recovery of efficacy markers, the pharmacologic effect should be present in animals designated for recovery. This is of particular concern in studies with biologic test articles that induce immunogenicity in the test species. Depending on the level of development and the effects on test article exposure of antitest article antibodies, not all animals will have similar pharmacologic effects that can be monitored during the recovery phase.
Conclusions
Regulatory guidelines provide the flexibility required for adapting the recovery phase to the needs of the program, but offer limited direction for the evaluation of clinical pathology in the recovery phase. The mandate of this STP/ASVCP working group was to consider the assessment of and to provide considerations and recommendations on the appropriate inclusion of clinical pathology biomarkers in the recovery phase of nonclinical toxicology studies.
The following recommendations represent the “best practice” for appropriate evaluation of clinical pathology biomarkers in the recovery phase of nonclinical toxicity studies. The clinical pathology recovery assessment strategy should be determined after consultation with a clinical pathologist, ideally the one who will be interpreting the data. Data from concurrent controls are recommended for assessment of recovery data in rodent studies and complicated nonrodent studies but in simple short-term (<28 days) nonrodent studies, comparisons of recovery data in test article–treated animals to baseline may be adequate. Recovery of biomarkers of toxicity should be assessed when the timing or ability of the organ to recover is of concern (e.g., loss of architecture in a labile tissue or injury to a stable tissue while normal architecture is retained). An evaluation of biomarkers is not required after the recovery phase if an associated group of biomarkers is unchanged after the dosing phase, the recovery of test article–related clinical pathology changes are well understood, there is no expectation of recovery, the changes are secondary effects, and/or the changes have no relevance to recovery. Assessment of clinical pathology during the recovery phase should be considered when clinical pathology changes indicate a functional effect with no morphologic correlates or when clinical pathology changes are more sensitive predictors of toxicity than morphologic changes. Reassignments of dosing and recovery phase animals are only recommended if they will make the data set more interpretable. Evaluation of clinical pathology biomarkers of delayed toxicity should be considered on a case-by-case basis in development programs with specific concerns for delayed toxicity that can be monitored using clinical pathology. In addition to the guidelines for clinical pathology assessment of recovery from vaccine administration, evaluation of acute phase proteins (CRP in dogs and monkeys, α 2-macroglobulin or α 1-acidic glycoprotein in rats, and serum amyloid A or haptoglobin in mice) and biomarkers of skeletal muscle injury is recommended. Evaluation of novel exploratory or nontraditional clinical pathology biomarkers during the recovery phase should be used within a clearly defined context. For the evaluation of recovery of efficacy markers, the pharmacologic effect should be present in animals intended for the recovery phase.
These recommendations are intended to complement rather than replace recommendations currently available in the literature and to provide additional clarification for the clinical pathology evaluation in the recovery phase of nonclinical toxicity studies.
Footnotes
Acknowledgments
The working group acknowledges the input from members of STP and ASVCP.
Author Contributions
Authors contributed to conception or design (LT, DE, and DB); data acquisition, analysis, or interpretation (LT, DE, LR, NT, VB, DB, FP, and AV); drafting the manuscript (LT, DE, LR, NT, VB, DB, FP, and AV); and critically revising the manuscript (LT, DE, and AV). All authors gave final approval and agreed to be accountable for all aspects of work in ensuring that questions relating to the accuracy or integrity of any part of the work are appropriately investigated and resolved.
Authors’ Note
This article is a product of a Society of Toxicologic Pathology (STP) Working Group involving the Scientific and Regulatory Policy Committee (SRPC) and Clinical Pathology Interest Group (CPIG) of the STP in collaboration with the Regulatory Affairs Committee (RAC) of the American Society for Veterinary Clinical Pathology (ASVCP) and has been reviewed and approved by the SRPC and Executive Committee of the STP.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.