
Abstract
Substances historically thought to cause direct vascular injury in laboratory animals are a heterogeneous group of toxic agents with varied mechanisms of action. Morphologically, the reviewed agents can be broadly categorized into those targeting endothelial cell (ECs) and those targeting smooth muscle cells (SMCs). Anticancer drugs, immunosuppressants, and heavy metals are targeting primarily ECs while allylamine, β-aminopropionitrile, and mitogen-activated protein kinase kinase inhibitors affect mainly SMCs. It is now recognized that the pathogenicity of some of these agents is often mediated through intermediary events, particularly vasoconstriction. There are clear similarities in the clinical and microscopic findings associated with many of these agents in animals and man, allowing the use of animal models to investigate mechanisms and pathogenesis. The molecular pathogenic mechanisms and comparative morphology in animals and humans will be reviewed.
Introduction
Drug-induced vascular injury has long been an obstacle for drug development due to our inability to monitor it in man or laboratory animal species. Vascular injury (VI) has been associated with compounds with vasoactive or hemodynamic properties, antibodies and other large molecules (Frazier et al. 2015), oligonucleotides (Engelhardt et al. 2015), and toxic agents causing “direct vascular toxicity.” Historically, vasoactive compounds received the most attention, and considerable efforts were made to understand their mode of action (Kerns et al. 2005). In addition, a number of papers covering biologicals and oligonucleotides have been published in the last 2 years. These well-researched vasoactive compounds (conventional vasodilators like fenoldopam and conventional vasoconstrictors like isoprenaline), biologicals and oligonucleotides, will not be covered by this publication. The subject of this review is a heterogeneous mix of toxic agents including toxins, toxicants including pharmaceuticals, and heavy metals that were originally thought to cause direct pharmacological or physicochemical damage to components of the vascular wall (Kerns et al. 2005; Louden and Morgan 2001). The main aims of this article are to review current knowledge on these drugs in order to complement and complete information published on VI during the last years, to discuss relevant molecular mechanisms of these toxic agents, and to present information about comparative morphology of vascular lesions in animals and effects in humans.
The list of toxic agents reviewed here includes pharmaceuticals like oncology and immunosuppressant drugs, as well as chemical and environmental agents like heavy metals and the industrial reagent allylamine. They can be broadly categorized as those targeting endothelial cells (ECs) or smooth muscle cells (SMCs) based on mechanism of action and morphology. Accordingly, the sections of this article are based on primary target cell and mechanism of action. There is now evidence that primary EC injury is caused by interference with cell division, by vasoconstriction, and by inhibition of EC receptors. These mechanisms will be discussed using typical example agents; but because most of them act through more than one mechanism, the combination of effects will be described. Toxic agents targeting smooth muscle frequently interfere with specific local enzyme systems. Nonconventional vasoconstriction (i.e., not related to adrenergic or ion channel drugs) is emerging as a mechanism of action in a wide variety of agents causing VI. An overview of mechanisms of VI and examples is presented in Table 1.
Overview of mechanisms and agents causing VI.
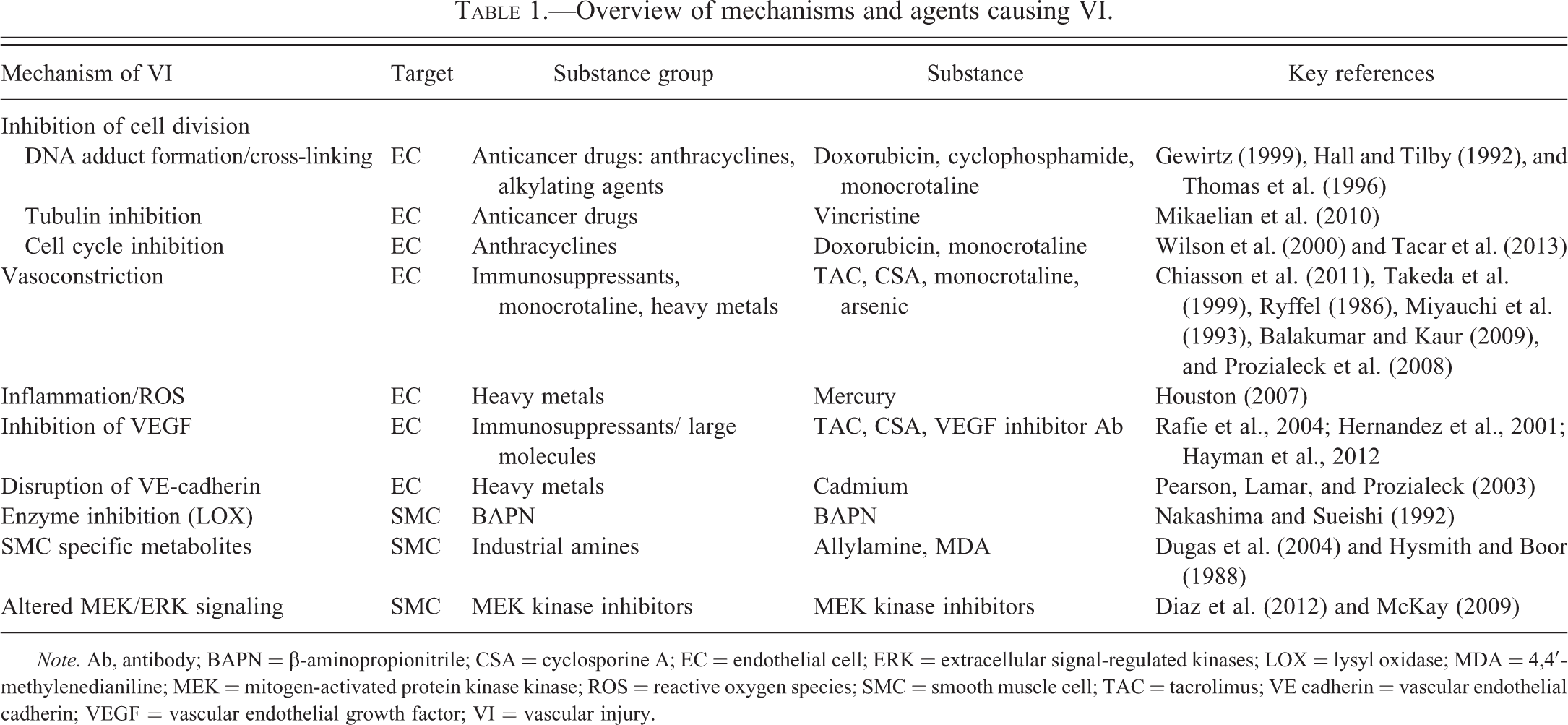
Note. Ab, antibody; BAPN = β-aminopropionitrile; CSA = cyclosporine A; EC = endothelial cell; ERK = extracellular signal-regulated kinases; LOX = lysyl oxidase; MDA = 4,4′-methylenedianiline; MEK = mitogen-activated protein kinase kinase; ROS = reactive oxygen species; SMC = smooth muscle cell; TAC = tacrolimus; VE cadherin = vascular endothelial cadherin; VEGF = vascular endothelial growth factor; VI = vascular injury.
In terms of morphology, there are no changes in preclinical laboratory animals that are characteristic for the entire group. There is, however, a clear divide between toxic agents that cause primary EC injury and primary SMC injury. Microscopically, the acute stage of EC death appears as apoptosis/necrosis, EC loss/desquamation, and exposure of basement membrane. Exposure of the basement membrane can lead to microthrombi and perivascular edema (e.g., following treatment with alkylating agents).
Endothelium targeting compounds produce lesions in a wide range of blood vessels that appear to be randomly distributed, targeting major organs like lung, heart, intestine, and kidney. Arteries, veins, and capillaries can be affected. Organs like the lung that have extensive capillary beds are more often affected. This is in sharp contrast to vasodilator drugs, which typically affect small to medium size muscular arteries in specific vascular beds, particularly in the mesentery and abdominal organs in rodents, and coronary blood vessels in dogs and monkeys (Mikaelian et al. 2014; Kerns et al. 2005). Primary SMC injury frequently appears as fibrinoid necrosis of the vascular media in the acute stage. The subacute and chronic stages are often dominated by proliferative and fibrotic changes of the vascular wall. It has been pointed out that, regardless of the initial insult, all microscopic vascular lesions look similar over time due to a limited repertoire of response (Greaves 2000), and therefore the morphologic differences between primary EC and SMC damage become blurred over time.
It will also become apparent that there are more morphologic and mechanistic similarities between findings in animals and humans in the presented agents of VI than in other groups of agents causing VI (e.g., antibodies or vasodilator drugs). This is of relevance for safety issues detected in preclinical studies, as they can be expected to translate more closely than findings with biologicals, oligonucleotides, and vasodilators. At the same time, this similarity allows the use of preclinical species for mechanistic and pathogenetic studies. As stated earlier, the toxic agents discussed in this article are grouped by the primary target cell (EC or SMC) and the most important mechanism of action. For each compound, known molecular mechanisms and the comparative morphology in man and preclinical animals are presented.
Mechanisms of Vascular Injury
The purpose of this section is to explain the primary and secondary effects of primary endothelial targeting agents and their interplay with SMC; special reference will be made to nonconventional vasoconstriction. ECs are capable of producing vasoconstricting and vasodilating agents (Bernatova 2014), including the potent vasoconstrictors endothelin-1 (ET-1) and angiotensin II (AngII; reviewed in Sprague and Khalil 2009). This may happen in the context of EC activation and lead, among other things, to increases in vascular permeability and changes in vascular tone. Both ET-1 and AngII are signaling through the G-protein coupled receptors angiotensin receptor-1 (AT-1) and endothelin receptor A (ETA) on SMCs. These signaling cascades result acutely in calcium influx and activation of Rho-associated coiled-coil-containing protein kinase (Rho kinase or ROCK) and subsequent vasoconstriction (Ivey, Osman, and Little 2008; Montezano et al. 2014; Nguyen Dinh Cat and Touyz 2011). In addition, AngII is thought to cause hyperplasia in SMCs through increases in platelet-derived growth factor and basic fibroblast growth factor and to cause fibrosis through increased accumulation of extracellular matrix (Ruiz-Ortega et al. 2003; Montezano et al. 2014). Therefore, primary EC activation also results in secondary SMC modification. AT-1 signaling also stimulates cell proliferation through activation of the mitogen-activated protein kinases (MAPK) and extracellular signal-regulated kinases (ERK). Similarly, ET-1 is known to stimulate MAPK, phosphatidylinositol 3-kinase, and protein kinase B in vascular SMCs, all of which can lead to SMC proliferation (Bouallegue, Daou, and Srivastava 2007; Ivey, Osman, and Little 2008). Cumulatively, these mechanisms cause vascular SMC hypertrophy and proliferation as well as fibrotic changes that are the morphologic hallmarks of chronic VI. Cytokines can also mediate EC dysfunction, causing ECs to be less responsive to vasodilator stimuli than normal (Bernatova 2014; Sprague and Khalil 2009).
Physiologic control of vascular tone depends on balancing vasoconstrictor and vasodilator influences (Figure 1). While there are numerous endogenous agents that can cause vasoconstriction, vasodilation is mainly caused by the short-lived second messenger nitric oxide (NO). Cardiovascular homeostasis is regulated by NO produced by various nitric oxide synthase (NOS) isoforms; constitutively expressed endothelial NOS is the most important for this review. Endothelial nitrous oxide synthase (eNOS) regulation is under the control of Ca2+/calmodulin, as well as numerous kinases (Mount, Kemp, and Power 2007). Phosphorylation at several loci of eNOS has been recognized as a mechanism for both activation and inhibition. There is increasing evidence suggesting that vascular endothelium reacts with NO production to stimulation of β-adrenoreceptors, in particular of the β2- and newly described β3-type (Conti et al. 2013). As the main factor opposing vasoconstrictive agents like ET-1, AngII, and others, NO protects the vasculature from their inflammatory and apoptosis-inducing effects. However, excessive levels of NO can also cause inflammation and cell death by reacting with reactive oxygen species (ROS) and superoxide dismutase (Choy et al. 2001). The net effect of NO—vasoprotective or harmful—therefore depends on local levels and the cells exposed. Toxic agents in this review can cause damage by decreasing NO levels, which reinforces vasoconstriction, and also by increasing NO levels, which contributes to inflammation and cell death.
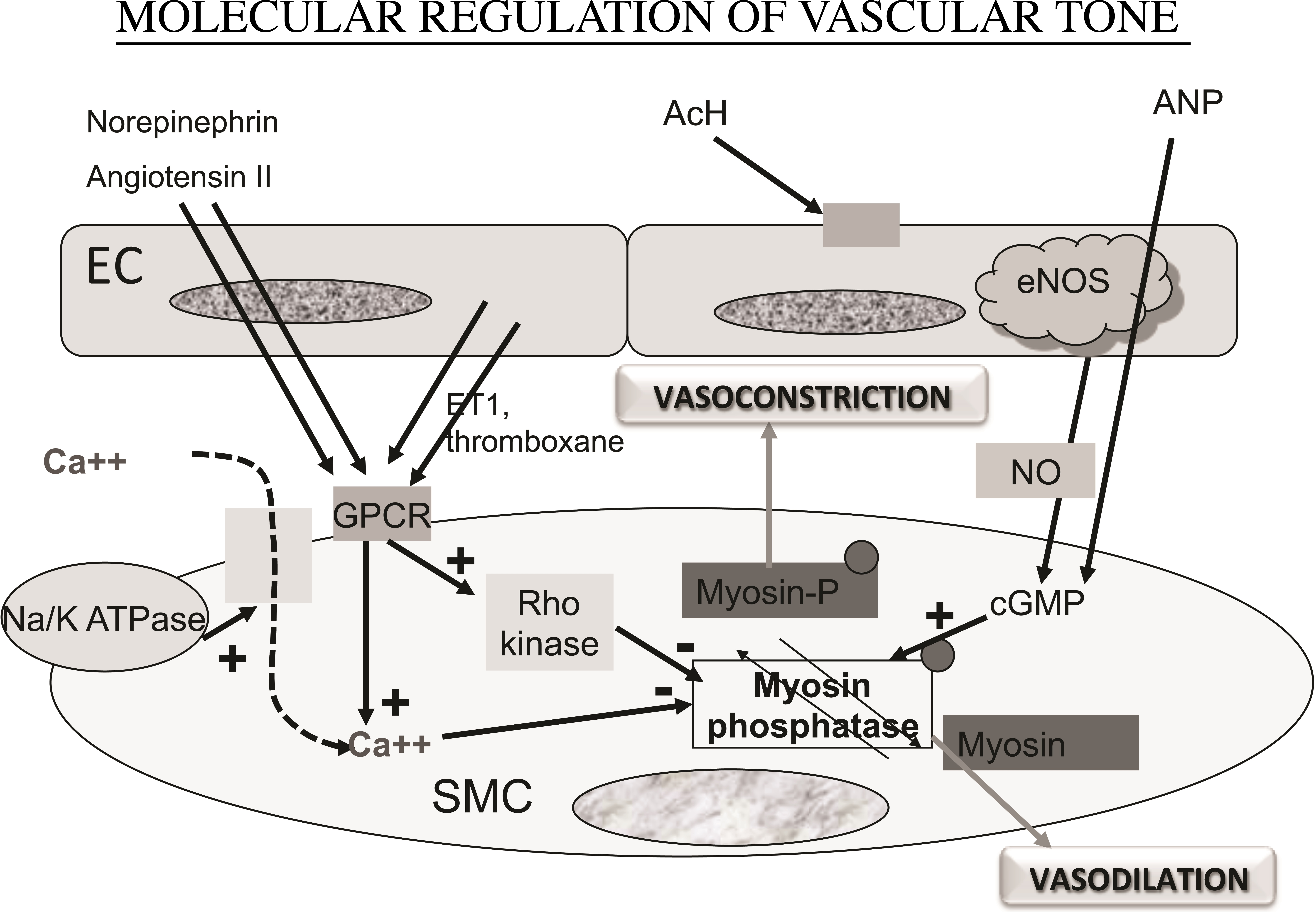
Molecular regulation of vascular tone. Vascular tone is regulated by circulating vasoactive agents, like the vasoconstrictors angiotensin II and norepinephrine (Montezano et al. 2014), and vasodilators acetylcholine (AcH) and atrial natriuretic peptide (ANP). Norepinephrine can stimulate endothelial nitric oxide synthase eNOS through β2 and 3 adrenoreceptors. Endothelial cells (ECs) produce the vasoconstricting endothelin-1(ET1) and the vasodilating nitric oxide (NO; Galley and Webster, 2004). Vasoconstrictors signal through G-protein coupled receptors (GPCRs) to increase intracellular calcium (Ca2+) and Rho-associated coiled-coil- containing protein kinase (Rho) kinase activity in smooth muscle cells (SMCs) (Ivey, Osman, and Little 2008; Higuchi et al. 2007), leading to inhibition of the enzyme myosin phosphatase. On the other hand, NO increases cyclic guanosine monophosphate in SMCs, leading to an activation of myosin phosphatase phosphorylation of the regulatory light chain of myosin (Myosin) results in vasoconstriction (Loirand and Pacaud 2010), while dephosphorylation leads to muscular relaxation and vasodilation.
Nonconventional vasoconstriction is an important toxic mechanism for the agents covered in this review. The main mechanisms by which these agents cause vasoconstriction are increases in ET-1 and decrease in NO production. Vasoconstriction and the induction of an inflammatory state are closely related. Both AngII and ET-1 are capable of causing cell damage through oxidative stress and the perpetuation of an inflammatory state. They are known to regulate the production of cytokines interleukin-1 (IL-1), tumor necrosis factor α (TNFα), and interleukin-6 through activation of nuclear factor kappa B (NFkB; Ruiz-Ortega et al. 2001; Callera et al. 2007; Sprague and Khalil 2009). AngII can also induce oxidative stress by induction of nicotinamide adenine dinucleotide phosphate (NADPH) oxidase (Callera et al. 2007; Loirand and Pacaud 2010; Nguyen Dinh Cat and Touyz 2011), and there is evidence for ET-1 acting in the same way (Fellner and Arendshorst 2007).
Both AngII and ET-1 signaling can result in activation of Rho kinase with profound short- and long-term effects on the cardiovascular system. Its importance in cardiovascular disease has been recognized in recent years, and Rho kinase has been proposed as drug target for diseases like hypertension (Loirand and Pacaud 2010; Surma, Wei, and Shi 2011).
Rho kinase is involved in acute vasoconstriction via regulation of myosin light chain phosphatase and by increasing Ca2+ sensitization in SMCs (Surma, Wei, and Shi 2011). It also promotes remodeling and inflammation through the production of pro-inflammatory cytokines like monocyte chemoattractant protein-1 (MCP-1), upregulation of NADPH oxidase, and paracrine growth mechanisms (Loirand and Pacaud 2010; Surma, Wei, and Shi 2011). In addition, NO production in ECs and NO mediated signaling in SMCs are negatively regulated by Ras homolog gene family, member A (RhoA), impeding vascular relaxation (Loirand and Pacaud 2010). Chronically, Rho kinase activation results in proliferation and fibrosis typical of long-term vasoconstrictor effects. Activation of Rho kinase by AngII is via protein kinase Cδ, proline-rich tyrosine kinase-2, and Rho-guanine nucleotide-exchange factors (Higuchi et al. 2007), some of which are controlled by other kinases (e.g., janus kinase-2). There are 20 immediate downstream targets of Rho kinase, many of which can also be activated by other serine–threonine kinases (Surma, Wei, and Shi 2011). It becomes apparent that Rho kinase is at the center of a dense network of related kinases involved in a multitude of cell functions with significance for VI (Burridge and Wennerberg 2004); this carries the risk that toxic agents acting on other kinases modulate Rho kinase, with unintended vascular effects.
In summary, toxic agents that activate ECs, decrease NO levels, and modulate Rho kinase cause nonconventional vasoconstriction and initiate apoptotic, inflammatory, and proliferative changes typical for all stages of VI.
Primary Endothelial Cell Injury
Inhibition of Cell Division
Inhibition of cell division in EC causes cell death, as in other tissues. Pharmacologic inhibition of cell division is a feature of anticancer agents. These drugs are typically administered to patients via intravenous injection, reaching high concentrations in contact with endothelium. The main mechanisms involved include interaction with DNA (anthracyclines and alkylating agents) or interference with the anaphase of cell mitosis (tubulin polymerization inhibitors like vincristine; Mikaelian et al. 2010). Cell cycle arrest and induction of apoptosis have also been reported (Thomas et al. 1998a, 1998b). An overview of primary mechanisms of EC damage is presented in Figure 2.
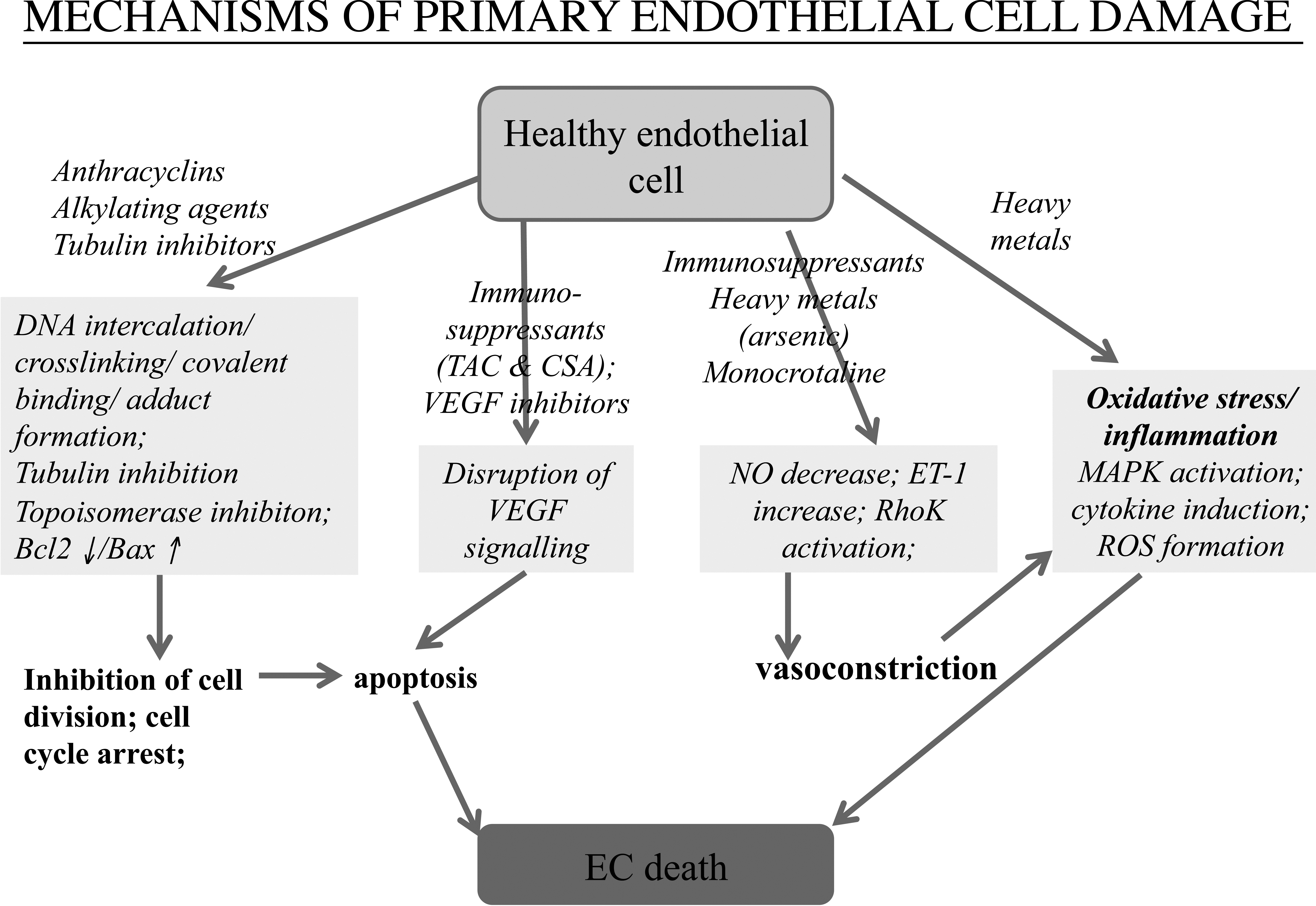
Mechanisms of primary endothelial cell damage. Four mechanisms of EC damage are presented together with exemplary vasotoxic agents and characteristic downstream events. TAC, tacrolimus; CSA, cyclosporine; Bcl2, B-cell lymphoma 2 protein; Bax, Bcl2 associated X protein; VEGF, vascular endothelial growth factor; NO, nitric oxide; ET-1, endothelin 1; RhoK, Rho-associated coiled-coil-containing protein kinase; MAPK, mitogen activated protein kinase; ROS, reactive oxigen species.
Interaction with DNA can occur in the form of intercalation, adduct formation, and covalent binding, all of which interfere with DNA transcription. These mechanisms have been described for the anthracycline doxorubicin (Tacar et al. 2013; Gewirtz 1999) and the alkylating agents cyclophosphamide and monocrotaline (Hall and Tilby 1992; Thomas et al. 1996; McCarroll et al. 2008). After enzymatic activation, cyclophosphamide causes DNA cross-linking via alkylation of guanine (McCarrol et al., 2008). DNA cross-linking has also been described for doxorubicin; however, this was only seen in vitro at concentrations exceeding clinical levels (Gewirtz 1999). The experimental drug monocrotaline is an alkylating agent like mitomycin C and cyclophosphamide and has been shown to form covalent bonds and cross-links with DNA (Thomas et al. 1996; Wang et al. 2005).
In addition to direct interaction with DNA, other pathways contribute to cell death in EC damage through cell division inhibition. The enzyme topoisomerase protects DNA integrity during transcription; topoisomerase inhibition is thought to be as important for the cytostatic activity of doxorubicin as DNA intercalation (Tacar et al. 2013). In addition, disturbances of the cell cycle may induce apoptosis, for example, via decrease in Bcl-2/increase in Bax or alterations of cell cycle check points. While the first mechanism has been demonstrated for doxorubicin (Takar et al., 2012, cell), persistent cell cycle arrests in G2 with persistent cyclin B1 expression were seen in experiments with monocrotaline (Wilson et al., 2000).
Endothelial damage has been recognized as a critical event in patients treated with oncology agents, contributing to efficacy as well as toxicity of cytostatic drugs. Some have in fact been termed “accidental anti-angiogens” (Soultati et al. 2012), as their antitumor activity is in part thought to depend on the disruption of tumor microvasculature. Cardiotoxicity has long been recognized as a major dose limiting effect in cancer patients, particularly following treatment with anthracyclines (Lipshultz, Alvarez, and Scully 2008; Soultati et al. 2012), and induction of apoptosis in cardiomyocytes and cardiac EC is thought to be the cause of dose-limiting cardiac toxicity seen in patients (Kalyanaraman et al. 2002; Wu et al. 2002). Similarly, cardiac (Buzdar et al. 1980; Godfrey and Wilbur 1972) and pulmonary (Cooper, White, and Matthay 1986; Gunstream et al. 1983; Lenci et al. 1994) toxicities are recognized side effects of cancer treatment with the alkylating agents cyclophosphamide and mitomycin, and endothelial damage is a contributing factor. Patients treated with cyclophosphamide develop pulmonary endothelial lesions with increased inflammatory cell adhesion, vascular sclerosis, pulmonary fibrosis, and pulmonary hypertension (Segura et al. 2001; Musiatowicz et al. 1997). Vascular complications due to both anthracyclines and alkylating drugs (alone or as part of combination treatments) include thrombotic microangiopathy, myocardial infarction, thrombosis/thromboembolic events, and hepatic veno-occlusive disease (Doll and Yarbro 1992; Meinardi et al. 2000).
Similar to man, EC death due to anthracyclines and alkylating agents particularly affecting the lung and heart has been documented in rats. Treatment of rats with doxorubicin resulted in increased EC apoptosis and caspase-3 immunostaining (a marker for apoptosis) in small myocardial blood vessels (Wu et al. 2002; Zhang et al. 1996; S. Wang et al. 2004). Besides direct induction of cell death, toxicity is thought to be also due to ROS formation and activation of MAP kinases (S. Wang et al. 2004; Grethe et al. 2006). Secondary changes in Sprague-Dawley (SD) rats included arteritis involving the kidney, pancreas, heart, skeletal muscle, and mesentery was seen after a single intravenous injection of the anthracycline daunomycin; the kidney was particularly affected (Sternberg, Philips, and Cronin 1972).
Short-term combination treatment of the 2 alkylating agents cyclophosphamide and busulfan in Balb/c mice resulted in EC degeneration and detachment, interendothelial gaps, and increased diameter of mesenteric arteries (Al-Hashmi et al. 2012). Mitomycin C and cyclophosphamide have been associated with vascular damage in rats, frequently involving the vasculature of the lung (Bregman et al. 1989; Kachel and Martin 1994). In Sprague- Dawley (SD) rats, single or multiple dose injection of BMY-25282, a mitomycin derivate, caused pulmonary changes that progressed from focal EC destruction and proliferation with subintimal fibrin accumulation, to hypertrophy of the tunica media and focal disruption of the elastic lamina. At later stages, fibrosis of the vascular intima and adventitia and mixed inflammatory infiltrates were also present. Necrotizing arteritis was present mainly in the lung but also in large intestine, kidney, pancreas, and testes. In the kidney and large intestine, intense mixed perivascular inflammation including eosinophils was characteristic in addition to fibrinoid necrosis of the media (Bregman et al. 1987).
Endothelial damage due to treatment with cytostatic drugs like mitomycin, cyclophosphamide, and doxorubicin can contribute to organ toxicity, particularly to the heart (Subbiah, Lenihan, and Tsimberidou 2011). This is increasingly becoming an issue in long-term cancer survivors, where mortality due to heart failure often is the main nonneoplastic cause of death (Green et al. 1999; Schlitt et al. 2014; Lindsey et al. 2014; Reulen et al. 2010). Vascular toxicity and cardiotoxicity due to cyclophosphamide can become apparent decades after treatment was initiated (Yeh et al. 2004; Santos et al. 1971; Storb et al. 1970; X. Wang, Zhang, and Xu 2009; Zver et al. 2007; Albini et al. 2010). In rodent hearts, a population of actively dividing ECs has been demonstrated (Mikaelian et al. 2010; Fernandez, Siddiquee, and Shohet 2001; Heron and Rakusan 1995). These ECs have a higher proliferation rate than ECs in other organs, which makes them more vulnerable to cytostatic therapy. Primary endothelial toxicity with mitotic arrest and induction of apoptosis has been established as the cause of cardiotoxicity for tubulin-binding drugs in rodents (Mikaelian et al. 2010). Similarly, doxorubicin has been shown to trigger apoptosis in proliferating ECs (Grethe et al. 2006). In conclusion, toxic agents interfering with cell division have been documented to cause primary EC damage, which is often particularly apparent in the heart and lung. Late stage organ toxicity in the heart may be in part explained by this mechanism. However, other mechanisms of damage like ROS formation have also been documented.
EC Damage Due to Vasoconstrictor Mechanisms
Apart from classic vasoconstrictor drugs that have long been known to have the potential to cause VI, nonconventional vasoconstriction is emerging as important mechanism in a variety of toxic agents. Anticancer agents cause vascular damage not only through direct interference with cell division. Reduced nitric oxide (NO) production and induction of inflammatory cytokines in EC contribute to thrombus formation, increased vascular permeability, decreased vascular relaxation, and arterial hypertension. Effects on the smooth muscles of the media reinforce vascular constriction, thus increasing hypertension. Neuropathic effects on the autonomic nervous system may contribute to vasoconstriction and hypertension through dysregulation of vasomotor function (Soultati et al. 2012). However, ET-1 increases, decreases in NO, and activation of Rho kinase are the main factors causing nonconventional vasoconstriction.
The experimental alkylating agent monocrotaline appears to cause vascular damage by vasoconstrictive mechanisms in addition to DNA binding. In man, the pyrrolizidine alkaloid monocrotaline causes pulmonary hypertension, cor pulmonale, and veno-occlusive disease in the liver (Huxtable 1990). Mechanistic studies in young rats demonstrated pulmonary hypertension following a single injection of 60 mg/kg, similar to the findings in humans. Hypertension was preceded by an increase in ET-1 levels (Miyauchi et al. 1993) and the ETA receptor antagonist BQ-123 significantly inhibited development of pulmonary hypertension in this model. ET-1 stimulates proliferation of vascular SMCs (Bouallegue, Daou, and Srivastava 2007; Ivey, Osman, and Little 2008), and BQ-123 inhibited monocrotaline-induced arterial medial thickening. Rho kinase activity also appears to be involved in monocrotaline toxicity. Abe et al. (2004) demonstrated that long-term treatment with the Rho kinase inhibitor fasudil improved pulmonary hypertension and ameliorated morphologic changes including vascular SMC hypercontraction, apoptosis and proliferation, and macrophage infiltration following administration of monocrotaline. These findings further underpin the role of vasoconstrictor activity not only in acute but also in chronic monocrotaline toxicity.
Morphologic changes in rats following a single subcutaneous injection include alveolar edema and the formation of fibrin thrombi with partial to complete occlusion in arteries, capillaries, and veins, demonstrating acute endothelial damage. The obliteration of vessels is also similar to humans. These findings are followed later by arteriolar medial hypertrophy and cellular proliferation and increased connective tissue in pulmonary septae (J. L. Lalich et al. 1977; Miyauchi et al. 1993). Endothelial hyperplasia and spreading of arterial smooth muscle have also been reported (Huxtable 1990). In mice, a severe inflammatory response is seen (Molteni et al. 1989). Pulmonary hypertension similar to rats and humans has also been induced in Stumptail monkeys (Macaca arctoides) by subcutaneous injection of monocrotaline (Chesney and Allen 1973). Lesions in monkeys were similar to the rat, with fibrin and platelet thrombi in capillary lumina, swelling and hypertrophy of arterial ECs, hypertrophy of medial smooth muscle, and panarteritis, culminating in vascular occlusion.
Some types of T-cell suppressors also appear to cause VI through vasoconstrictor mechanisms. The best characterized are tacrolimus (TAC) and cyclosporine A (CSA), which are used for maintenance immunosuppression in organ transplant recipients.
As in the case of monocrotaline, increased production of ET-1 appears to be one of the mechanisms contributing to vasoconstriction in TAC-treated transplant patients as well as in rats (Raina, Horn, and Benza 2012; Takeda et al. 1999; Textor et al. 2000). Takeda et al. (1993) described production of ET-1 in cultured ECs following treatment with TAC and CSA. Chronic treatment of rats with TAC significantly increased ET-1 and slightly decreased eNOS (Takeda et al. 1993). In addition, there is evidence of decreased NO production contributing to vasoconstriction in TAC-treated patients (Raina, Horn, and Benza 2012; Textor et al. 2000; Takeda et al. 1993). Both TAC and CSA suppress T cells by decreasing the transcription of interleukin-2 via inhibition of calcineurin. TAC forms a calcineurin-inhibiting complex with FK506-binding protein-12 (FKBP12). The TAC/FKBP12 complex also inhibits eNOS by phosphorylating its Thr495 residue, resulting in lower NO production and further moving the balance of soluble vasoactive factors toward vasoconstriction (Chiasson et al. 2011; Takeda et al. 1999). CSA has been shown to increase angiotensin levels in rats (Ryffel 1986), which may further contribute to vasoconstriction.
Hypertension, endothelial injury, and thrombosis are recurrent findings following treatment with TAC and CSA in man and animals. Patients treated with either TAC or CSA frequently experience hypertension and renal complications (Rezzani 2004; Lindenfeld et al. 2005; Jegasothy et al. 1992; Textor et al. 2000). Following treatment with CSA, acute vasoconstriction of afferent glomerular arteries causes endothelial injury and thrombotic microangiopathy, possibly in connection with EC apoptosis (Fortin et al. 2004). Histologically, EC vacuolation, SMC necrosis with nodular protein depositions, and mucoid degeneration have been observed (Kahan 1989; Rezzani 2004). Hyalinosis of renal arteries and arterioles (i.e., replacement of necrotic arterial smooth muscle by nodular protein deposits) has been seen following treatment with both CSA and TAC (Rezzani 2004; Textor et al. 2000).
In analogy to the situation in patients, hypertension has been noted in rodent models after treatment with TAC (Takeda et al. 1999) and CSA (Ryffel 1986) and may have contributed to the development of lesions. In Wistar/ST and Lewis rats, a single injection of TAC produced acute endothelial swelling, vacuolation, and detachment with subsequent thrombotic microangiopathy in the small intestine (Fujino, Kim, and Ito 2007), which is reminiscent of findings in man. Later, myxoid degeneration and necrosis of the media in submucosal arterioles were also described. Püschel et al. (2012) reported accelerated microvascular thrombus formation compared to saline in mice treated with TAC or CSA in a skinfold chamber experiment (Puschel et al. 2012). Mice treated with TAC developed renal arterial hyalinosis (Chiasson et al. 2012). In dogs used for experimental kidney transplants, TAC caused vasculitis characterized by fibrinoid necrosis, inflammation, and hypertrophy of the vessel wall in the heart, liver, and gastrointestinal tract (Collier, Thiru, and Calne 1987; Gunji et al. 1993).
CSA produced fibrinoid necrosis in the media and proliferative and obliterating changes in the intima of renal arterioles of various rat strains (Ryffel 1986; Ryffel et al. 1983). Lesions were easily induced in spontaneously hypertensive rats and more rarely seen in F344, Lewis, Wistar, and SD rats.
Vasoconstrictor mechanisms also play a role in the pathogenesis of arsenic toxicity. Exposure to arsenic, a heavy metal, has been linked to increased blood pressure in man (Balakumar and Kaur 2009; States et al. 2009; Prozialeck et al. 2008), possibly by increasing expression of ET-1. It also appears to induce endothelial dysfunction (an imbalance between vasodilation and vasoconstriction), by inactivating eNOS, decreasing NO, and inducing ROS (Balakumar and Kaur 2009). Increased expression of inflammatory cytokines like IL-1, TNF α, and MCP-1, increases in oxidized glutathione, and activation of NFkB were observed in human and animal ECs in vitro. In addition, arsenic induces a prothrombotic state by enhancing platelet aggregation, decreasing expression of tissue plasminogen activator and increasing plasminogen activator inhibitor type-1 (Balakumar and Kaur 2009). These factors induce endothelial injury, oxidative stress, and vascular proliferation (Balakumar and Kaur 2009; Prozialeck et al. 2008; States et al. 2009).
Hypertension, thrombosis, EC damage, and vascular occlusion appear as common findings in man and experimental animals following exposure to arsenic and other heavy metals. Heavy metals can occur as environmental pollutants, causing at times epidemics of toxicity. A well-known example is “blackfoot disease” in Taiwan, which is caused by high levels of arsenic in drinking water (Tseng 2005). Patients develop obliterating angiitis and arteriosclerosis, leading to progressive arterial occlusion and spontaneous amputation in the lower extremities.
Similar to humans, exposure to arsenic in domestic animals and rodents has been associated with capillary damage and increased vascular permeability (Tsai et al. 2005; Jubb and Huxtable 1993), thrombosis, and necrotizing and suppurative vasculitis (Pace et al. 1997). Exposure to arsenic has been shown to induce apoptosis in EC in vitro (Roboz et al. 2000), which is likely to be the initiating event. Administration of arsenic has been shown to increase blood pressure in rodents and rabbits (Yang et al. 2007; Carmignani, Boscolo, and Iannaccone 1983; Carmignani, Boscolo, and Castellino 1985; T. G. Park et al. 2005) and cause endothelial dysfunction and atherosclerosis in rodents (Sanchez-Soria et al. 2012; Carmignani, Boscolo, and Iannaccone 1983).
Similar effects have been attributed to the heavy metal cadmium, particularly cell death (Messner and Bernhard 2010) and the release of inflammatory cytokines, creation of a prothrombotic state, and proliferation of vascular SMCs.
As with other toxic agents, more than one mechanism is involved in VI due to heavy metal exposure. Oxidative stress, oxidized lipids, and low density lipoproteins can be produced not only by inflammation but also by the blocking of sulfhydryl groups by heavy metals like mercury (Jubb and Huxtable 1993). Many molecules belonging to the antioxidant defense system, such as glutathione, contain sulfhydryl groups and are therefore blocked by heavy metals, leading to secondary inflammation (Houston 2007; Anniko and Sarkady 1977; Patrick 2006).
Inhibition of EC Receptors
Inhibition of EC receptors like vascular endothelial growth factor (VEGF) and vascular endothelial (VE)-cadherin can contribute to EC death. VEGF signaling through nuclear factor of activated T cells is important for EC proliferation, migration, and vessel morphogenesis (Rafiee et al. 2004; Jang et al. 2010; Zaichuk et al. 2004) and can be specifically blocked by calcineurin inhibitors like TAC and CSA (Rafiee et al. 2004; Hernandez et al. 2001). VEGF is also a recognized trophic factor for ECs, and VEGF inhibition can potentially lead to EC apoptosis. Although not recognized in laboratory animals, treatment with large molecule VEGF inhibitors in cancer patients has been associated with hypertension, renal VI, and thrombotic microangiopathy (Hayman et al. 2012; Usui et al. 2014).
VE-cadherin is a component of endothelial adherens junctions and controls vascular permeability (Giannotta, Trani, and Dejana 2013). Cadmium has been shown to disrupt VE-cadherin in vitro in a process that precedes and is independent of the induction of necrosis (Prozialeck, Edwards, and Woods 2006). Loss of VE-cadherin has also been demonstrated in a murine in vivo model of cadmium-induced pulmonary injury (Pearson, Lamar, and Prozialeck 2003). This resulted in ultrastructurally visible gaps in adherens junctions between ECs and in increased vascular permeability and hemorrhage (reviewed in Messner and Bernhard 2010; Prozialeck, Edwards, and Woods 2006; Nolan and Shaikh 1986). Particular sensitivity of the testes to cadmium has been observed in multiple species. Dalton et al. (2005) showed that susceptibility to cadmium-induced testicular damage in mice was conferred by the Slc39a8 gene that encodes for zinc transporter 8, a transporter for Mn2+/HCO3−. This transporter can be hijacked by cadmium, increasing intracellular levels by up to 10 times. Macroscopically, hemorrhage and discoloration of the testes are observed in a range of species (rodents, rabbits, dogs, and cattle; reviewed in Prozialeck et al. 2008). Microscopically, this is consistent with perivascular edema, hemorrhage, microthrombosis, and ischemic necrosis. The lung, liver, placenta, uterus, and nervous system can also be affected. Primary EC damage is evident as endothelial vacuolation, necrosis, and sloughing into the vascular lumen, leaving the vascular wall exposed (Abe et al. 2004; Nolan and Shaikh 1986).
Inhibition of EC receptors therefore results in typical characteristics of primary EC damage including perivascular edema and thrombosis, while the organ distribution can be quite specific for the toxic agent and different from agents discussed in Inhibition of Cell Division and EC Damage Due to Vasoconstrictor Mechanisms subsections.
Agents with Direct Action on Vascular Smooth Muscle Cells
Compared with compounds acting directly on ECs, the number of agents directly affecting the vascular media is small. The mechanism of targeting the vascular media is not through spatial proximity but rather through interaction with specific characteristic enzymes of medial SMCs (compare Figure 3, mechanisms of primary SMC damage). In particular, vascular SMCs contain various oxidases (including semicarbazide-sensitive amino-oxidases [SSAOs], lysyl oxidase [LOX], cytochrome P450, and cyclooxygenase) which can be specifically inhibited or can activate xenobiotics locally (Zhao et al. 1998; Marnett, Reed, and Dennison 1978; Awasthi and Boor 1998; Trent and Boor 1994; Kumar, Hysmith and Boor 1990).
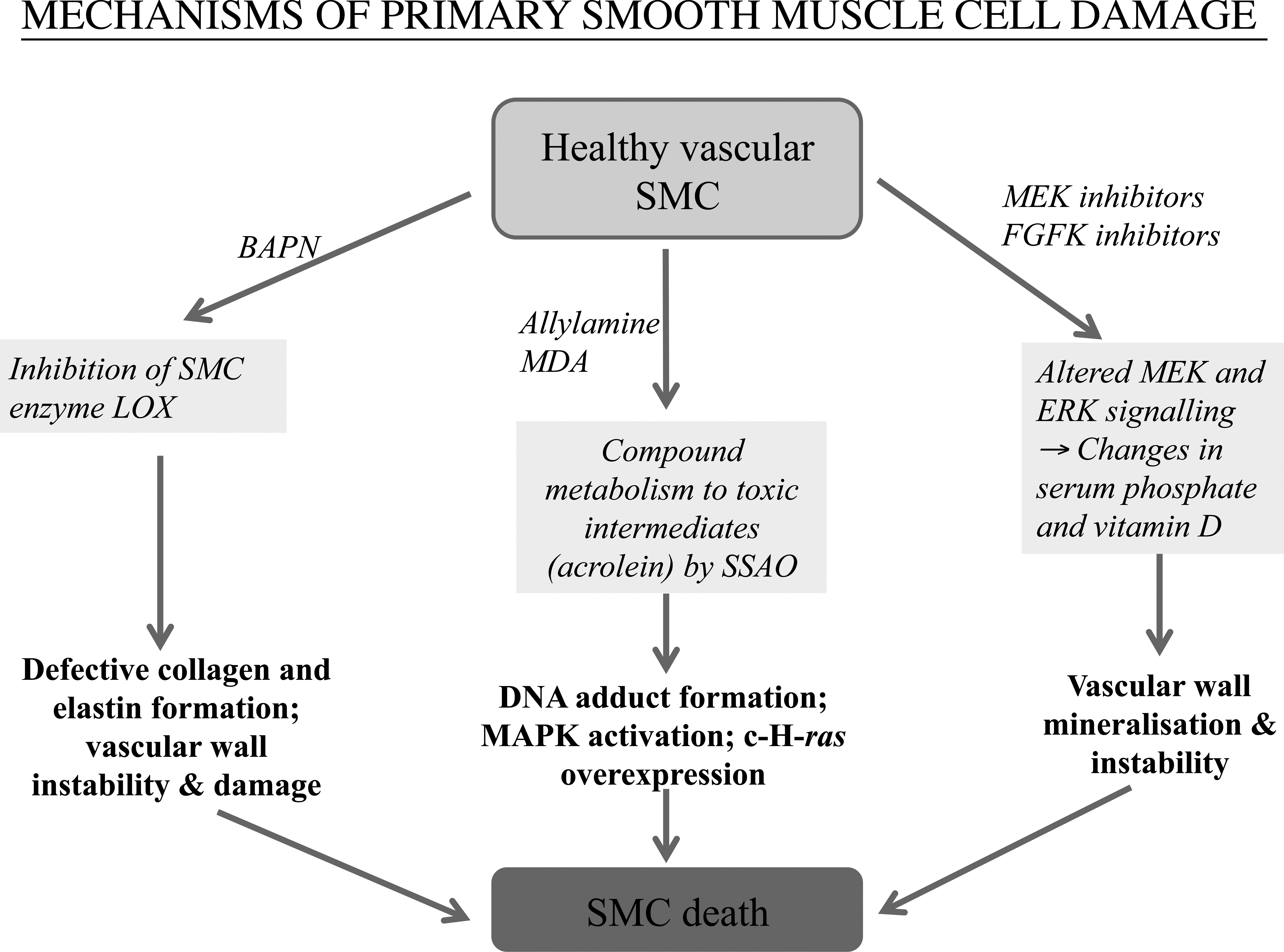
Mechanisms of primary smooth muscle cell (SCM) damage. Four mechanisms of vascular SCM damage are presented together with exemplary vasotoxic agents and characteristic downstream events. BAPN, β- aminopropionitrile; LOX, lysyl oxidase; MDA, 4,4′- methylendianiline; SSAO- semicarbazide-sensitive amino-oxidases; MAPK- mitogen activated protein kinases; c-H-ras, GTPase HRas; MEK, MAPK kinase; ERK, extracellular signal- regulated kinases; FGFK, fibroblast growth factor kinase.
Inhibition of LOX, which cross-links collagen and elastic fibers, making them more mechanically resistant, is caused by β-aminopropionitrile (BAPN). BAPN is a plant toxin known to cause aortic aneurysms in humans and animals (Boor 2001; Sherif 2010). A failure of this enzyme results in weakened arterial walls that over time are distended and distorted by blood pressure and may develop aneurysms. Administration of BAPN to rat pups caused a high incidence of fatal aortic aneurysms with prominent reduction of interlaminar elastic fibers (Walker and Wirtschafter 1956; Nakashima and Sueishi 1992) as well as high blood pressure (Nakashima and Sueishi 1992). The aneurysms were characterized by attenuation and disruption of elastic fibers, intramural hematomas, and by fibroblastic proliferation of the aortic wall. LOX is an essential factor in cardiovascular system development in mice (Hornstra et al. 2003; Maki et al. 2002). Besides a grossly abnormal, tortuous aorta, LOX knockout mice had discontinuous aortic SMCs, disrupted elastic fibers, aortic aneurysms, and hemothorax (Hornstra et al. 2003; Maki et al. 2002).
LOX is a member of the enzyme family of SSAOs (Sherif 2010), which have significant organ-specific distribution and activity (Boor and Nelson 1980). SSAOs can be hijacked by chemical agents to produce toxic metabolites, resulting in site-specific lesions. The toxic effect of allylamine, a highly reactive primary amine used in the chemical industry, depends mainly on its conversion to acrolein by SSAOs in vascular SMCs (Langford, Trent, and Boor 2002; Ramos, Grossman, and Cox 1988; Nelson and Boor 1982; Toraason et al. 1989; Hysmith and Boor 1988). Acrolein is capable of causing DNA adduct formation and activation of p38 mitogen-activated protein (MAP) kinase (Y. S. Park and Taniguchi 2008; Myers and Myers 2009). Proliferative changes associated with allylamine have also been attributed to c-Ha-ras overexpression (Bowes and Ramos 1993), which may explain the prominent proliferative response.
Early changes due to allylamine included perivascular edema, mononuclear infiltrates, and nuclear swelling. By 48 hours postdosing, perivascular fibroblast proliferation and adventitial thickening were present (Brady 2008). Chronic lesions include medial hyalinization, proliferation, and hypertrophy of the vascular wall, and increased adventitial collagen (Boor, Moslen, and Reynolds 1979; Boor, Nelson, and Chieco 1980; J. J. Lalich 1969; Waters 1948; Waters and Mc 1948; Boor and Hysmith, 1987).
Treatment of rats with 4,4-methylenedianiline, another aromatic amine used in industrial chemistry, resulted in increased SMC proliferation and hypertrophy of the media of pulmonary and hepatic arteries (Dugas et al. 2004), necrotizing inflammation of the portal vein (Bailie, Mullaney, and Roth 1993). Metabolisation to a toxic agent by cytochrome P450 enzymes in SMCs has been hypothesized.
Direct mineralization of the vascular media can occur as a result of mitogen-activated protein kinase kinase (MEK) inhibition. FGF23, a physiologic regulator of serum phosphate and 1, 25-dihydroxy vitamin D (Shimada et al. 2004), signals through MEK and ERK (Haussler et al. 2012; Perwad and Portale 2011). FGF23 knockout mice had mineralization of renal arteries, among other changes (Shimada et al. 2004). Fibroblast growth factor (FGF) suppresses vitamin D production, while inhibition of FGF or MEK increases it, leading to some of the symptoms of vitamin D toxicity like increased serum inorganic phosphorus and soft tissue mineralization (Haussler et al. 2012). Treatment of SD rats with MEK inhibitors resulted in mineralization of the aortic intima and media as well as arteries and veins in the heart (Diaz et al. 2012; McKay 2009). Similar changes without evidence of endothelial damage have been observed in young SD and mature Wistar rats after oral treatment with an FGF kinase inhibitor (Brown et al. 2005). Fatal hemorrhages secondary to aortic mineralization and rupture can be dose limiting.
Conclusion
A heterogeneous group of toxic agents that were in the past thought to cause “direct” VI are targeting primarily ECs or SMCs through different mechanisms. Anticancer drugs (alkylating agents and anthracyclines), immunosuppressants (TAC, CSA), and heavy metals are targeting mainly ECs while allylamine, BAPN, and MEK inhibitors affect mainly SMCs. DNA binding, interference with transcription, and cell cycle arrest can be found in cytotoxic anticancer drugs like the anthracyclines (doxorubicin and daunomycin), and alkylating agents (mitomycin C, cyclophosphamide) and tubulin inhibitors (vincristine). Nonconventional vasoconstriction (i.e., not related to adrenergic or ion channel drugs) is emerging as an important mechanism for a number of agents including monocrotaline, T-cell suppressors, and heavy metals. Increases in ET-1 and decreases in NO are frequent intermediate steps. Clinical hypertension and morphologic findings of EC damage/death, thrombus formation, and perivascular edema are a recurrent theme in EC targeting agents. EC targeting agents cause a more widespread organ distribution pattern of lesions, and lung and heart are often involved. Vasoconstriction also results in signaling cascades that drive inflammation and cell proliferation, exacerbating cell damage and ultimately creating the proliferative and fibrotic appearance characteristic for chronic VI. Agents targeting SMC often act locally via inhibition of enzyme systems (SSAO, LOX) or alteration of vitamin D metabolism. Most toxic agents act via more than one mechanism; ultimately, only heavy metals can truly be considered as direct acting by chemical interference with the sulfhydryl groups of antioxidant molecules, but in this group too, other mechanisms contribute to VI.
Interestingly, there is a large body of evidence supporting similar mechanisms both in man and animals for a large number of agents discussed in this review. In addition, where data are available, the microscopic lesions are very similar in man and animals. This contrasts sharply with pharmaceuticals causing VI where the mechanism observed in animals could not be identified in man (vasodilator drugs) or where pathology in preclinical animals is due to immunologic phenomena caused by the specific reactivity of a humanized compounds in animals (Frazier et al. 2015; Engelhart et al. 2015).
Interest in the toxic mechanisms of oncology drugs and immunosuppressants used in organ transplantation was limited in the past, possibly because of a considerable unmet need for effective treatment that would justify the risk of VI. A greater range of available treatments and longer survival times have changed the risk–benefit ratio and made the risk of VI less acceptable. For example, the long-term risk particularly to the ECs of the heart (Subbiah, Lenihan, and Tsimberidou 2011) is thought to contribute to cardiotoxicity that reach 65% in adult survivors of childhood cancer treated with anthracyclines (Volkova and Russell, 2011). A better understanding of the pathogenetic mechanism may improve safety assessment of VI and aid in finding both mitigating and preventative treatments. The mechanistic and morphologic similarities between findings in experimental animals and humans discussed in this review are relevant because they indicate that animal models are available to extend our understanding of these mechanisms.
Footnotes
Acknowledgment
The author wishes to acknowledge Dan Morton for valuable discussions.
Author Contribution
All authors (SG) contributed to conception or design; data acquisition, analysis, or interpretation; drafting the manuscript; and critically revising the manuscript. All authors gave final approval and agreed to be accountable for all aspects of work in ensuring that questions relating to the accuracy or integrity of any part of the work are appropriately investigated and resolved.
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
The author(s) received no financial support for the research, authorship, and/or publication of this article.