
Abstract
Drug-induced vascular injury (DIVI) is a nonclinical finding that often confounds the toxicological evaluation of investigational drugs, but there is an absence of qualified biomarkers that can be used to detect and monitor its appearance in animals and patients during drug development and clinical use. It is well known that endothelial cell (EC) activation plays a key role in the expression and evolution of DIVI, and the various immunological and inflammatory factors involved in its expression may serve as potential biomarker candidates. Activated ECs change their morphology and gene expression, generating endothelial adhesion molecules, pro-coagulant molecules, cytokines, chemokines, vasodilators, nitric oxide, and acute-phase reactants. This review provides a brief historical background of EC activation and the search for biomarkers of early EC activation for monitoring DIVI. At present, no biomarkers of EC activation have been qualified to predict DIVI in the nonclinical or clinical context, and a robust pathologic foundation for their use is still lacking. We propose three categories of EC activation biomarkers: recommended surrogate markers, potentially useful markers, and emerging candidate markers. This review alerts pharmaceutical companies, research institutions, and regulatory agencies to the continuing need for reliable biomarkers of EC activation in drug development.
Introduction
Drug-induced vascular injury (DIVI) is a nonclinical finding that often confounds the toxicological evaluation of investigational drugs. Despite uncertainties about the basic mechanisms and clinical implications of DIVI, and the absence of generally accepted biomarkers for monitoring DIVI in animals and patients, it is clear that endothelial cell (EC) activation plays an important role in its expression and evolution. Endothelial cell activation is an immunological and inflammatory response that promotes de novo expression of leukocyte adhesion molecules, the secretion of pro-inflammatory cytokines, the elaboration of chemokines, the de novo induction of procoagulant molecules, and the elicitation of T-cell–mediated immune responses (Ballermann 1998; Cockwell et al. 1997; Cotran 1987; Hunt and Jurd 1998; Pober 1988, 1998, 1999). Endothelial cell activation also plays a critical role in ischemia-reperfusion injury, thrombosis, immune responses, atherosclerosis, and transplant rejection, as well as in the initiation and progression of vasculitis, thrombosis, and vascular leak syndrome (Bach et al. 1994, 1997; Ballermann 1998; Cockwell et al. 1997; Cotran 1989; Hunt and Jurd 1998; Leung et al. 1989; Leung 1991; Oshima et al. 2001).
Currently, there are no biomarkers of EC activation that can be used to detect and monitor DIVI with both specificity and sensitivity. However, biomarkers of EC activation associated with DIVI remain of great interest to pharmaceutical companies, research institutions, and regulatory agencies. In this review, we propose the use of three categories of EC activation biomarkers—recommended surrogate markers, potentially useful markers, and emerging candidate markers intended to provide a framework for assessing the appearance of DIVI in preclinical and clinical studies.
Endothelial Cell Activation
The original concept of EC activation is based on the ultrastructural and light microscopic alterations of the vascular endothelium seen in the tuberculin reaction and in the delayed hypersensitivity and contact dermatitis reactions of guinea pigs treated with either azobenzenearsonate-insulin or 2,4-dinitrochlobenzene (Willms-Kretschmer et al. 1967). The concept of EC activation, without overt evidence of endothelial injury or division, has been expanded to include enhanced expression of specific gene products (i.e., proteins) and specific elements involved with inflammation, coagulation, and immunity (Cotran et al. 1986; Cotran 1987; Cotran et al. 1987; Pober 1988; Pober and Cotran 1990a; Pober 1998).
Endothelial cell activation is divided into two stages, “EC stimulation” (an early event) and “EC activation” (a later event), and they are referred to as “Type I EC activation” and “Type II EC activation,” respectively (Bach et al. 1994; Pober and Cotran 1990a, 1990b). Type I EC activation occurs immediately following stimulation and does not require de novo protein synthesis or gene transcription. The surface of the type I-activated ECs is capable of shedding endothelial adhesion and antithrombotic molecules, such as P-selectin, thrombin, heparin, antithrombin III, and thrombomodulin (Bach et al. 1994). In Type I EC activation, the endothelium in the venules and small veins rapidly retracts. Loss of EC contact can lead to hemorrhage, edema, and markedly increased vascular permeability (Bach et al. 1994).
Type II EC activation is a delayed response that is dependent on the activation of gene transcription and the de novo synthesis of proteins (Pober and Cotran 1990a, 1990b). The genes involved in Type II EC activation encode adhesion molecules, cytokines, chemokines, and procoagulant factors (Bach et al. 1994; Goepfert et al. 2000). In Type II EC activation, the endothelium expresses E-selectin on its surface and releases von Willebrand factor (vWF), chemokines (IL-8), and platelet-activating factor (PAF) (Bach et al. 1994; Hunt and Jurd 1998). The endothelium also secretes nitric oxide (NO) and prostacyclin (Goepfert et al. 2000). The ultrastructural alterations of the type II EC activation are characterized by protrusion of ECs into the lumen of blood vessels, hypertrophy of ECs (plump cuboidal appearance), increased biosynthetic organelles (Golgi complex, rough endoplasmic reticulum, and ribosomes), and increased permeability (pinocytotic vesicles) (Willms-Sretschmer et al., 1967). Monocytes and lymphocytes appear in the vicinity of the activated ECs, and harboring Fc receptors (FcR), may be exposed to the basement membranes, extravasated through the interaction of selectins and integrins, and further activated by binding of FcR to deposited immune complex (Kevil and Bullard 1999; Willms-Kretschmer et al. 1967).
Endothelial cell activation is distinct from endothelial injury; however, the two phenomena are likely to overlap in the activation process (Pober and Cotran 1990b). Endothelial cell activation may be associated with, and precede, vascular injury in vivo (Cotran 1989). Endothelial cell activation represents a reversible endothelial alteration resulting in morphological rearrangement (increase in cell size and cytoplasmic organelles) and inducible new functions, but without loss of endothelial integrity (Cotran 1989). The phenotype of activated ECs may return to the quiescent, nonactivated phenotype (Ballermann 1998; Blann 2000). However, the EC activation process, if uncontrolled, can progress to EC apoptosis (Bach et al. 1997). In contrast, EC apoptosis represents irreversible endothelial injury, endothelial fragmentation, and EC separation from the intima.
The terms “EC activation,” “EC injury/EC damage,” “and “EC dysfunction” are not interchangeable and should not be used without a clear definition of each (Blann 2000). The term “EC activation” is distinct from sublethal injury with consequent EC dysfunction (Pober 1988). Electron microscopy shows that the morphological changes of EC activation (increased biosynthetic organelles) are reversible upon the withdrawal of cytokines, tumor necrosis factor, and interferon (Pober 1988). Endothelial cell activation may also lead to EC dysfunction without evidence of vascular injury, as seen in the vascular leak syndrome induced by interleukin-2 (Pober 1988). Dysfunction of EC can manifest as imbalance between relaxing and contracting factors, for example, nitric oxide (NO) and endothelin; between procoagulant and anticoagulant mediators; or between growth-inhibiting and growth-promoting substances (De Meyer and Herman 1997). Endothelial cell dysfunction involves disruption of its vasoactive role in regulating tissue perfusion (Szmitko, Wang, Weisel, de Almeida et al. 2003), and such dysfunction may play an important role in the development and maintenance of high blood pressure via the phosphatidylinositol/Ca2+ signaling pathway for nitric synthase (Hedner et al. 2000). Therefore, the process of EC activation leading to EC dysfunction and EC injury evidently involves a series of immediate and delayed events. The first event may be a very early, immediate immunological activation of EC (type I EC activation) involving the release of stored proteins independent of de novo protein synthesis, followed by an early delayed activation (type II EC activation) that does involve de novo synthesis and secretion of the proteins. Endothelial cell dysfunction with irreversible EC injury can be produced by uncontrolled chronic and persistent EC activation. Persistent EC activation can result in critical local levels of endothelial adhesion molecules, procoagulant molecules, vasodilators, cytokines, chemokines, vasculitis thrombosis EC necrosis, and other mural cell injury. Figure 1 demonstrates the close relationship between EC activation, EC dysfunction, and EC injury.
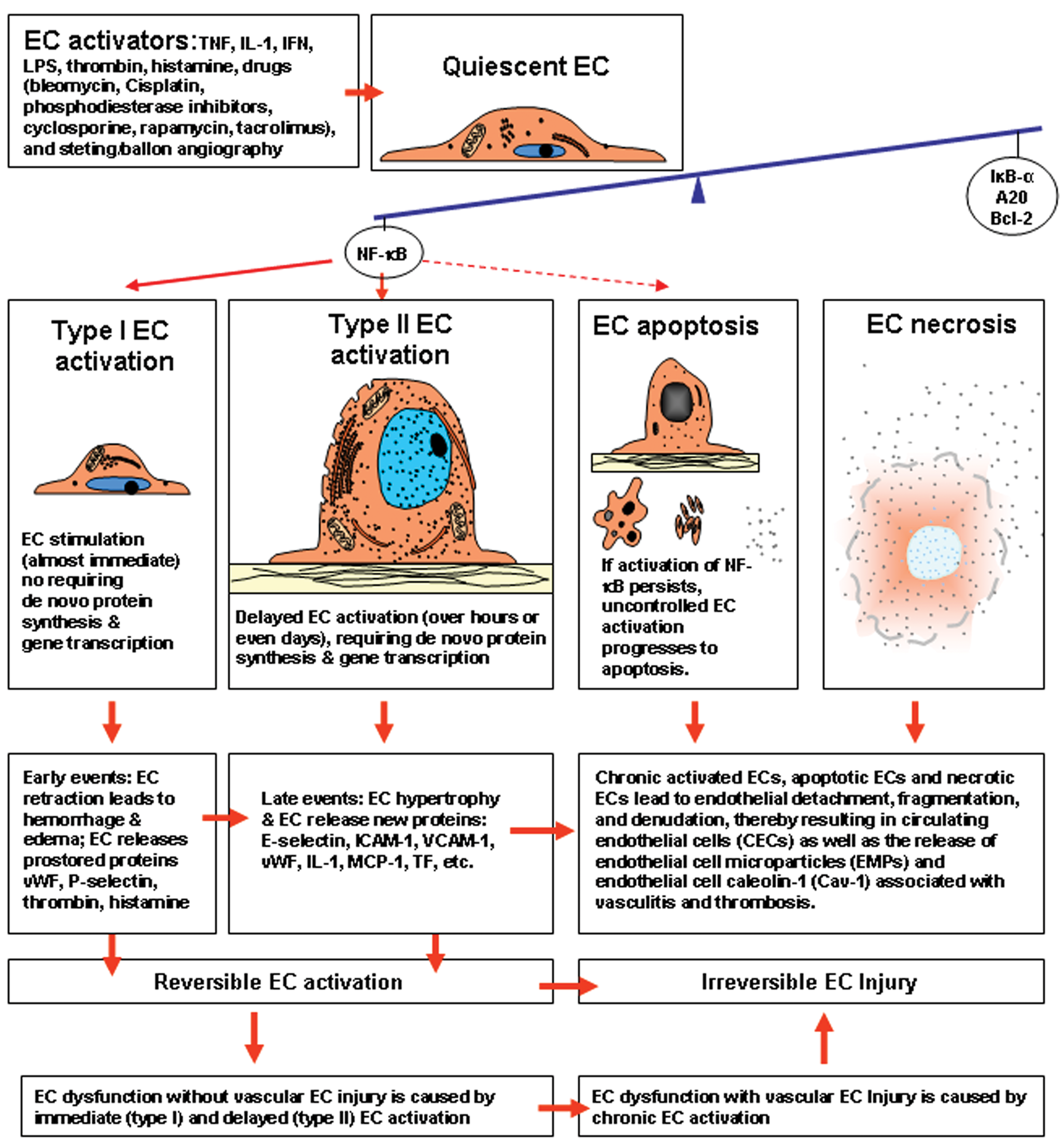
Schematic representation of the relationship of endothelial cell (EC) activation, EC apoptosis, EC dysfunction, and EC injury. Endothelial cell activation and EC apoptosis are strictly controlled through the expression of a set of protective genes (IκκB-a, A20, Bcl-2) that down-regulate the expression of the transcription factor NF-κB. Activation of NF-κB triggers EC activation and renders the endothelium more susceptible to apoptosis. Activated ECs enhance expression of specific gene products (i.e., proteins), with resultant new capacities and new functions in inflammation, coagulation, and immunity, but no visible evidence of EC injury is present. The EC activation process, if uncontrolled, can progress to EC apoptosis. Endothelial cell activation is distinct from EC injury; however, the two phenomena are likely to overlap in the activation process. Endothelial cell activation may be associated with vascular EC injury in the development of vasculitis, thrombosis, and other vascular diseases that are mediated by leukocyte adhesion molecules, vasoactive mediators, pro-inflammatory cytokines, chemokines, and procoagulant molecules released by activated ECs. Type I EC activation (immediate event) and Type II EC activation (over hours or days) are reversible when EC activators are withdrawn, whereas chronic EC activation (over months) leads to EC injury with EC detachment from the underlying basement membrane and denudation of the vessel wall, resulting in circulating endothelial cells (CECs) and the release of EC microparticles (EMPs) and EC caveolin-1 (Cav-1) from the plasmalemmal membranes. TNF, tumor necrosis factor; IL-1, interleukin-1; IFN, interferon; LPS, lipopolysaccharide.
Nuclear factor-κB (NF-κB) is a major regulator of EC activation and EC apoptosis, both of which are strictly controlled through the expression of a set of anti-inflammatory genes (protective genes) that down-regulate the expression of pro-inflammatory genes (Bach et al. 1997). The activation of NF-κB plays a key role in the induction of the pro-inflammatory genes, the products of which activate EC (Bach et al. 1997; Pober 1998). Nuclear factor-κB not only triggers EC activation but also renders the endothelium more susceptible to apoptosis (Aoki et al. 2001). Upon activation, NF-κB binds to promoter regions of several genes involved in EC activation (Bach et al. 1994 Bach et al. 1997; Hunt and Jurd 1998; Pober 1998). The constitutively expressed protective genes (e.g., NF-κB inhibitor-α, IκB-α, A20, and Bcl-2) within ECs guard against EC activation and apoptosis under normal conditions (Bach et al. 1997). The gene NF-κB inhibitor-α (IκB-α) is a specific inhibitor of NF-κB, and the genes A20 and Bcl-2 are anti-apoptotic (Bach et al. 1997). The gene A20 is expressed on activated ECs and is dependent on the gene NF-κB for its induction (Bach et al. 1997). However, if ECs are critically stimulated, both the constitutively expressed and induced protective genes are insufficient to counteract the stimulation, and uncontrolled EC activation may ensue to subsequently induce EC apoptosis (Bach et al. 1997).
As noted, NF-κB can also activate pro-inflammatory genes, including genes for inducible nitric oxide synthase (iNOS). Nitric oxide (NO) and derived peroxynitrite can contribute to EC apoptosis (Xu et al. 2001). Cluster of differentiation CD39 (the vascular nucleoside triphosphate diphosphohydrolase [NTPDase]) is expressed on the luminal surface and caveolar microdomains of ECs (Goepfert et al. 2000). Molecule CD39 can modulate EC activation and EC apoptosis through blockage of adenosine triphosphate (ATP)-induced translocation of the NF-κB (Goepfert et al. 2000). The CD40/CD40 ligand (CD40L) signaling, which is enhanced by activation of NF-κB, is a potent EC activator and inducer of EC apoptosis. It also affects E-selectin and tissue factor (TF) activity (Longo et al. 2003). Overexpression of A20 (an anti-inflammatory and anti-apoptotic gene in EC) protects against CD40-induced EC activation and apoptosis by inhibiting NF-κB indirectly via upregulation of IκB-α and inhibition of E-selectin, but not TF, activity (Longo et al. 2003). A recent study has shown that activated signal transducer and activator of transcription 3 (STAT3) is a mediator and biomarker of EC activation stimulated by vascular endothelial growth factor (VEGF) (Chen et al. 2008). Activated STAT3 mediates VEGF induction of EC Bcl-2 (a protective gene for apoptosis) and contributes to VEGF protection of EC from apoptosis (Chen et al. 2008).
Drug-induced Endothelial Cell Activation
The vascular endothelium can be activated by cytokines, adipocytokines, endotoxins, thrombin, complement components, neutrophils and EC autoantibodies (Ballernman 1998; Contran et al. 1986; Contran et al. 1987; Kevil and Bulllard 1999; Kougias et al. 2005), inflammatory markers (C-reactive protein, CD40 ligand, and interleukin-18) (Szmitko, Wang, Weisel, de Almeida et al. 2003), and mechnical stimuli during balloon antgiography and stenting (Tesfamarian and DeFelice 2007). Endothelial cells can also be activated by toxic pharmacologic agents such as bleomycin and cisplatin (Nuver et al. 2010); immunosuppressive drugs such as cyclosporine, rapamycin, and tacrolimus (Trapp and Weis 2005); and vasoactive agents such as phosphodiesterase (PDE) inhibitors (Zhang, Herman, Knapton et al. 2002; Zhang, Herman, Holt et al. 2002; Zhang et al. 2006; Zhang et al. 2008).
Our laboratory has observed EC activation in the mesenteric blood vessels in rats treated with SK&F 95654, a PDE III inhibitor (Zhang, Herman, Knapton et al. 2002; Zhang, Herman, Holt et al. 2002; Zhang et al. 2006). SK&F 95654 (±-5-methyl-6-[4-94-oxo-1,4-4-dihydropyridin-l-yl)phenyl]-4,5-dihydro-3(2H)-pyridazinone) (SmithKline Beecham Pharmaceuticals, now GlaxoSmithKline) belongs to the cyclic 3′,5′-adenosine guanosine monophosphate (cGMP)-inhibited PDE family of enzymes that hydrolyze intracellular cyclic adenosine monophosphate (cAMP) (Zhang, Herman, Knapton et al. 2002; Zhang et al. 2006). The activated ECs in these studies appear plump and cuboidal in shape, protrude into the lumen, and contain prominent organelles, that is, well-developed Golgi complexes, prominent rough endoplasmic reticulum, abundant ribosomes, vacuoles, and pinocytic vesicles (Zhang, Herman, Knapton et al. 2002).
In a further study of SK&F 95654 (Zhang, Herman, Holt et al. 2002), electron microscopy revealed activated ECs free in the lumina of the blood vessels. These ECs have long cytoplasmic processes, numerous plasmalemmal caveolae (flask-shaped invaginations) on the surface of the plasmalemmal membrane, and enlarged Weibel-Palade bodies (Zhang, Herman, Holt et al. 2002). In contrast, in control rats treated with the dimethyl sulfoxide (DMSO) vehicle, the endothelium displays a normal histological pattern. In particular, there are only a few plasmalemmal caveolae on the surface of the luminal plasma membrane in normal ECs.
Activation of ECs, mast cells, and macrophages was identified morphologically in our previous studies of PDE III and IV inhibitors in rats (Zhang, Herman, Knapton et al. 2002; Zhang, Herman, Holt et al. 2002; Zhang et al. 2006; Zhang et al. 2008). Upon activation, mast cells and macrophages release a wide variety of pro-inflammatory mediators such as cytokines, chemokines, and NO, which may reinforce EC activation. The interaction of activated ECs with activated neutrophils, mast cells, and macrophages may amplify vascular inflammatory responses to produce a cascade of other reactions. The close association of EC activation with that of other cells suggests that some biomarkers of EC activation are not exclusively produced by activated ECs but are also produced by other activated cells. Endothelial cell activation is not an independent cellular event, but an orchestrated phenomenon involving other activated cells. Furthermore, some of the events that make up activation of host effector cells by various antigenic stimuli can be considered to be integral to immune surveillance.
Biomarkers of Endothelial Cell Activation
Tables 1-3 present profiles of EC activation biomarkers considered in this review. Because some of the biomarkers can reflect activation of EC as well as other cells, a range of actions should be expected. Furthermore, these actions may coincide with EC dysfunction, EC apoptosis, or EC injury, depending on the stage of activation.
Recommended surrogate biomarkers of endothelial cell activation for drug-induced vascular injury.
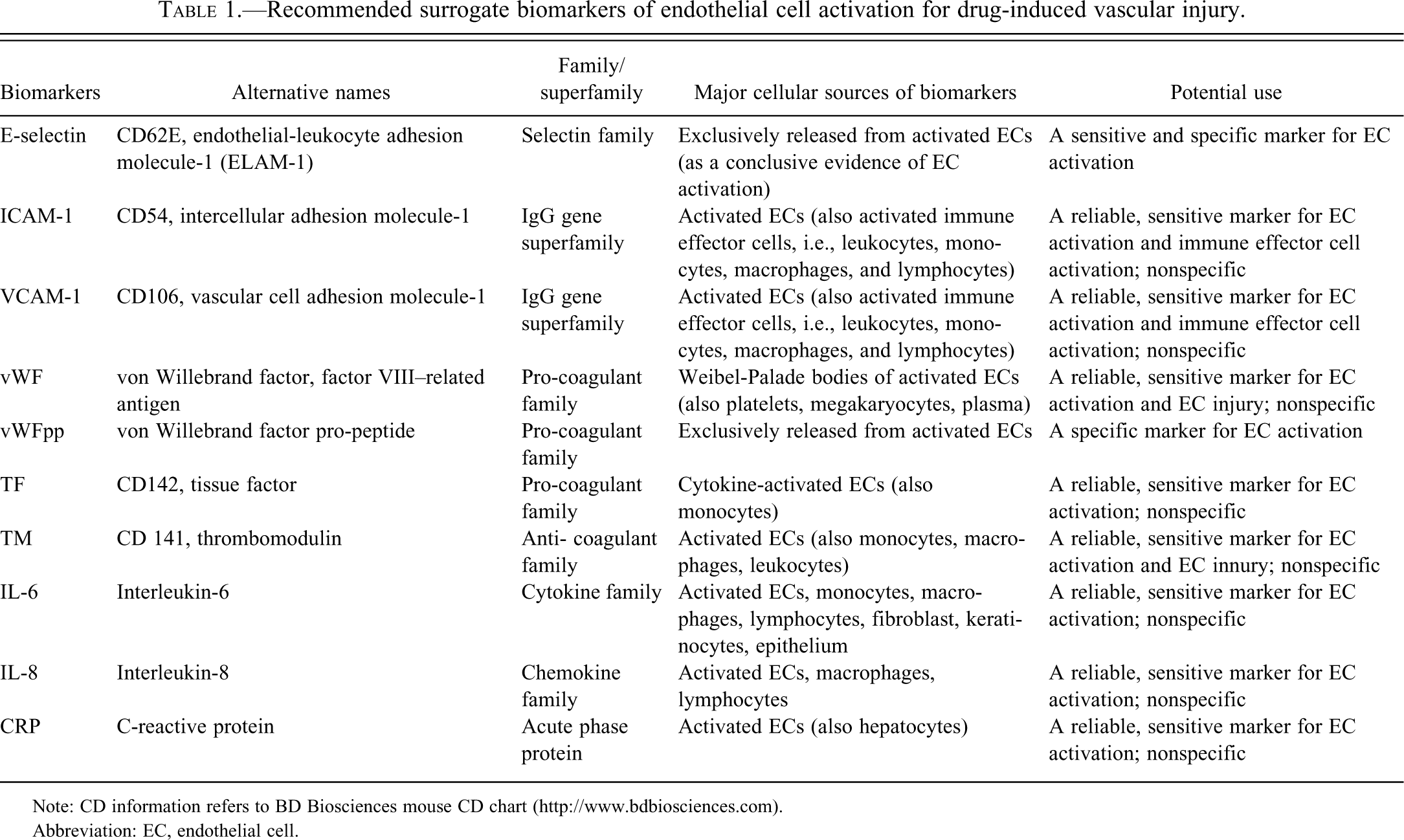
Note: CD information refers to BD Biosciences mouse CD chart (http://www.bdbiosciences.com).
Abbreviation: EC, endothelial cell.
Potentially useful biomarkers of endothelial cell activation for drug-induced vascular injury.

Note: CD information refers to BD Biosciences mouse CD chart (http://www.bdbiosciences.com).
Abbreviations: DIVI, drug-induced vascular injury; EC, endothelial cell; GMP-140, granule membrane protein-140; PADGEM, platelet activation-dependent granular-external membrane protein.
Emerging candidate endothelial cell activation biomarkers for drug-induced vascular injury.
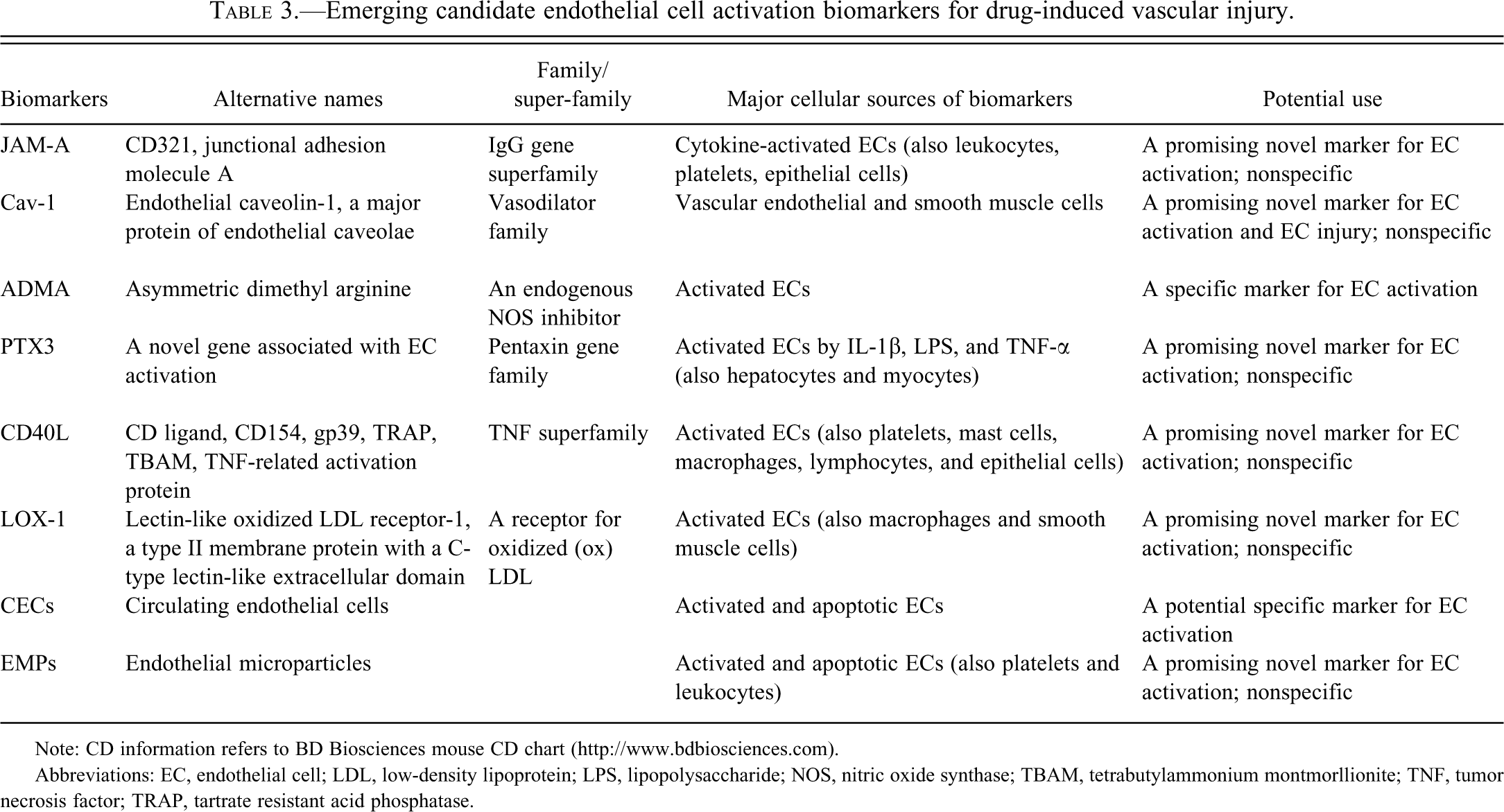
Note: CD information refers to BD Biosciences mouse CD chart (http://www.bdbiosciences.com).
Abbreviations: EC, endothelial cell; LDL, low-density lipoprotein; LPS, lipopolysaccharide; NOS, nitric oxide synthase; TBAM, tetrabutylammonium montmorllionite; TNF, tumor necrosis factor; TRAP, tartrate resistant acid phosphatase.
The selectin family is composed of three proteins named according to their sources: E- (endothelial), P- (platelet), and L- (leukocyte) selectin (Gonzalez-Amaro and Sanchez-Madrid 1999). E-selectin is a specific marker of EC activation (Kerns W. 2005; Tesfamariam and De Felice 2007). E-selectin (alternative names: CD62E and endothelial–leukocyte adhesion molecule 1, or ELAM-1) is induced in vivo and reflects functional alterations of EC (Cotran et al. 1986; Cotran 1987; Cotran et al. 1987). Because E-selectin is expressed solely on the activated ECs (Gearing and Newman 1993; Pober and Cotran 1991), its presence in the blood is taken as conclusive evidence of EC activation (Ara et al. 2001; Furui 2001; Gearing and Newman 1993; Kim and Lee 1994) (Table 1), and the titer reflects the extent of EC activation (Burrows et al. 1994).
E-selectin upregulation has been studied in vitro and in vivo. In vitro expression of cytokine-induced E-selectin is rapid and transient, appears within one to two hours, peaks at four to six hours, and then declines to baseline by twenty-four to forty-eight hours (Pober, Bevilacqua et al. 1986). Further in vitro studies confirmed that expression of E-selectin in cytokine-activated ECs rises to maximal levels at six to twelve hours and then decreases to low basal levels at twenty-four hours (Leeuwenberg et al. 1992). Expression is more persistent in vivo and extends beyond twenty-four hours (Cotran et al. 1986). In xenograft rejection–associated EC activation, E-selectin is first seen at four hours in focal ECs, persists for twelve to twenty-four hours, and is absent by forty-eight to seventy-two hours (Bach et al. 1994). In an in vivo model of endotoxin-stimulated EC activation, E-selectin and VCAM-1 are detected at one and a half hours, reach a maximum at one and a half hours to three hours, and persist at low levels for longer than seventy-two hours (Fries et al. 1993).
E-selectin induced by cytokines is expressed on the ECs of venules or capillaries, but not the ECs of arterioles or arteries (Pober and Cotran 1990a). In a mouse model of endotoxin-stimulated EC activation, E-selectin is expressed on the ECs of medium- or small-sized veins, but not on the ECs of the aorta (Fries et al. 1993). These results are perhaps not surprising, given the evidence of regional differences in EC “phenotype” (Garlandla and Dejana 1997). A high degree of structural and functional homology of human E-selectin with murine and rabbit E-selectin exists (Carlos and Harlan 1994), raising the possibility of similar roles in toxic and idiopathic vasculopathies and the relevance of studies of this selectin in mouse and rabbit models of DIVI.
P-selectin (alternative names: CD62P, granule membrane protein-140 [GMP 140], and platelet activation–dependent granular-external membrane protein [PADGEM]) is an adhesion glycoprotein that is stored in the Weibel-Palade bodies of endothelial cytoplasm and in the α-granules of platelets (Bach et al. 1994). The release of Weibel-Palade bodies is a form of EC activation (Yang et al. 2004). When ECs or platelets are activated, P-selectin is rapidly translocated to the cell surface through the fusion of vesicles and the plasma membranes and is exposed on the external plasma membranes of ECs and platelets (Ara et al. 2001; Bach et al. 1994). Therefore, increased soluble P-selectin may be a marker for EC activation and/or platelet activation (Furui 2001) (Table 2). As with E-selectin, there is a high degree of P-selectin structural and functional homology between mice and humans (Carlos and Harlan 1994), which, if it predicts a similar pathophysiological role, would enhance the usefulness of P-selectin in animal studies of vascular injury. In xenograft rejection–associated EC activation, P-selectin first appears within four hours in focal ECs and platelets and persists for up to seventy-two hours, whereas E-selectin is first seen at four hours in focal ECs and persists for twelve to twenty-four hours (Bach et al. 1994). In our previous study with SK&F 95156 in rats (Zhang, Herman, Knapton et al. 2002; Zhang, Herman, Holt et al. 2002), electron microscopy revealed enlarged Weibei-Palade bodies in the cytoplasm of activated ECs of the mesenteric vessels compared with those in the cytoplasm of normal ECs. These findings suggest that enhanced levels of P-selectin are available for release from ECs following activation.
The immunoglobulin (IgG) gene superfamily consists of seven important adhesion molecules, namely, intercellular adhesion molecule-1 (ICAM-1); intercellular adhesion molecule-2 (ICAM-2); intercellular adhesion molecule-3 (ICAM-3); junctional adhesion molecule-A (JAM-A); vascular cell adhesion molecule-1 (VCAM-1); platelet EC adhesion molecule-1 (PECAM-1); and mucosal addressin cell adhesion molecule-1 (MAdCAM-1) (Carlos and Harlan 1994). These adhesion molecules participate in the cascade and in the coordination of adhesion events between leukocytes and the activated ECs. Mucosal addressin cell adhesion molecule-1 (MAdCAM-1) is involved in leukocyte/lymphocyte rolling; ICAM-1, ICAM-2, VCAM-1, and MAdCAM-1 are involved in the firm adhesion of leukocytes; and PECAM-1, ICAM-1, and VCAM-1 are involved in transendothelial migration (Carlos and Harlan 1994). Leukocyte transmigration is also mediated by EC junctional adhesion molecule-A (JAM-A) (Woodfin et al. 2009). These molecules facilitate sequential interaction of the ligands on the activated endothelial surface with the integrin receptors on the leukocyte surface (Carlos and Harlan 1994).
Knowledge of how endothelial junctional adhesion molecules control leukocyte transmigration and vascular permeability is rapidly accumulating. These endothelial junctional adhesion molecules consist of vascular endothelium cadherin (VE-cadherin), junctional adhesion molecules (JAMs), endothelial selective adhesion molecule-1 (ESAM-1), platelet EC adhesion molecule-1 (PECAM-1), and CD99 (Pfeiffer et al. 2008; Sanchez-Madrid and Barreiro 2009). VE-cadherin is produced only by ECs and is found at the junctions of the ECs (Vestweber 2007; Vestweber 2008; Vestweber et al. 2009). Junctional adhesion molecules (JAMs) belong to the IgG superfamily and regulate leukocyte recruitment to the sites of inflammation and ischemia-reperfusion injury (Pfeiffer et al. 2008; Sanchez-Madrid and Barreiro 2009; Weber et al. 2007; Woodfin et al. 2009). With the exception of JAM-A, it is presently unknown what role, if any, these junctional adhesion molecules play in EC activation.
One important source of ICAM-1 is the activated ECs (Cotran et al. 1987). The ligand of ICAM-1 on the activated ECs interacts with Mac-1 (CD11b/CD18), a leukocyte integrin receptor (Carlos and Harlan 1994). In vitro, cytokines activate ECs to express ICAM-1, which appears at four to six hours, peaks at twelve to twenty-four hours, and persists for seventy-two hours (Leeuwenberg et al. 1992; Pober, Gimbrone et al. 1986; Pober et al. 1987; Pober and Cotran 1991). Activated immune effector cells, such as monocytes, macrophages, T-cells, and B-cells, are also sources of soluble ICAM-1 which, therefore, is a sentinel for immune activation (Coll-Vinent et al. 1997; Furukawa, Imai et al. 1992). Immunohistochemistry performed in rats treated with SK&F 95654 (a PDE-III inhibitor) showed that ICAM-1 expression is up-regulated in the activated ECs of the mesenteric vessels (Zhang, Herman, Knapton et al. 2002). Intercellular adhesion molecule-1, induced on ECs by IL-1 and IFN-g, appears to promote adhesion of T- and B-lymphocytes to ECs at sites of inflammation or a localized immune response and extravasation of these cells at such sites (Dustin et al. 1986). It is also expressed on thymic and mucosal epithelial cells, fibroblasts, tissue macrophages, lymphocytes, and dendritic cells in tonsils, lymph nodes, and Peyer’s patches (Coulpier et al. 1995; Dittman and Majerus 1990). Intense staining with ICAM-1 monoclonal antibody is found on vascular ECs in T-lymphocyte areas of tonsils and lymph nodes that have reactive hyperplasia (Dustin et al. 1986). Increased levels of soluble intercellular adhesion molecules may reflect EC activation and/or leukocyte activation (Coll-Vinent et al. 1999) (Table 1).
Increased levels of soluble VCAM-1 primarily reflect EC activation (Ara et al. 2001). However, VCAM-1, like ICAM-1, is also expressed on dendritic cells, lymphocytes, and macrophages and can be released when these immune effector cells are activated (Ara et al. 2001; Carlos and Harlan 1994; Dustin et al. 1986). Therefore, increased titers of soluble forms of ICAM-1 or VCAM-1 may reflect EC activation or activation of immune effector cells (Ara et al. 2001; Nash et al. 1995) (Table 1).
In vitro, expression of VCAM-1 by cytokine-activated ECs appears at two hours and persists for at least seventy-two hours (Osborn et al. 1989; Pober and Cotran 1991). Soluble forms of VCAM-1 as well as ICAM-1 and E-selectin are present at eighteen hours in supernatants of the cytokine-activated ECs (Pigott et al. 1992). The ligand of VCAM-1 on the activated ECs interacts with very late antigen-4 (VLA-4, CD49d/CD29), leukocyte integrins of the α4 β1 subfamily (Carlos and Harlan 1994; Gonzalez-Amaro and Sanchez-Madrid 1999), thereby promoting firm attachment and subsequent transendothelial migration of leukocytes (Gearing and Newman 1993).
Platelet EC adhesion molecule-1, PECAM-1 (alternative name CD31), is expressed on ECs, platelets, and megakaryocytes. Cluster of differentiation 31 is a widely distributed glycoprotein on these cells (Carlos and Harlan 1994). Platelet EC adhesion molecule-1/CD31 is a 100kD glycoprotein that participates in the adhesion between platelets and ECs (Ferrer et al. 1995). Thus, PECAM-1/CD31 expression could be an endothelial and/or platelet marker (Miettinen et al. 1994; Muller et al. 2002) (Table 2). Platelet EC adhesion molecule-1 is involved in leukocyte adhesion and transmigration (Carlos and Harlan 1994). Unlike ICAM-1 which is expressed over the entire EC surface, PECAM-1 expression is localized at the intercellular junctions of EC; thus, the protein is also referred to as an EC junction molecule. Intercellular localization of PECAM-1 may facilitate binding of leukocyte CD31 receptors to endothelial CD31 and contribute to the transendothelial migration of leukocytes (Carlos and Harlan 1994). Surface expression of endothelial CD31 is not increased by TNF-α, IL-1, or interferon-γ (IFN-g) (Carlos and Harlan 1994). However, TNF-α and IFN-g induce redistribution of PECAM-1 (CD31) on human ECs (Romer et al. 1995). It is possible that PECAM-1 redistribution may have a role in transendothelial migration and leukocyte emigration (Romer et al. 1995). Immunohistochemical staining of canine tissues with the monoclonal antibody JC7 (anti-CD31) detects CD31 antigen on the cell membrane and in the cytoplasm of ECs (Ferrer et al. 1995). The CD31 antigen is regarded in the beagle dog as a useful marker for ECs (Ferrer et al. 1995).
Junctional adhesion molecule-A (JAM-A) is an adhesion protein expressed on IL-1β–activated ECs (Woodfin et al. 2009). This protein also occurs in leukocytes, platelets, and epithelial cells (Huang et al. 2006). Junctional adhesion molecule-A on the activated ECs plays a role in the process of neutrophil transmigration and in mediating the host immune response (Sanchez-Madrid and Barreiro 2009; Woodfin et al. 2007; Woodfin et al. 2009). In the process of transendothelial migration of neutrophils, JAM-A cooperates with ICAM-1 and PECAM-1, but each of the endothelial junctional molecules performs a distinct function. Three-dimensional imaging of the venules has revealed that neutrophil transmigration and extravasation is a coordinated, multistage process. Entry and penetration of neutrophils into the EC junction, through the basement membranes, and into the extravascular tissues is mediated by ICAM-1, JAM-A, and PECAM-1 acting sequentially (Sanchez-Madrid and Barreiro 2009; Woodfin et al. 2009). The IL-1β–activated ECs can provide all three of the critical adhesion molecules (ICAM-1, JAM-1, and PECAM-1) that act in concert to effect local attachment and extravasation of neutrophils in local inflammatory and antigen-processing events (Woodfin et al. 2009). It is hypothesized that JAM-A might serve as a promising novel EC activation marker (Table 3) for DIVI pending further evaluation of the extent to which its expression in other cells (e.g., platelets, epithelial cells) might compromise the sensitivity and specificity of its use.
Murine mucosal lymphoid addressin-1 (MAdCAM-1) is an EC adhesion molecule that plays a role in trafficking of lymphocytes to gut-associated lymphoid tissues and to sites of inflammation (Fujisaki et al. 2002; Takeuchi and Baichwal 1995). Immunohistochemistry demonstrates that it is located in the venular endothelium in the small intestine and in Peyer’s patches both in the rat (Fujisaki et al. 2002) and in the human, especially when such tissue is inflamed (Briskin et al. 1997). In addition, MAdCAM-1 expression is also invoked as a mediator of lymphocyte recruitment in acute and chronic inflammation (Connor et al. 1999). In the rat, α4 integrins on the surface of neutrophils interact with MAdCAM-1 on the endothelium and mediate selective recruitment of neutrophils to sites of inflammation in vivo (Davenpeck et al. 1998). In nonobese diabetic mice, MAdCAM-1 expression is localized to the vessels within and adjacent to the inflamed pancreatic islets, but not within normal islets (Hanninen et al. 1993). When provoked by TNF-α, IL-1β, and lipopolysaccharide (LPS), a murine EC line (Takeuchi and Baichwal 1995), murine lymphatic ECs (Oshima et al. 2001), murine hepatic ECs (Ando et al. 2007), and human esophageal microvascular ECs (Rafiee et al. 2003) all expressed MAdCAM-1, and in the human cells, E-selectin, ICAM-1 and VCAM-1 were expressed as well (Rafiee et al. 2003). Tumor necrosis factor-α–enhanced expression of MAdCAM-1 in the intestine and colonic tissues occurs within three hours, peaks at eighteen hours, and persists for at least forty-eight hours (Connor et al. 1999). Like other endothelial adhesion molecules (e.g., E-selectin, ICAM-1, and VCAM-1), transcription induction of MAdCAM-1 by TNF-α also requires activated NF-κB proteins that play a central role in cytokine-induced expression of the MAdCAM-1 gene (Takeuchi and Baichwal 1995). These findings suggest that MAdCAM-1 may serve as a potentially useful marker for DIVI involving EC activation and EC injury (Table 2) occurring in the microvasculature of the intestine, pancreas, liver, and mesenteric lymphoid tissues. However, further work is needed to explore this prospect.
The integrin family is composed of heterodimeric glycoproteins consisting of α and β subunits, which mediate EC–leukocyte and EC–extracellular matrix interactions (Davenpeck et al. 1998). Of these integrins, only the αvβ3 heterodimer (CD51/CD61) is an endothelial adhesion molecule, whereas other integrins are receptors for leukocytes. The αvβ3 heterodimer is expressed by the activated ECs and, to a lesser extent, on leukocytes and macrophages (Gonzalez-Amaro and Sanchez-Madrid 1999). The integrin family functions to integrate the intracellular with the extracellular environment, thus the avb3 heterodimer plays an important role in the interaction of intracellular proteins with a variety of extracellular matrix proteins, such as vitronectin, fibronectin, laminin, and collagen (Gonzalez-Amaro and Sanchez-Madrid 1999; Saito et al. 2002). The αvβ3 heterodimer also promotes recognition of apoptotic cells (Gonzalez-Amaro and Sanchez-Madrid 1999). The αvβ3 heterodimer might serve as a marker for EC activation (Table 1). However, further work is needed to assess its appropriateness before it can be used for this purpose.
The coagulant family, which promotes thrombosis or fibrinolysis, includes anticoagulant molecules, thrombomodulin (TM), tissue plasminogen activator (tPA), heparin sulfate, antithrombin III (AT-III) and procoagulant molecules, thrombin, plasminogen-activating inhibitor-1 (PAI-1), von Willebrand factor (vWF), tissue factor (TF) and platelet-activating factor (PAF) (Cockwell et al. 1997; Cotran 1987). The anticoagulant (antithrombotic) activity and procoagulant (prothrombotic) activity are balanced in quiescent (nonactivated) ECs. The procoagulant activity can be augmented by EC activation. Endothelial cell activation can switch the endothelial function from anti-thrombotic activity to prothrombotic potential. The loss of anticoagulant molecules and the synthesis of procoagulant molecules on the activated ECs convert the EC environment to one favoring thrombosis (Bach et al. 1994; Cotran 1987). The type I EC activation results in the loss of anticoagulant molecules (e.g., heparin sulfate, TM, and tPA), whereas the type II EC activation leads to de novo synthesis of procoagulant molecules (e.g., PAI-1, vWF, TF, and PAF) (Bach et al. 1994; Ballermann 1998).
The activity of the fibrinolytic system depends on the cleavage of plasminogen to plasmin (a fibrin-digesting enzyme) by tPA that can be inhibited by PAI-1 (Cockwell et al. 1997). Endothelial cell activation by IL-1α, IL-1β, and TNF-α decreases tPA anticoagulant activity and increases PAI-1 procoagulant activity (Cotran 1987; Pober 1988). Thus, the reduced tPA expression and the increased PAI-1 expression on the activated ECs favors thrombosis (Ballermann 1998). Because EC activation by inflammatory cytokines greatly changes the balance between tPA and PAI-1 synthesis through pathways involving the reduction of tPA and the increase of PAI-1 expression and synthesis, elevated levels of PAI-1 can be regarded as a biomarker of EC activation (Ballermann 1998) (Table 2). A more recent in vitro study using a human dermal microvascular EC line (HMEC-1) showed that bleomycin and cisplatin induce EC activation and EC apoptosis, in association with upregulation of ICAM-1, tPA, and PAI-1 (Nuver et al. 2010).
Endothelial cells express vWF (previously named factor VIII–related antigen). The molecule vWF is synthesized by the ECs and megakaryocytes and is stored in the cytoplasmic Weibel-Palade bodies of the ECs where it co-localizes with P-selectin (Ballermann 1998). The activated ECs can release vWF into the subendothelial matrix and the plasma through exocytosis of the cytoplasmic Weibel-Palade bodies (Hunt and Jurd 1998). Exocytosis of vWF from ECs is usually coupled with the mobilization of P-selectin to the surface of ECs (Ballermann 1998). At sites of EC activation or EC injury, vWF binds to platelet glycoprotein receptors, promotes platelet aggregation, and mediates platelet adhesion to the subendothelium (Cockwell et al. 1997). In rats treated with SK&F 95654 (a PDE III inhibitor), immunohistochemical staining of sections of mesenteric tissue with DIVI showed that expression of vWF is up-regulated on the activated ECs (Zhang, Herman, Knapton et al. 2002). Electron microscopy also showed that Weibel-Palade bodies in the cytoplasm of activated ECs were well developed (Zhang, Herman, Knapton et al. 2002). In endotoxin-treated or balloon-injured rats, immunohistochemical staining for intracellular vWF is increased threefold, indicating that EC injury leads to increased vWF (Reidy et al. 1989). Thus, soluble vWF may be a sentinel for EC activation and/or EC injury (Jurd et al. 1996; Kerns et al. 2005; Tesfamariam and DeFelice 2007) (Table 1). However, a limitation of the use of soluble vWF as a circulating marker of vascular endothelial damage is that vWF, as it is expressed in platelets, is not EC specific (Kim and Lee 1994). Both endothelial and platelet-derived vWF act as carriers for factor VIII in plasma (Ballermann 1998). In addition, the main argument against the use of vWF as a specific marker of DIVI is that vWF is raised post surgery, after exercise, in pregnancy, in non-inflammatory peripheral vascular disease, and in infectious disease (Nash et al. 1995; Woolf et al. 1987). Unlike E-selectin, which is derived exclusively from activated ECs, vWF may be derived from platelets, megakaryocytes, and even plasma. In rats treated with fenoldopam, the increased level of soluble vWF is considered to be an acute phase response rather than a direct consequence of DIVI (Newsholme et al. 2000). Thus, vWF may not be a suitable biomarker to monitor progression of vascular damage in a preclinical safety study, because the increase in circulating vWF is transient, that is, it occurs at two to six hours, and returns to normal levels by forty-eight hours post-treatment (Brott et al. 2005; Louden et al. 2006).
Two proteins, mature vWF and vWF pro-peptide, originate from pro-vWF. Unlike vWF, which is released from multiple sources, vWFpp is derived solely from ECs. The virtual absence of vWFpp in normal plasma, its rapid turnover, and its short half-life make it attractive as a specific biomarker of EC activation for monitoring initiation and progression of DIVI (Kerns et al. 2005; Louden et al. 2006) (Table 1). The ratio of vWF to vWFpp may distinguish acute from chronic or progressive vascular injury (Louden et al. 2006).
Tissue factor is a procoagulant that interacts with factor VIII to mediate coagulation (Cockwell et al. 1997). Tissue factor is not normally expressed by quiescent ECs (Ballermann 1998). Endothelial cell activation by IL-1α, IL-1β, and TNF-α increases TF procoagulant activity (Cotran 1987; Pober 1988). Monocytes are also capable of expressing TF (Bach et al. 1994). The activity of procoagulants is inhibited by anti-procoagulant molecules such as antithrombin III and heparin sulfate which catalyzes the antithrombin III pathway (Cockwell et al. 1997). Endothelial cell activation induces the up-regulation of TF expression and the down-regulation of antithrombin III expression forty-eight hours after xenograft rejection (Bach et al. 1994). An expert working group on DIVI has proposed TF as a marker of EC activation (Kerns et al. 2005) (Table 1).
Thrombomodulin (TM) is an EC surface glycoprotein that promotes anticoagulant protein C activation and binds thrombin, thus it has potent anticoagulant activity (Cockwell et al. 1997; Dittman and Majerus 1990). When thrombin is bound to TM, the latter is no longer thrombogenic; rather, the complex prevents fibrin formation (Cockwell et al. 1997; Dittman and Majerus 1990). Thrombomodulin is not EC specific, as it is also formed in monocytes, macrophages, and neutrophils (Boffa and Karmochkine 1998). Thus, TM may also be directly involved in inflammation. Expression of TM on the surface of IL-1α–, IL-1β–, and TNF-α–activated ECs is markedly suppressed through rapid internalization and degradation of TM and via reduced TM transcription, thus decreasing its potent inhibition of coagulation (Ballermann 1998; Boffa and Karmochkine 1998; Pober 1988). By suppressing TM, IL-1α, IL-1β, and TNF-α are prothrombotic and markedly inhibit the anticoagulant effects of TM-dependent activation of protein S and protein C (Cotran 1987). The effects of pro-inflammatory cytokines (i.e., IL-1, TNF-α) on TM expression depend on the types of cells synthesizing TM. Expression of TM is down-regulated on TNF-α– and IL-1–activated ECs, but up-regulated on cytokine-activated macrophages (Aoki et al. 2001), which may then release soluble fragments of TM into the blood (Boffa and Karmochkine 1998). An in vitro model of the kinetics of soluble endothelial adhesion molecule demonstrates that the TNF-α–activated ECs result in a significant increase in the levels of E-selectin, ICAM-1, and VCAM-1 at twenty-four hours, but not TM (Boehme et al. 2000). A significant increase in TM levels occurs only following co-incubation of the TNF-α–activated ECs with the TNF-α–activated neutrophils at forty-eight hours, indicating a role for neutrophil activation with resultant EC injury induced by the release of TM (Boehme et al. 2000). Several heterogeneous fragments of degraded TM are present in the circulation, and these fragments enable soluble TM to be detected by immunosorbent assay (Boehme et al. 2000), which demonstrates an advantage of TM over the three monitored adhesion molecules (E-selectin, ICAM-1, and VCAM-1) as a marker of overt endothelial injury. Although TM is a nonspecific EC activation marker, it can serve as a circulating marker of vascular EC damage (Kim and Lee 1994; Kerns et al. 2005) (Table 1). Boehme, et al. (2000) have proposed it as a reliable marker of disease activity in systemic lupus erythematosu (SLE).
Endothelial cell activation promotes sustained release of NO (Ballermann 1998; Cotran 1989). The role of endothelial Cav-1, Cav-1/eNOS, iNOS, and nitrotyrosine in the pathogenesis of vascular diseases is now recognized (Feng et al. 2001; Frank et al. 2003; Mathew et al. 2007; Slim et al. 2003; van der Meer et al. 2009). Endothelial cells contain abundant, multifunctional caveolae (plasmalemmal vesicles) associated with the luminal and abluminal plasma membranes (Feng et al. 2001; Frank et al. 2003). In rat lung microvasculature, they are flask-shaped vesicles measuring 50–100 nm in diameter (Frank et al. 2009; Schnitzer, Liu et al. 1995). Because they transport macromolecules such as drugs, insulin, and albumin from the circulating blood across the EC barrier to the underlying tissue cells or the interstitium, they are also called transendothelial transport vesicles or carrier vesicles (Schnitzer, Liu et al. 1995; Schnitzer, Oh et al. 1995; Massey and Schnitzer 2010). Transcytosis by endothelial caveolae is involved in increased vascular permeability (Cybulsky et al. 2001) and in the development of atherosclerosis (Frank et al. 2009). In addition, endothelial caveolae have been implicated in vesicular trafficking and signal transduction (Frank et al. 2003), as well as mechanosignal transduction in response to shear stress (van der Meer et al. 2009). A role of endothelial Cav-1, Cav-1/eNOS, iNOS, and nitrotyrosine in the pathogenesis of vascular diseases is now recognized (Feng et al. 2001; Frank et al. 2003; Mathew et al. 2007; Slim et al. 2003; van der Meer et al. 2009).
Cav-1 is the principal structural protein component of caveolae in vascular endothelial and smooth muscle cells (Brott et al. 2005; Frank et al. 2009; Murata et al. 2007) and serves as a marker for these organelles (Frank et al. 2003). Endothelial cells express the highest levels of Cav-1 in keeping with its function to mediate endocytosis and transcytosis as well as mediating scavenger endocytosis to endosomes and lysosomes for degradation (Schnitzer, Liu et al. 1995; Schnitzer, Oh et al. 1995). Endothelial Cav-1 is capable of inhibiting eNOS as a negative regulator of agonist-mediated eNOS activation (Frank et al. 2003; Murata et al. 2007). Constitutive expression of eNOS can be modulated by increased shear stress (Wilcox et al. 1997); however, Cav-1 has the ability to respond to shear stress (van der Meer et al. 2009). When ECs are exposed to shear stress, the ECs rapidly release NO, possibly through dissociation of eNOS from Cav-1 (Frank et al. 2003), because the binding of eNOS to Cav-1 can inhibit eNOS activity (Cybulsky et al. 2001). In the activated ECs, regulated NO synthesis from eNOS is down-regulated, whereas NO production from iNOS tends to be high and sustained (Ballermann 1998).
At present, the relationship of expression of Cav-1 with DIVI and EC activation is not established. In ECs of the mesenteric artery in fenoldopam-treated rats and coronary ECs in dogs treated with an endothelin-receptor antagonist or a potassium channel opener, Cav-1 expression at both sites of DIVI is down-regulated (Brott et al. 2005). Therefore, Cav-1 has been proposed as a potential biomarker of DIVI (Brott et al. 2005; Louden et al. 2006). In our study with SK&F 95654, a PDE III inhibitor, electron microscopy revealed that numerous plasmalemmal caveolae on the luminal surface are discharged from the circulating activated ECs (Zhang, Herman, Holt et al. 2002). These findings may prompt researchers to further investigate endothelial Cav-1 for its potential use as an EC activation marker (Table 3).
Nitric oxide is not only a potent local vasodilator, but also an important activator of ECs. This oxide can interact rapidly with a free radical superoxide (O2 -). The interaction results in the formation of a highly toxic oxidant, peroxynitrite (OONO-) (Viera et al. 1999; Xu et al. 2001). Peroxynitrite radicals induce cellular damage via their strong oxidant properties and release of hydroxyl radicals upon nitration of tyrosine residues on proteins (Viera et al. 1999; Wilcox et al. 1997). There is in vitro evidence that local peroxynitrite formation also plays a crucial role in neutrophil-induced EC activation (Sohn et al. 2003). The footprint left by peroxynitrite in vivo can be detected by using antibodies directed against nitrotyrosine, an intracellular marker of oxidative stress (Viera et al. 1999). In a study of patients with decompensated heart failure, activated ECs in forearm veins displayed enhanced nitrotyrosine immunoreactivity, increased cyclooxygenase-2 (COX-2) activity, and up-regulated iNOS expression, whereas venous eNOS expression did not change (Colombo et al. 2005). In eNOS knockout mice, an effect of eNOS-derived NO on EC activation has been studied. Evidence from eNOS knockout EC showed that EC activation does not result from eNOS deficiency and that eNOS-derived NO plays a role in the counterbalance of signals leading to EC activation (Kuhlencordt et al. 2004). In rats treated with PDE VI inhibitors, SCH 351591 and 534385, immunohistochemical staining of rat mesenteric tissue sections revealed up-regulated expression of nitrotyrosine and iNOS on the activated ECs (Zhang et al. 2006). Accordingly, nitrotyrosine and iNOS may serve as tissue markers of EC activation and mechanistic markers for DIVI (Table 2).
Asymmetric dimethyl arginine (ADMA) is an endogenous nitric oxide synthase (NOS) inhibitor (Goonasekera et al. 2000), which may be associated with vascular EC activation. In children and young adults with hypertension, increased plasma ADMA is accompanied by increased VCAM-1, a marker of EC activation (Goonasekera et al. 2000). Because the endothelium-derived NO, when inhibited, can induce vascular EC activation, the raised plasma endogenous NO inhibitor ADMA suggests that EC activation is triggered by eNOS inhibition (Goonasekera et al. 2000). These findings indicate that ADMA may serve as a specific marker of EC activation (Table 3).
Vascular ECs are not only a primary cellular target for the action of pro-inflammatory cytokines but also a source of cytokines and chemokines (Chan et al. 2001; Mantovani et al. 1998; Pober 1998). Endothelial cells activated by bacterial endotoxin, cytokines, and thrombin can produce various pro-inflammatory cytokines, for example, TNF-α, IL-1α, IL-β, and IL-6 (Ballermann 1998; Cotran 1987, 1989; Pober and Cotran 1990b). Interleukin-1β and IL-6 can also be synthesized by activated monocytes, macrophages, T lymphocytes, fibroblasts, keratinocytes, and epithelial cells (Kim 1992; Noris et al. 1999; Ueno et al. 1989). The presence of these cytokines may signal EC activation and/or mononuclear cell activation (particularly T-cell activation). In vitro studies reveal distinct time-dependent profiles of cytokine synthesis. Activated ECs express TNF-α, associated with endothelial NK-κB activation, at 60 min following lipopolysaccharide (LPS) stimulation (Chan et al. 2001). Interleukin-6 should be considered as a general marker of inflammation (Table 1), the levels are increased not only by bacterial endotoxin, but also in vasculitis and in many other inflammatory diseases. Its release from activated ECs triggers synthesis of acute-phase proteins in the liver via binding to IL-6 receptors on hepatocytes (Castell et al. 1988; Geiger et al. 1988). The acute-phase response proteins CRP and serum amyloid P component (SAP) belong to the pentaxin or pentraxin gene family, in which pentaxins present a common pentameric structure (Breviario et al. 1992; Introna et al. 1996). Both are synthesized by hepatocytes.
In response to pro-inflammatory cytokines (IL-1β, TNF-α, and IFN-g) and fibrin, activated ECs can synthesize and secrete interleukin-8 (IL-8) (Pober and Cotran 1990a, 1990b; 1991; Qi and Kreutzer 1995). Chemokines play an important role in the stimulation of integrin expression and the increase of integrin affinity on leukocytes, thereby promoting the adhesion of leukocytes and ECs (Ballermann 1998). Although elevated levels of IL-8 have been regarded as a marker for EC activation (Cockwell et al. 1997; Kerns et al. 2005), IL-8 can also be synthesized by macrophages and T lymphocytes (Noris et al. 1999). Thus, IL-8 should be considered as a marker for either EC activation or mononuclear cell activation or both (Table 1). Weaver et al. (2008) identified lesion severity–related increases in circulating IL-6, IL-8 (alternative name GRO/CINC-1), CRP, and nitrite in rats treated with the PDE IV inhibitors (SCH 351591 and SCH 534385), which caused mesenteric vascular injury.
The activated EC can directly produce PTX3, an acute-phase protein and a new member of the pentraxin gene family (CRP and SAP) (Breviario et al. 1992; Introna et al. 1996). The gene PTX3 may serve as a promising novel marker for EC activation (Table 3). Expression of this gene was associated with IL-1β– and LPS–induced activation of human umbilical vein ECs in vitro (Breviario et al. 1992; Introna et al. 1996). In response to the pro-inflammatory cytokines (IL-1β, TNF-α), mouse ECs in vitro express mouse PTX3 mRNA which could serve as an indicator of inflammatory reactions (Introna et al. 1996; Mantovani et al. 1998). Mouse PTX3 is also expressed on vascular ECs following injection of endotoxin in vivo (Introna et al. 1996). Moreover, human PTX3 has been reported in IL-1β–activated ECs (Breviario et al. 1992). The gene PTX3 is strongly induced in human umbilical vein ECs, hepatocytes, and monocytes (Breviario et al. 1992). Messenger ribonucleic acid (mRNA) of PTX3 is strongly induced by IL-1β and TNF-α, but not by IL-6 and IFN-g, whereas CRP and serum amyloid A component (SAA) are directly induced by IL-6 (Breviario et al. 1992).
Cluster of differentiation 40 ligand (CD40L) is a member of the TNF family and is primarily expressed on ECs, platelets, mast cells, macrophages, lymphocytes, and epithelial cells (Schönbeck and Libby 2001). Ligation of CD40 mediates immune and inflammatory responses as well as prothrombotic activity (Schönbeck and Libby 2001). The ligand CD40 and its receptor (CD40/CD40L, also referred to as CD40/CD154 receptor/ligand dyad) are now recognized as signaling pathways that play an important role in immunity, inflammation, and apoptosis (Schönbeck and Libby 2001). It is also expressed on vascular endothelium and antigen-presenting cells (Rushworth et al., 2001). Constitutive basal expression of CD40/CD40L is markedly increased on ECs activated by IL-1, TNF-α, IL-4, or IFN-γ (Schönbeck and Libby 2001). The CD40/CD40L signaling, via activation of NF-κB, plays a role in EC activation (Longo et al. 2003). Endothelial cells activated by CD40L produce reactive oxygen species, chemokines (monocyte chemoattractant protein-1, MCP-1), and cytokines (IL-1, IL-6, and TNF-α) and up-regulation of expression of adhesion molecules, such as EC activation markers E-selectin, ICAM-1, and VCAM-1 (Longo et al. 2003; Rushworth et al. 2001; Szmitko, Wang, Weisel, de Almeida et al. 2003). Signaling by CD40/CD40L also induces TF expression on the ECs and smooth muscle cells and enhances their thrombogenic potential (Szmitko, Wang, Weisel, de Almeida et al. 2003). Because CD40L controls EC adhesion molecules, procoagulant molecules, cytokines, and chemokines, it holds promise as a novel EC activation biomarker (Table 3).
Lectin-like oxidized-low-density lipoprotein (LDL) receptor-1 (LOX-1) is the major receptor for oxidized LDL (oxLDL), which is expressed on ECs, macrophages, and smooth muscle cells (Szmitko, Wang, Weisel, Jeffries et al. 2003). Oxidized LDL activates ECs by binding to LOX-1 (Szmitko, Wang, Weisel, Jeffries et al. 2003). Such binding can induce EC apoptosis via NF-κB activation, promote vascular inflammation by up-regulating the adhesion molecules (ICAM-1,VCAM-1) and amplify EC dysfunction by promoting intracellular reactive oxygen species, which inactivate and reduce the intracellular concentration of NO (Szmitko, Wang, Weisel, Jeffries et al. 2003). In addition, oxLDL, through LOX-1, triggers the CD40/CD40L signaling pathway, and thus reinforces local inflammation (Szmitko, Wang, Weisel, Jeffries et al. 2003). Because LOX-1 can activate ECs, leading to apoptosis and dysfunction, it could serve as a promising novel EC activation biomarker (Table 3).
Circulating ECs (CECs) are ECs detached from the basement membrane at sites of vascular injury. They are approximately 10–50 μm in size (Al-Massarani et al. 2008) and can be detected under a variety of physiologic and pathological conditions, for example, EC activation and apoptosis, mechanical injury during angioplasty, and elevated cytokine levels (Sabatier et al. 2009; Shantsila et al. 2008). Elevated CEC levels are associated with, and may be sentinels for, antineutrophil cytoplasmic antibody (ANCA)–associated vasculitis (Al-Massarani et al. 2008), atherosclerotic complications, transplantation, and SLE (Sabatier et al. 2009). These cells express EC activation markers including EC adhesion molecules, such as E-selectin, ICAM-1, and VCAM-1, and procoagulant molecules, such as TF and vWF (Sabatier et al. 2009). This phenotypic evidence of an endothelial derivation supports the use of CECs as a sentinel for EC activation and injury and/or vascular dysfunction (Al-Massarani et al. 2008; Kerns et al. 2005; Sabatier et al. 2009; Strijbos et al. 2009) (Table 3).
An Expert Working Group on DIVI has proposed pursuing endothelial microparticles (EMPs) as a marker of EC activation in preclinical IND trials (Kerns et al. 2005) (Table 3). Endothelial microparticles have also been proposed as a biomarker of vascular injury in human patients with cardiovascular disease such as vasculitis, shock, and sickle cell disease (Strijbos et al. 2009; Wills et al. 2009). These particles, which play a role in vasculitis, thrombosis and atherosclerosis, as well as in vascular reactivity and angiogenesis (Ardoin et al. 2007; Chironi et al. 2009; Erdbruegger et al. 2008; Sabatier et al. 2009), are shed from the plasma membrane of activated ECs, apoptotic ECs, or CECs detached from injured vessels (Al-Massarani et al. 2008; Chironi et al. 2009; Sabatier et al. 2009). The EMPs vary in procoagulant activity as well as in phenotype, expressing CD62E+/E-selectin, CD54+/ICAM-1, CD106+/VCAM-1, CD31+/ PECAM-1, and CD144+/Ve-cadherin (Sabatier et al. 2009). These particles amplify EC dysfunction by impairing the NO pathway (Sabatier et al. 2009). We have observed numerous long and slender cytoplasmic processes, the length and width of which varied greatly, on the surface of circulating activated ECs (Zhang, Herman, Holt et al. 2002). Some of the processes were broken into membrane-bound vesicles. Our observations are consistent with the morphological features of these EMPs as small, membrane-bound vesicles shed from the plasma membrane of ECs (Ardoin et al. 2007; Chironi et al. 2009).
The phenotype of EMPs not only confirms their endothelial origin, but it also provides a basis to distinguish between EC activation and apoptosis (Chironi et al. 2009; Sabatier et al. 2009; Wills et al. 2009). Cluster of differentiation 146 (an endothelial junctional protein) and UEA-1 (Ulex europacus agglutinin-1, a marker of vascular endothelium) can be used to confirm endothelial origin of EMPs and to enumerate canine CECs/EMPs in whole blood (Wills et al. 2009). Cluster of differentiation 62E and CD31 are used to distinguish EMPs associated with EC activation as opposed to apoptosis. Increased expression of CD62E (a hallmark of E-selectin) was found in EMPs derived from activated ECs, whereas increased expression of CD31 (an indicator of PECAM-1) was found in EMPs from apoptotic ECs (Chironi et al. 2009). The ratio of EMPs/CD62E+ and EMPs/CD31+ was used as an index of EC activation (high ratio) and EC apoptosis (low ratio) (Esposito et al. 2008). A low ratio was found in patients with diabetes and erectile dysfunction, suggesting that EC apoptosis predominates in these patients (Esposito et al. 2008). A monoclonal antibody against CD51 has been used to measure EMPs from microvascular EC. Cluster of differentiation 51+/EMPs are increased after TNF-α-activation, and thus CD51 can serve as a marker for EMPs (Jimenez et al. 2001). Endothelial microparticles are elevated in the plasma of patients with thrombotic thrombocytopenic purpura, and correlated with increased expression of ICAM-1 and VCAM-1 (Jimenez et al. 2001). Endothelial microparticles derive from ECs, platelets, and leukocytes, and thus CD51+/EMPs and CD31+/ EMPs can be expressed on ECs and platelets, respectively. Endothelial microparticles could serve as a marker of EC activation and platelet activation (Ferrer et al. 1995; Gonzalez-Amaro and Sanchez-Madrid 1999; Jimenez et al. 2001). Currently EMPs can be identified and enumerated with flow cytometry, using Annexin V, CD62E, and CD 105 antibodies (Ardoin et al. 2007; Erdbruegger et al. 2008).
Given the different types of EC phenotype, the specific EC patterns for individual vessel types, and the different anatomic compartments within the same organ (Garlandla and Dejana 1997; Kawanami et al. 2000; Pusztaszeri et al. 2006), it should be expected that biomarkers of EC activation will include a heterogeneous group of candidate molecules. Although ECs of various organs possess many common functional and morphological features, ECs clearly exhibit significant heterogeneity of constitutive and inducible molecules across organs and, in the same organ, across vessels (post-capillaries, venules, arterioles, arteries, small vessels, and large vessels) (Garlandla and Dejana 1997). Not all activated ECs are alike (Pober 1988), and it should not be expected that any single biomarker can accommodate the variety of manifestations of DIVI, especially considering that hemodynamic, immunopathic, or direct cytotoxic pathogenic mechanisms could underlie such injury. It remains to be seen which of the biomarkers of EC activation mentioned in this review will have the greatest general utility in the area of DIVI.
Summary
The cardinal role of EC activation in the natural history of drug-induced as well as spontaneous vascular disease is now being increasingly recognized and appreciated. As a result, numerous biomarkers of EC activation have been explored and proposed. Of these biomarkers, only E-selectin, von Willebrand factor pro-peptide (vWFpp), MadCAM-1, ADMA, and CECs are considered to be endothelial specific in that their only source is the activated ECs. In contrast, the other biomarkers, although reliable and sensitive for vascular inflammation and injury, are not endothelial specific and may derive from multiple types of activated cells (neutrophils, platelets, mast cells, macrophages, antigen representing cells, T lymphocytes, etc.). It is now generally accepted that activation of immune cells, neutrophils, and especially ECs is an early critical event in the natural history of DIVI. Subsequent events may include apoptosis, dysfunction, and necrosis of EC, as well as varying degrees of reversible or irreversible injury to other mural cells and the vascular wall. Biomarkers of EC activation can also serve as sentinels for EC injury/damage or EC dysfunction. At present, our knowledge of these biomarkers remains limited, and their reliability in detecting and monitoring vascular injury remains uncertain or even speculative within any given context of use. Regardless of the biochemical pathways responsible for generating biomarkers of EC activation, some may have promise as sentinels of EC activation in preclinical or clinical studies of investigational new drugs, especially where DIVI has occurred or may be encountered. However, qualification of these biomarkers will require further study to establish their utility in the development of new therapeutics.
Footnotes
The findings and conclusions in this article have not been formally disseminated by the Food and Drug Administration and should not be construed to represent any agency determination or policy.
Acknowledgments
This work was supported by The Office of Testing and Research, Center for Drug Evaluation and Research, Food and Drug Administration. The authors wish to thank Dr. Vincent Vilker (OTR, CDER, FDA) for his encouragement and support in preparation for the manuscript and for critical review of this article. The authors also thank Joanne Berger (FDA Biosciences Library) for technical assistance with database searching and reference management and Alan D. Knapton (Division of Applied Pharmacology research, FDA) for help in ![]() preparation.
preparation.