
Abstract
Tissue localization of immune cells is critical to the study of disease processes in mouse models of human diseases. However, immunohistochemistry (IHC) for immune cell phenotyping in mouse tissue sections presents specific technical challenges. For example, CD4 and CD8 have been difficult to detect using IHC on formalin-fixed and paraffin-embedded mouse tissue, prompting alternative methods. We investigated the use of formalin-free zinc-salt fixation (ZN) and optimized IHC protocols for detecting a panel of immune cell–related markers (CD3, CD4, CD8, Foxp3, B220, F4/80, CD68, and major histocompatibility complex [MHC] class-I, MHC class-II, and Gr-1). The IHC results for these markers were compared on mouse spleen tissue treated with neutral buffered formalin (NBF) or ZN with or ZN without antigen retrieval (AR). Whereas CD4 and CD8 were not detected in NBF-treated tissue, all markers were detected in ZN-treated tissue without AR. Thus, the use of ZN treatment for IHC staining can be a good tool for studying immunoreactive lesions in tissues.
Introduction
Tissue localization of immune cells is critical to the study of disease processes in mouse models of human diseases. For example, the role of immune cells in cancer suppression and progression depends on analysis of intratumoral versus peritumoral immune cell infiltrates, localized macrophage polarization, and direct tumor cell–immune cell interactions (Coussens and Pollard 2011). Antibody reagents useful in flow cytometry and Western blot analyses do not always perform well in immunohistochemistry (IHC), and immune cell phenotypes are defined primarily by cluster of differentiation (CD) markers, themselves originally defined by mouse monoclonal antibodies recognizing leukocyte surface epitopes. Use of mouse monoclonal antibodies on mouse tissue for IHC is difficult due to the need for anti-mouse secondary antibody detection. Cell surface epitopes are often more difficult for IHC detection due to relatively inadequate levels of target proteins and limited epitope access in conventionally formalin-fixed paraffin-embedded (FFPE) tissue sections. Whereas the distribution of immune cells in tissue has been performed by IHC, not all immune cell markers can be detected in tissue section (Cardiff et al. 2013; Whiteland et al. 1995). For example, most of the studies have shown that T-cell lineage markers, CD4 and CD8, were not detectable with IHC on neutral buffer formalin (NBF)-treated tissue. However, some studies have successfully detected these markers on tissues treated with zinc fixative (Beckstead 1994; Hicks et al. 2006; Wester et al. 2003), paraformaldehyde (Tingstedt et al. 2003), or periodate-lysine-paraformaldehyde (Whiteland et al. 1995). Detecting other markers on tissue sections treated with different fixative reagents including NBF, ZN, and paraformaldehyde was also performed previously, which showed that non-NBF fixatives have advantages in IHC (Mikaelian et al. 2004). In these fixatives, ZN has been especially suggested as an alternate fixative for mouse immune cell markers that has previously been unable to stain for histology sections for CD4 and CD8 (Whiteland et al. 1995). In this study, we sought a practical solution to these problems and report the results of ZN fixation and optimized protocols for IHC for a panel of immune cell markers. Our results indicate that this ZN method is useful to detect immune cell–related markers including CD4 and CD8, which will support studies to decipher the differences in normal and tumor microenvironments.
Materials and Methods
Preparation of Tissues from Mice
Spleen was isolated from FVB/NJ (JAX Labs, Bar Harbor, ME) and used as positive control for some immune cell–related markers. Mice were housed in a vivarium under National Institute of Health guidelines, and all animal experiments followed protocols approved by the UC Davis Institutional Animal Care and Use Committee. Animals were fed LabDiet (PicoLab #5058, St. Louis, MO), ad lib water was autoclaved deionized water, and they were housed in a 12-hr/12-hr light–dark cycle at 21°C. Pathogenic agents were routinely monitored both by histopathological and by serogenic profile (UC Davis mouse level2 serogenic profile: Mouse Hepatitis Virus (MHV), Sendai, Pneumonia Virus of Mice Reo-3 (PVM), MPV, Minute Virus of Mice (parvovirus type) NS-1 (MVM), M.pul and arth, Theiler’s Muine Encephalomyelitis Virus part of GDVII strain (TMEV) [GDVII], Reo-3, Lymphocytic ChorioMeningitis Virus (LCM), Ectro, Epidemic Diarrhea of Infant Mice Virus (EDIM), Mouse Adeno DNA Virus (MAD) 1 and 2, Mouse Noro Virus (MNV)). Bacterial pathogens were tested on cecum or nasopharynx. Pinworms or fur mites were also checked. No pathogens were detected during this study.
ZN
Tissues were cut into 2 to 3 mm slices and fixed in IHC zinc fixative solution (Beckstead 1994; BD Biosciences, San Jose, CA) for 24 hr at room temperature (RT). After rinsing with tap water for 45 min, tissues were dehydrated at RT for 45 min each with 70% ethanol, 95% ethanol, 100% ethanol, and xylene, respectively. Tissues were infiltrated in paraffin at 58°C for 45 min using a Sakura Tissue-Tek®IV Embedding center (Sakura, Mars, PA). Tissue sections were prepared by cutting at 4 μm and floated out on a water bath at 43°C and collected on coated glass slides (SuperFrost/Plus; Fisher Scientific, Pittsburgh, PA). The slides were dried at RT for overnight.
IHC
Sections were deparaffinized in 3 times changes of xylene for 5 min each, followed by 3 times changes of 100% ethanol for 2 min each. They were rehydrated through 95% and 70% ethanol to tap water, then antigen retrieval (AR) procedure was performed with a decloaking chamber (Biocare Medical LLC, Concord, CA) with citrate buffer (10 mM sodium citrate, pH 6) for 45 min constantly heating at 125°C at 15 psi. using a digital decloaking chamber (Biocare Medical LLC), if it is required (see Table 1). Tissue section was washed in EnVision™ FLEX wash buffer (Dako, Carpinteria, CA) for 2 min followed by blocking with a 10-min incubation in 10% goat serum in phosphate buffered saline (PBS; pH 7.4). Goat serum was used because any antibody used in this study was not raised in the species. The primary antibody (listed in Table 2) was diluted in 0.5% bovine serum albumin (BSA) in PBS (pH 7.4) and incubated with tissue sections for 1 hr at RT. The slides were then rinsed twice with EnVision™ FLEX wash buffer (Dako) for 5 min each. Immunohistochemical reaction was performed by using a VECTASTAIN Elite ABC kit (PK-6100; Vector Laboratories, Burlingame, CA) with biotinylated anti-rabbit or anti-rat IgG (BA-9400 and BA-9401 respectively; Vector Laboratories) following manufacturer’s protocol. Shortly, the slides were incubated with a biotinylated anti-rabbit at 1:1,000 dilution or an anti-rat IgG at 1:500 dilution in 5% goat serum in PBS (pH 7.4) for 60 min. Then sections were washed twice with PBS for 5 min each followed by incubating with VECTASTAIN Elite ABC reagent for 30 min. After the slides were washed twice with PBS, samples were then incubated in freshly prepared 0.1% 3,3′-diaminobenzidine (DAB) solution (Dako) following the manufacturer’s directions. After incubation with DAB for less than 5 min to prevent background staining, the sections were rinsed in tap water for 2 min. Counterstaining was performed with Mayer’s hematoxylin for 30 sec, then rinsed in tap water for 2 min. The slides were dehydrated in 70% and 95% ethanol, then 3 times changes of 100% ethanol and xylene for 2 min each and mounted with Clear Mount (American MasterTech Scientific, Lodi, CA). If an automated procedure was desired, a Dako Autostainer Plus (Dako) was programmed and operated with the following: a set of a VECTASTAIN Elite ABC kit with biotinylated anti-rat IgG for detecting CD4 and CD8, horse radish peroxidase (HRP)-labeled polymer anti-rabbit (K4003; Dako) for other markers, respectively. Counterstaining was performed with liquid DAB + substrate chromogen system (K3468; Dako). The procedures for the Autostainer Plus were performed by applying same protocols as described in manual IHC. Images were taken either by the Aperio Leica ScanScope XT (Leica Biosystems, Richmond, IL) or by Zeiss Axioskop with Zeiss AxioCam color CCD camera (Carl Zeiss, Pleasanton, CA) using the 20× objective lens.
The summary of detecting immune cell–related markers in tissues treated with NBF or ZN with or without AR.
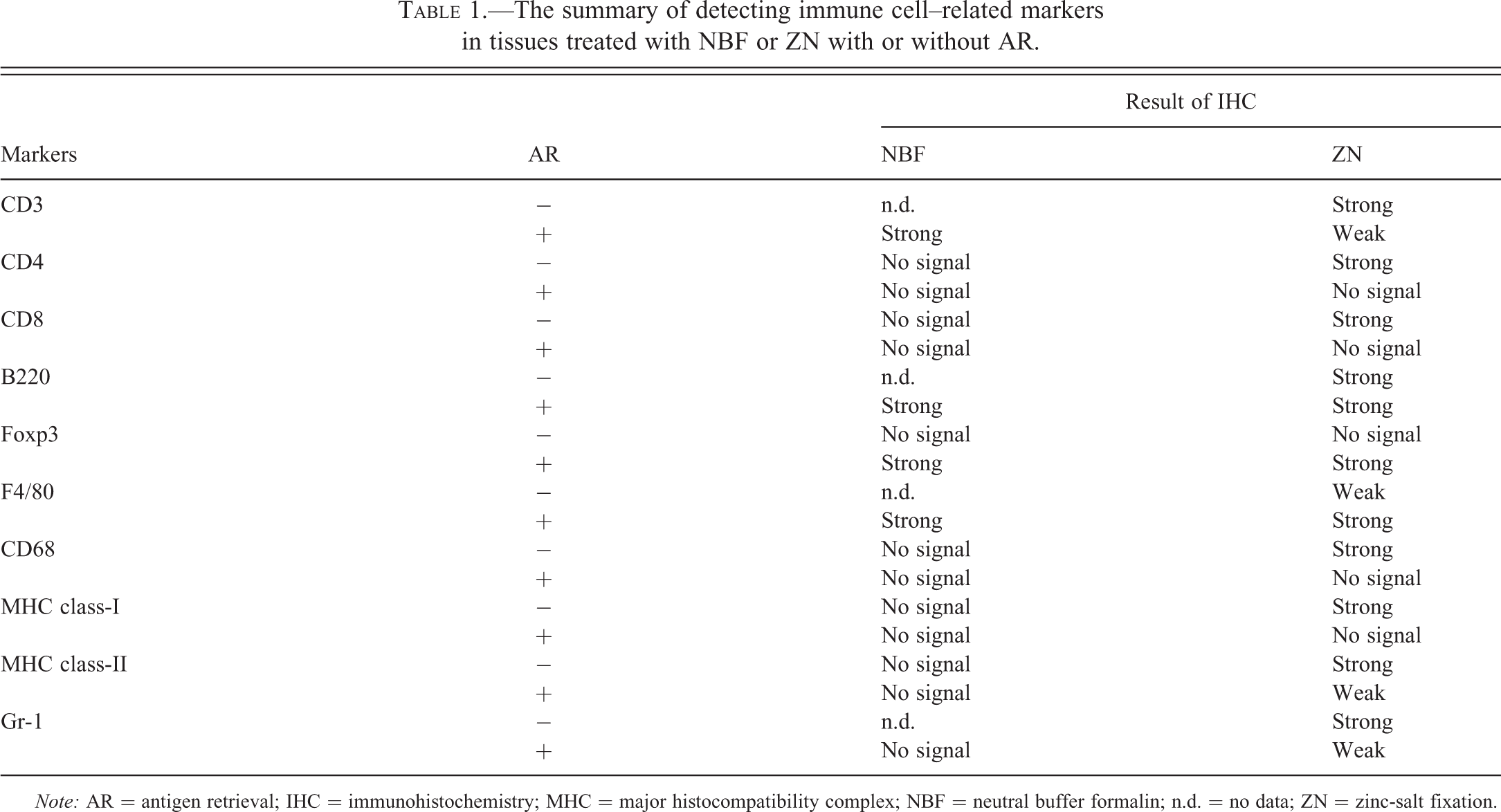
Note: AR = antigen retrieval; IHC = immunohistochemistry; MHC = major histocompatibility complex; NBF = neutral buffer formalin; n.d. = no data; ZN = zinc-salt fixation.
The list of antibodies used for detecting immune cell–related markers.
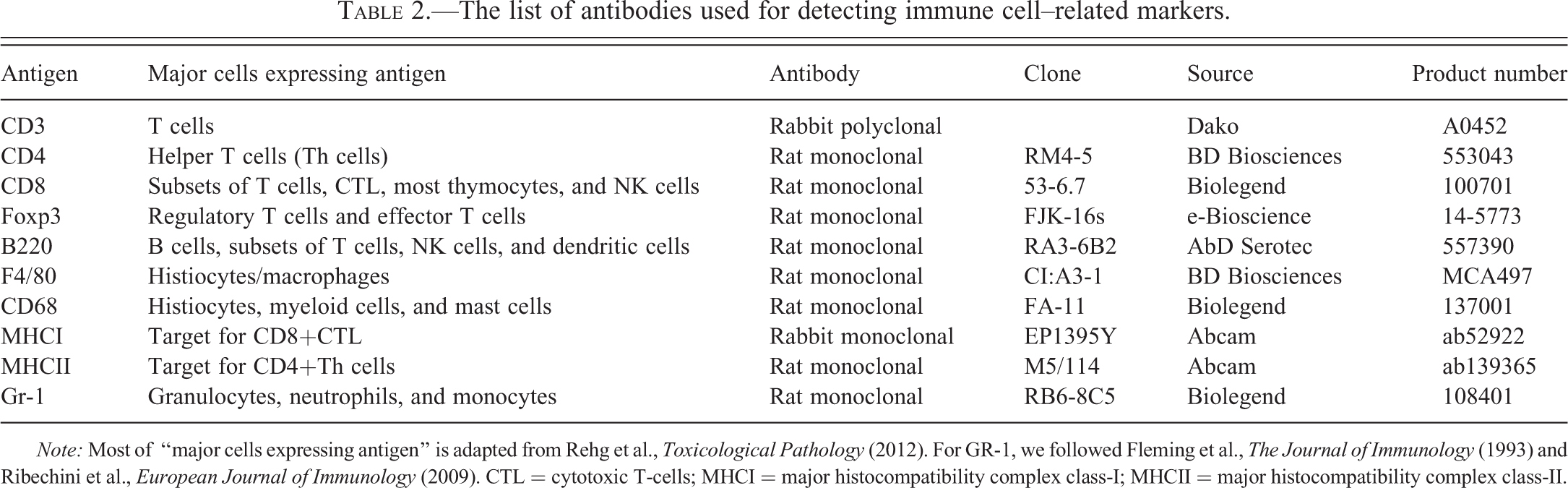
Note: Most of “major cells expressing antigen” is adapted from Rehg et al., Toxicological Pathology (2012). For GR-1, we followed Fleming et al., The Journal of Immunology (1993) and Ribechini et al., European Journal of Immunology (2009). CTL = cytotoxic T-cells; MHCI = major histocompatibility complex class-I; MHCII = major histocompatibility complex class-II.
Results
Comparison of Immune Cell–related Markers between NBF- and ZN-treated Mouse Spleen Tissues
We performed IHC on mouse spleen tissues treated with NBF and ZN to detect CD3, CD4, CD8, B220, and Foxp3 (a list for all antibodies tested in this work is indicated in Table 2). Whereas positive staining of CD4 and CD8 in periarteriolar lymphoid sheaths (PALS) were not seen in NBF-treated tissue sections as expected (Figure 1A [c,e]), CD3 (Figure 1A [a]) and Foxp3 (Figure 1B [c]) in PALS, and B220 in lymphoid follicles (Figure 1B [a]) were successfully detected. On the other hand, IHC staining for these markers on ZN-treated spleen sections were all positive (Figures 1A [b,d,f] and B [b,d]). These results indicate that ZN fixation on tissue samples is useful to detect lymphocytic lineage markers.
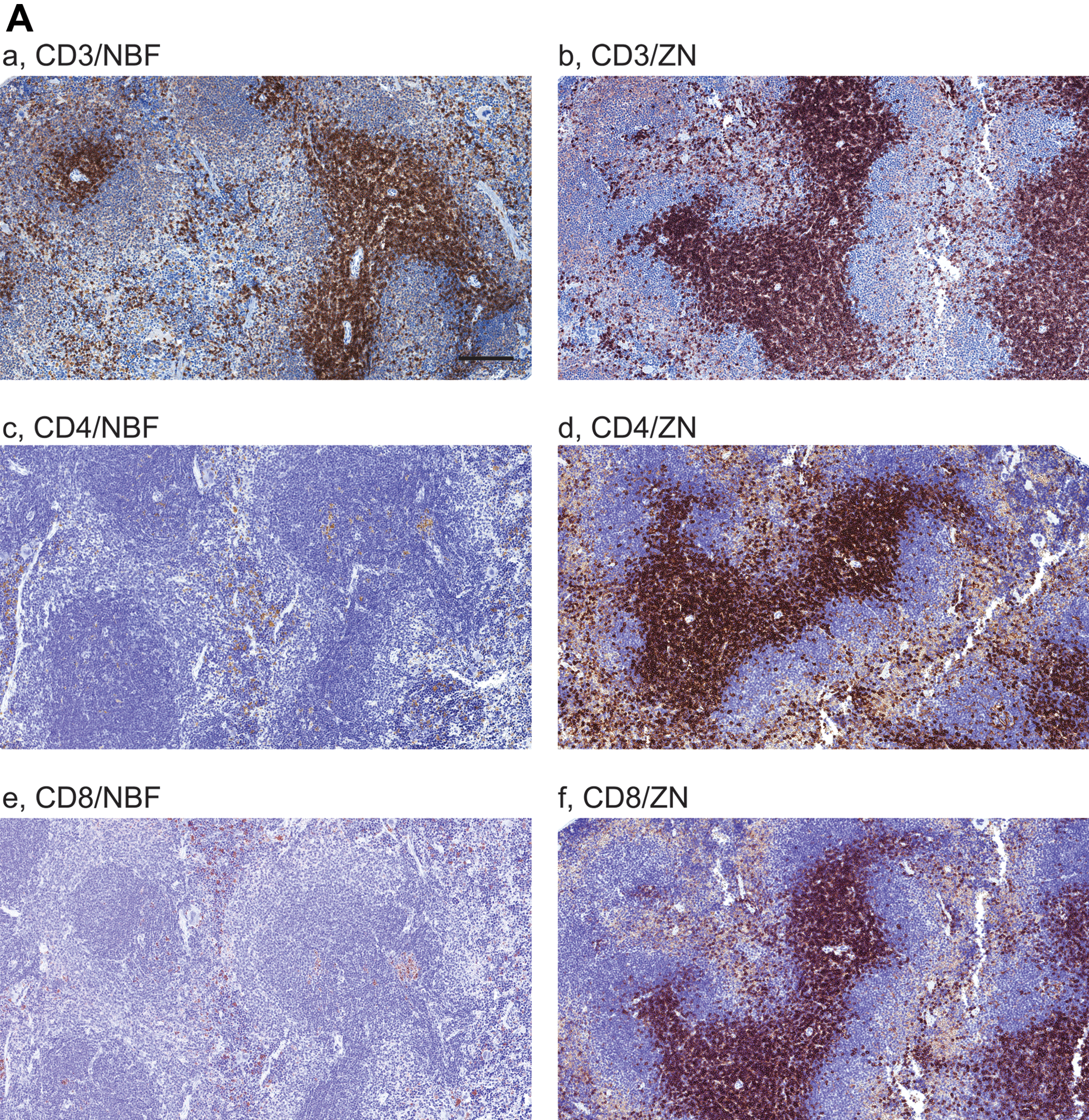
Comparison of immune cell–related markers between neutral buffer formalin (NBF) and zinc-salt fixation (ZN)-treated mouse spleen tissues. Images are showing immunohistochemistry (IHC) with 10 immune cell–related markers on NBF- or ZN-treated murine spleen tissue sections. (A and B) IHC images are indicating the staining with antibody for A. (a,b) CD3, (c,d) CD4, (e,f) CD8; B. (a,b) B220 and (c,d) Foxp3, respectively. Whereas CD4- and CD8-positive cells were not detected in NBF-treated spleen tissues, the IHC in ZN-treated tissue sections shows staining-positive cells. Scale bar indicates 200 μm (a–h) and 50 μm (i–j). (C and D) IHC images are indicating the staining with antibody for C. (a,b) F4/80, (c,d) CD68; and D. (a,b) major histocompatibility complex (MHC) class-I, (c,d) MHC class-II, (e,f) Gr-1, respectively. IHC with MHC class-I, -II, and Gr-1 did not detect staining-positive cells on NBF-treated tissue sections. However, all markers were detected in ZN tissue sections. These indicate that ZN treatment on tissue is better for detecting various cell surface markers. Counterstaining is performed with hematoxylin. Whereas all NBF-treated tissues were treated with antigen retrieval (AR), most of ZN-treated tissues were not treated except for Foxp3 and F4/80. Scale bar indicates 50 μm. Please see Table 1 for the requirement of AR.
Other immune cell–related markers (F4/80, CD68, MHC class-I, MHC class-II, and Gr-1) were also tested (Figure 1C and D). As far as we tested, whereas F4/80 (Figure 1C[a]) and CD68 (Figure 1C[c]) were positively detected in IHC on NBF-treated tissue sections, the staining of these markers were much clearer on ZN-treated samples (Figure 1C[b,d]). Staining positive cells for MHC class-I (Figure 1D[a]), MHC class-II (Figure 1D[c]), and Gr-1 (Figure 1D[e]) were not detected in NBF-treated tissue sections with antibody sets indicated in Table 2. In contrast, IHC for these markers on ZN-treated tissue section detected marker positive cells (Figure 1D[b,d,f]). These results suggest that ZN treatment on tissue is also useful to study various immune cell–related markers in addition to lymphocytes. We also validated the necessity of AR procedure in IHC on both NBF- and ZN-treated tissues (Table 1). NBF-treated tissue needed AR procedure to detect marker proteins. However, marker positive cells were clearly detected in most of ZN-treated tissues without AR, except for F4/80 and Foxp3. We also observed that IHC signals on ZN-treated tissue with AR were weaker than the condition without AR (Table 1; data not shown).
Discussion
The study of a variety of immune cells within normal and tumor tissues from patients or animal models can elucidate the involvement of immune cells in tumor progression or regression. However, limitations using current formalin-based fixation and standard IHC techniques prevent proper analysis of target immune cells or proteins in tissue samples. Previous reports indicate that IHC detection of CD4 and CD8 in formalin-fixed tissues was unsuccessful perhaps due to epitope masking of antibody recognition (Beckstead 1994; Cardiff et al. 2013; Hicks et al. 2006; Wester et al. 2003). Here we demonstrated that IHC on ZN-treated tissues successfully allowed for visualizing 10 different immune cell–related markers compared to NBF-treated tissues. As far as we observed cellular morphology from H&S tissues, ZN-treated tissue exhibited marginal shrinkage, which might be due to its hypertrophic effects encountered within the dehydration procedure (data not shown). Since the ZN buffer formulation used in this study is formalin free, ZN treatment on tissue might have less cross-linking reactivity compared to cross-linking formalin, consequently generating better epitope exposure of target proteins in tissues when compared with NBF-treated tissues. For this reason, ZN-treated tissue might not need AR treatment for IHC on various immune markers. In fact, IHC for detecting CD4 and CD8 markers did not need AR treatment in this study. However, detecting some other markers (Foxp3 and F4/80) needed AR treatment for IHC, suggesting that detecting each marker in ZN-treated tissue section may need optimization to verify if AR treatment is necessary or required. Our results further demonstrated that IHC on ZN-treated tissue could be performed even with antibodies which are not suitable for general NBF-fixed tissues. These information of method and antibodies indicated in this work might shed a light on new applications for histological analysis for diagnosis and basic research with any other antibodies.
We predicted that tumors cancer model mice might be enriched for immune cell populations. The comparison between different cancer model mice or human tissue biopsies in different cancer progression series to decipher the involvement of immune cells and its lineages will be necessary in the future. Since IHC with chromogenic detection is limited and immune cells have various lineages with complex cell surface markers, future analyses will need an ability to detect multiple markers at once which could be performed by a multiplexed-IHC methods (Angelo et al. 2014).
Footnotes
Acknowledgments
The authors wish to thank Ms. Judith Walls and Herlina Sugandha for providing excellent histology support.
Author Contribution
Authors contributed to conception or design (HM, PS, LS, QC, NH, RC, AB); data acquisition, analysis, or interpretation (HM, PS, LS, QC, NH, RC, AB); drafting the manuscript (HM, LS, RC, AB); and critically revising the manuscript (HM, LS, RC, AB). Authors gave final approval (HM, LS, QC, NH, RC, AB). All authors agreed to be accountable for all aspects of work in ensuring that questions relating to the accuracy or integrity of any part of the work are appropriately investigated and resolved.
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This project was funded by grants from the National Cancer Institute’s Mouse Models of Human Cancers Consortium (U01 CA141582, U01 CA141541, U01 CA105490-01).