
Abstract
This investigation examined microRNA-208a (miR-208a) as a potential biomarker of isoproterenol (ISO)-induced cardiac injury in superoxide dismutase-2 (Sod2+/− ) and the wild-type mice, and the potential sensitivity of Sod2+/− mice to ISO-induced toxicity. A single intraperitoneal injection of ISO was administered to age-matched wild-type and Sod2+/− mice at 0, 80, or 160 mg/kg. Plasma miR-208a, cardiac troponin I (cTnI), and ISO systemic exposure were measured at various time points postdose. Hearts were collected for histopathology examination and for tissue expression of miR-208a and myosin heavy chain 7. ISO administration caused increases in cTnI and miR-208a plasma levels that correlated with myocardial damage; however, the magnitude of increase differed according to the types of mice. At similar ISO systemic exposure, the magnitude of cTnI was greater in wild-type mice compared to Sod2+/ − mice; however, the magnitude of miR-208a was greater in Sod2+/− mice than that of the wild-type mice. Myocardial degeneration occurred at ≥3 hr in the wild-type and ≥6 hr in Sod2+/ − mice. At ≥24 hr after ISO administration, miR-208a appeared superior to cTnI in indicating myocardial injury in both wild-type and Sod2+/− mice. Sod2+/− mice were not more sensitive than wild-type mice to ISO-induced toxicity.
Introduction
Drug-induced cardiotoxicity is a major cause for attrition of drug candidates in the pharmaceutical industry (Laverty et al. 2011), often occurring in later stages of clinical trials or after postmarket approval (Shah 2006). Attrition of potential drug candidates due to cardiac toxicity in later stages of drug development occurs due in part to the lack of early detection of risk in animal models. Myocardial cells may only have a limited capacity to regenerate and irreversible injury often occurs rapidly after insult (Schoen and Mitchell 2010). Early detection of myocardial injury is of paramount importance to identify myocardial effects within a reversible time frame or to mitigate the extent of irreversible damage. Therefore, sensitive biomarkers predictive of early signs of cardiac toxicity are needed. The most commonly used biomarker for diagnosing cardiac injury are blood troponins (Mikaelian et al. 2009; Reagan 2010; Wallace et al. 2004; Wu et al. 2009); cardiac troponin I (cTnI) specifically has gained acceptance for use in animals (O’Brien et al. 2006). Although cTnI has the ability to detect severely damaged cardiac muscle, certain cardiac insults such as ischemic damage, hypertrophy, inflammation, and apoptosis may go undetected (Aragno et al. 2008; Margulies 2009; Van Rooij et al. 2007; Zhang et al. 2008). In addition, cTns have been endorsed internationally as biomarkers for myocardial injury and for risk stratification in patients with acute coronary syndrome (Apple and Collinson 2012), but their ability to identify myocardial damage in more chronic phases of cardiac disease remains less clear.
The discovery of a heart-specific microRNA (miRs), miR-208a, in the systemic circulation after myocardial infarction suggests that miR-208a holds promise as a noninvasive biomarker of myocardial injury (D’Alessandra et al. 2010). miR-208a appears to play a role in cardiac hypertrophy and heart failure in humans (Han, Toli, and Abdellatif 2011). miR-208a is also upregulated in cardiac hypertrophy in mice (Callis et al. 2009; Wang et al. 2010). Although the biological functions of miRs are not fully understood, it has been reported that many miRs exhibit a tissue- or cell-specific distribution and are remarkably stable in the circulation (Creemers, Tijsen, and Pinto 2012; Gilad et al. 2008); thus, miRs appear as ideal biomarkers for specific target organs. In a study of myocardial-specific injury in Sprague-Dawley rats, Ji et al. (2009) investigated miR-208a as a potential biomarker of myocardial injury in rats treated with a high dose of isoproterenol (ISO), demonstrating a similar time course to the plasma concentration of cTnI, suggesting that miR-208a may be an indicator of myocardial injury. Although the mechanism by which miRs are released into the circulation is unclear, it is established that cTnI escapes from the heart when cardiomyocyte membrane is compromised during injury (Brady et al. 2010; Engle et al. 2009; Reagan 2010) making this protein suitable for the diagnosis of cardiac damage in humans and animals. miR-208a might also exhibit different release profiles in various animal models and/or under various pathologic conditions (Wang et al. 2010; D’Alessandra et al. 2010), thus warranting additional miR-208a-related investigative work to elucidate its utility.
Oxidative stress is one of the known mechanisms of cardiac injury (Schimmel et al. 2004). A major contributor to the injury is the inability to detoxify reactive oxygen species produced by mitochondrial respiration. Manganese superoxide dismutase (MnSOD), coded for by the Sod2 gene, serves as the primary defense against mitochondrial superoxide. The importance of Sod2 function in mammalian organisms was confirmed by disruption of the Sod2 gene, which was lethal for Sod2 knockout mice due to neurodegeneration and cardiac dysfunction (Lebovitz et al. 1996). Sod2 deficiency causes increased susceptibility to oxidative mitochondrial injury in central nervous system neurons and cardiac myocytes. The Sod2+/− mice have been used to investigate the aging process associated with increased oxidative stress (Miwa, Beckman, and Muller 2008; Van Remmen et al. 2003) as well as hepatic injury (Hsiao, Younis, and Boelsterli 2010); however, the Sod2+/− mice have not been evaluated for detecting drug-induced cardiac toxicity. Given the higher level of superoxide radicals present in the heart of Sod2+/− mice (Ibrahim et al. 2013), they may exhibit greater sensitivity to drug-induced injury through the formation of reactive oxygen species.
ISO is an adrenergic (β1, β2) agonist that induces tachycardia, arrhythmias, and palpitations (Rona et al. 1959; Zhang et al. 2008; Jimenez et al. 2011) at suprapharmacological doses. These β-adrenergic effects can result in cardiac “infarct-like” lesions in experimental animals (York et al. 2007), similar to those in patients with myocardial infarction (Zhang et al. 2008). At high doses in rats, ISO induces cardiac oxidative stress, decreased blood pressure, hypoxia, and myocardial necrosis (Rona et al. 1959). Because these characteristics can be observed after a single acute dose in rodents, ISO was selected as the model cardiac toxicant for this study.
Other muscle-enriched miRs (miR-1, miR-133a, and miR-499) have also been tested in acute myocardial infarction (AMI) patients and animal models of myocardial infarction (D’Alessandra et al. 2010; Wang et al. 2010). Data from those investigations indicated that miR-1, miR-133a, and miR-499 were present with very low abundance in plasma, and miR-208a was absent from the plasma of healthy people; whereas, all 4 miR levels were higher in plasma from AMI patients compared to healthy subjects. Peak miR-1 and miR-133a plasma concentrations occurred at a similar time as cTnI, whereas miR-499 exhibited a slower time course (D’Alessandra et al. 2010; Wang et al. 2010). Of the miR biomarkers, miR-208a was suggested to be more sensitive and specific for diagnosing AMI.
The objectives of this investigation were to assess the released plasma profiles of miR-208a and cTnI in ISO-induced cardiac injury in the C57BL/6J mice (wild-type) and the C57BL/6J heterozygous for Sod2 (Sod2+/− ) mice, as well as to compare the potential effects in the putative mouse model (Sod2+/− ) of oxidative stress (Hsiao, Younis, and Boelsterli 2010) with those of the wild-type. This work demonstrated that miR-208a correlated more frequently than cTnI with ISO-induced myocardial injury in Sod2 +/− and the wild-type mice.
Materials and Methods
Animals
Heterozygous Sod2 tm1Leb/J mice (breeding pairs), congenic in the C57BL/6J background, were originally obtained from Jackson Laboratory (Sacramento, CA). A breeding colony was established by crossing male Sod2 +/− with female wild-type mice. The F1 littermates were genotyped before weaning as described (Ong et al. 2006) and subsequently used for the experiments. Mice were placed (up to 5/cage) in solid bottom polycarbonate caging housed in an environmentally controlled room on a 12-hr light/12-hr dark cycle and provided ad libitum Certified Rodent Diet 5002 and water that was purified by reverse osmosis. This study was conducted in accordance with the current guidelines for animal welfare (as amended in 1970, 1976, and 1985, and 1990, and the Animal Welfare Act [AWA] implementing regulations in title 9, CFR chapter 1, subchapter A, parts 1–3). The procedures used in this study were reviewed and approved by the Institutional Animal Care and Use Committee.
In Vivo Study Designs
ISO toxicokinetic (TK) study
For ISO TK profiling, a single dose of ISO was administered to male wild-type mice (N = 4/group) at 0, 20, 50, 80, or 160 mg/kg and male Sod2+/− mice at 160 mg/kg by intraperitoneal (IP) injection. Blood was collected into tubes containing dipotassium ethylenediaminetetraacetic acid (K2EDTA) at 1, 3, 6, and 24 hr postdosing on day 1 and processed to obtain plasma samples for measuring ISO systemic exposure. A liquid chromatography tandem mass spectrometer (LC-MS/MS) system that consisted of Waters Acquity binary solvent manager, sample manager, and column manager (Milford, MA) was used for plasma sample analysis. TK parameters were analyzed in Watson LIMS (version 7.4, Thermo Scientific©, Pittsburgh, PA) using a noncompartmental approach. The area under the curve (AUC0–24) was calculated using the linear trapezoid method.
Dose range finding study in Sod2+/− mice
Initial dose range finding study was conducted in Sod2+/− mice in an effort to assess tolerability. In the dose range finding study, ISO was administered intraperitoneally to 6 Sod2+/− mice per group at dose levels of 0 (0.9% saline), 80, 160, or 320 mg/kg. All animals were observed daily for mortality, clinical signs, and weights prior to dosing (day 1) and on day 2 prior to study termination. Plasma (K2EDTA) samples were obtained at 6 and 24 hr postdose and assessed for miR-1, miR-133a, miR-208a, miR-499-5p, and cTnI concentrations. Hearts from these animals were also collected for histopathologic examination at 6 and 24 hr postdose.
Definitive studies in the wild-type and Sod2+/− mice
In subsequent studies, ISO was administered to age- and body weight–matched male wild-type and Sod2+/− mice (N = 10/group) at dose levels of 0 (0.9% saline), 80, or 160 mg/kg. Group mean body weights of the wild-type and Sod2+/− mice were 36.01 and 34.50 g, respectively. Plasma samples for measuring miR-208a and cTnI were collected at 3, 6, and 24 hr postdose. Hearts were also collected on day 1 (3, 6, and 24 hr) and day 10 postdose from the same animals bled for biomarker assessment and submitted for histopathology examination or for gene expression analysis (3-hr time point only).
cTnI Measurement
cTnI was measured in K2EDTA plasma using Cardiac Injury Panel 2 (kit # K15155C; Meso Scale Discovery [MSD], Whitehouse Station, NJ), according to the instructions of the manufacturer. It was originally developed for a rat assay and was adapted for use with mouse plasma. In an MSD® assay, 25 μl of neat plasma from each sample were tested on the Cardiac Injury Panel 2 kit. The lower limit of detection (LLOD) was the calculated concentration of the signal that was 2.5 standard deviations over the zero calibrator. The cTnI plasma concentrations were expressed in pg/ml.
Brain Natriuretic Peptide (BNP) Measurement
BNP was measured in mouse K2EDTA plasma using RayBio® Human/Mouse/Rat BNP Enzyme Immunoassay kit (RayBiotech, Inc., Norcross, GA), according to the manufacturer’s instructions. This kit was intended for use in mouse, human, or rat plasma. In a RayBio assay, 100 μl of neat plasma from each sample were added to each well. The minimum detectable concentration of BNP was 1.02 pg/ml. The BNP plasma concentrations were expressed in pg/ml.
Quantitative Real-time PCR (qRT-PCR)
For measuring circulating miRs, total RNA was isolated from plasma with 300 µl of TRIzol® reagent (InvitrogenTM, Carlsbad, CA) and added to 100 µl of plasma followed by phase separation. RNA was precipitated by adding 30 μg of glycogen (Ambion®, Austin, TX) and resuspended in 50 µl of RNase-free water (Qiagen, Valencia, CA). miR-1, miR-133a, miR-208a, and miR-499-5p were measured using TaqMan® MicroRNA Assays (Applied Biosystems, Carlsbad, CA). For tissue RNA measurement, heart tissues were homogenized in TRIzol reagent (Invitrogen), followed by phase separation and RNA precipitation. miR-208a and myosin heavy chain gene (Myh7) were measured using TaqMan MicroRNA Assays. Approximately 5 μl of RNA was used in 15 μl of reverse transcriptase (RT) reaction. Duplicate qRT-PCR was run using 1.33 μl of RT product. Copies of miRs per reaction were determined using a standard curve of synthetic miR (Integrated DNA Technologies, Coralville, IA). The LLOD was 50 copies. miR plasma concentrations were expressed as copies/μl.
Histology Processing and Assessment
Entire hearts were immersion fixed in 10% neutral buffered formalin (NBF) and divided in half by cutting from the base of the heart through atria, ventricles, and apex prior to standard tissue processing procedures. The samples were embedded in paraffin blocks and 4-μm thick sections prepared using an automated microtome (Leica Microsystems, IL). All slides were dried overnight at 37°C and then stained using a standard hematoxylin and eosin (H&E) procedure, dehydrated, and mounted in Micromount mounting media (Surgipath, IL) before microscopic evaluation.
While evaluating the heart sections, a subjective grading system was used to ascribe a level of severity to lesions observed. The severity of each finding was determined by estimating the percentage of tissue affected or effaced by each lesion, and ascribing a point grade to the finding from a 5-point system in the following manner: 1+ (minimal) = ≤10%; 2+ (mild) = >10 to 30%; 3+ (moderate) = >30 to 50%; 4+ (marked) = >50 to 70%; 5+ (severe) = >70%.
Immunohistochemical Detection of Caspase 3
Selected heart sections were placed on a Leica Bond automated staining system (Leica Microsystems), dewaxed, and antigen retrieved with epitope retrieval 2 (ER2) for 20 min. This was followed by blocking endogenous peroxidases with hydrogen peroxide for 10 min and nonspecific immunolabeling was blocked with protein block (Dako) for 10 min. After washing in Bond buffer, the slides were immunolabeled using a specific polyclonal cleaved caspase 3 (Asp175) antiserum from Cell Signaling Technology that was diluted to 0.1µg/ml. After a 20-min incubation and wash, the immunolabeling was detected using the Bond Polymer Refine kit (Leica Microsystems). The slides were dehydrated and mounted in Micromount mounting media (Surgipath, IL) before histological evaluation.
Statistical Analysis
Each biomarker (cTnI, miR-208a, and BNP) was analyzed separately for each strain and each time point using the nonparametric Wilcoxon test to investigate differences due to treatment (vehicle vs. ISO). To study the effect on these 2 types of mice, an analysis of variance (ANOVA) was used. For comparison of wild-type versus Sod2+/− mice, only the ISO-treated animal data were used since almost all the vehicle controls had biomarker measurements below the LLOD and were therefore treated as constant. The end point in the ANOVA model was the fold change from the LLOD on the logarithmic scale. The 5% (p < .05) level of statistical significance were used to assess the data.
Results
Dose Range Finding Study in Sod2 +/− Mice
The initial dose range finding study revealed that ISO at 320 mg/kg was not tolerated in Sod2+/− mice as death occurred at 320 mg/kg, while 80 mg/kg appeared to be too low to induce a cardiac histologic change. The dose of 160 mg/kg not only caused significant plasma elevations in cTnI and miR-208a at 6 and 24 hr postdose but also resulted in a myocardial degeneration in the heart. At 160 mg/kg, ISO was well tolerated and there were no remarkable clinical signs of toxicity or changes in body weight. Therefore, the 160-mg/kg dose was selected for subsequent studies in Sod2+/− mice. As part of the initial dose range finding study, miR-1, miR-133a, miR-208a, and miR-499-5p were assessed as potential biomarkers of early cardiac injury. The concentrations of these miRs were compared to those of cTnI (Table 1). Among all the biomarkers tested in this study, miR-208a was comparable to that of cTnI; however, the magnitude (up to 353-fold) of the increases compared to control levels was far greater than that of cTnI (up to 171-fold). A lower magnitude of response compared to that of miR-208a was noted for miR-499-5p (up to 11.7-fold), while miR-1 (up to 2-fold) and miR-133a (up to 3.4-fold) appeared to be the least sensitive biomarkers of cardiac injury (Table 1). miR-208a was selected for further characterization since miR-208a exhibited the most robust increase and its releasing profile appeared similar or better than that of cTnI.
Summary of plasma levels of various miRs following ISO administration to Sod2+/− mice.a
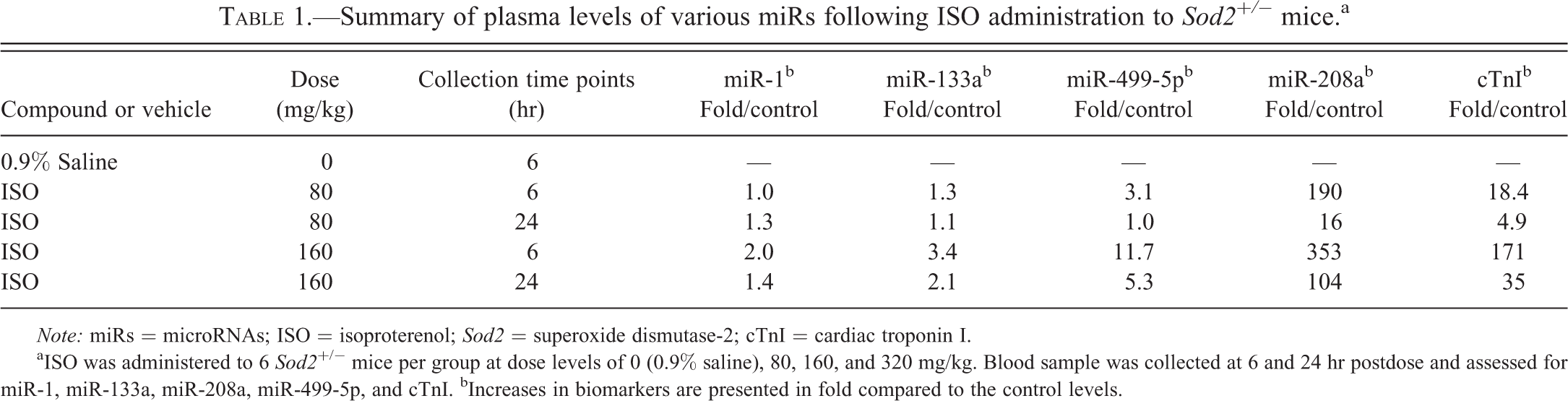
Note: miRs = microRNAs; ISO = isoproterenol; Sod2 = superoxide dismutase-2; cTnI = cardiac troponin I.
aISO was administered to 6 Sod2 +/− mice per group at dose levels of 0 (0.9% saline), 80, 160, and 320 mg/kg. Blood sample was collected at 6 and 24 hr postdose and assessed for miR-1, miR-133a, miR-208a, miR-499-5p, and cTnI. bIncreases in biomarkers are presented in fold compared to the control levels.
Plasma Cardiac-specific Biomarkers
ISO given at the same dose level (160 mg/kg) in both wild-type and Sod2+/− mice
One dose of ISO administered at 160 mg/kg to wild-type and Sod2+/− mice led to increases in cTnI and miR-208a in both types of mice. Although the magnitude of increases compared to the limits of their respective controls and the release profiles of these biomarkers differed between the types of mice, the incidence of mice with elevated miR-208a or cTnI was generally comparable between the 2 types of mice (Table 2). In the wild-type mice, the profiles and magnitudes of increases of cTnI and miR-208a were similar, and cTnI and miR-208a reached the highest plasma values at 3 hr after dosing (Figure 1A). In contrast, the magnitude of the miR-208a elevation compared to control levels was much greater than that of cTnI in Sod2+/− mice, and both biomarkers reached the highest values at 6 hr postdose (Figure 1B). Notably, mean plasma concentrations of both cTnI and miR-208a at 24 hr were about 2-fold higher in Sod2+/− mice than those in the wild-type. Furthermore, there were differences between miR-208a and cTnI within individual animals. Concurrent increases in both miR-208a and cTnI were observed in 78% wild-type mice at 24 hr postdose, but only 44% Sod2+/− mice with an elevation in miR-208a had an corresponding increase in cTnI (Table 2). Additionally at day 10, 80% of Sod2+/− and 90% of wild-type mice had elevated miR-208a levels, compared to only 20% of Sod2+/− and 30% of wild-type with increased cTnI levels (Table 2).
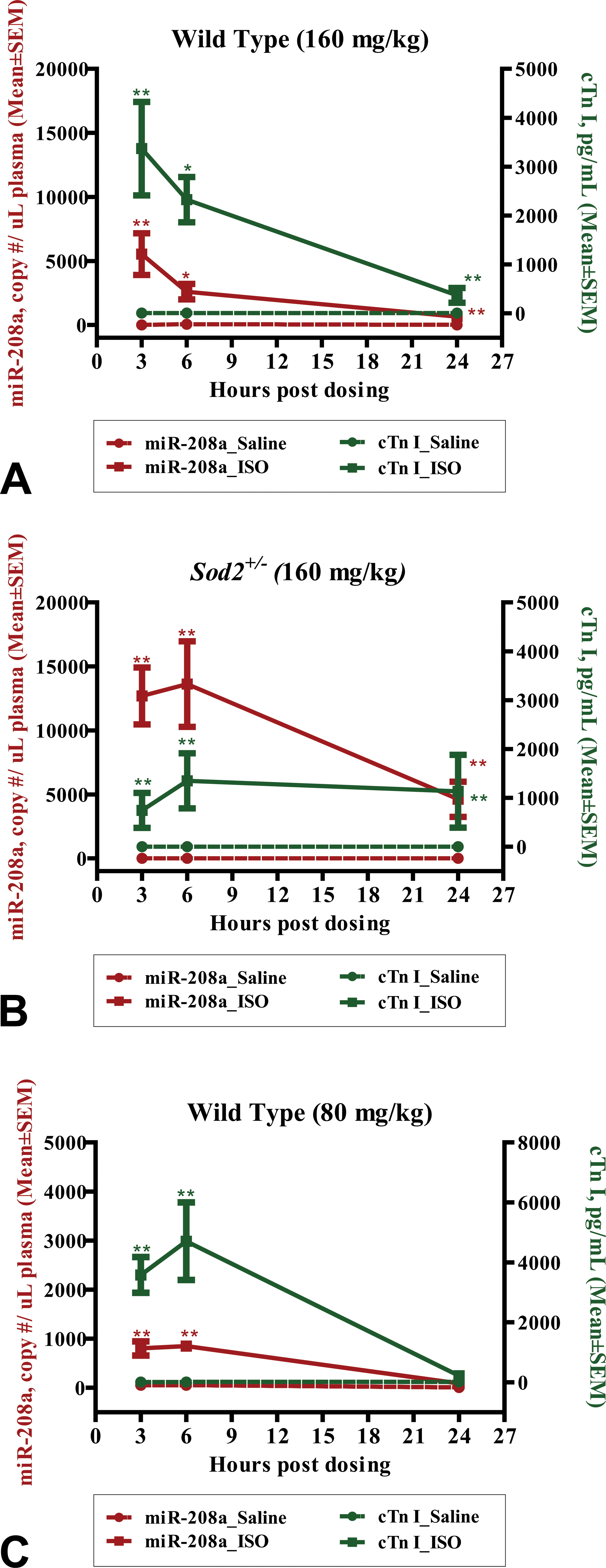
Mouse type-dependent effects of ISO on the release profiles of miR-208a and cTnI in wild-type and Sod2+/−
mice. Plasma was collected from male wild-type and Sod2+/−
mice at 3, 6, or 24 hr after ISO administration. Group mean plasma levels of cTnI and
Incidencesa in Sod2+/− and the wild-type mice of ISO-induced increases in miR-208a and cTnI plasma levels with ventricular and atrial effects.
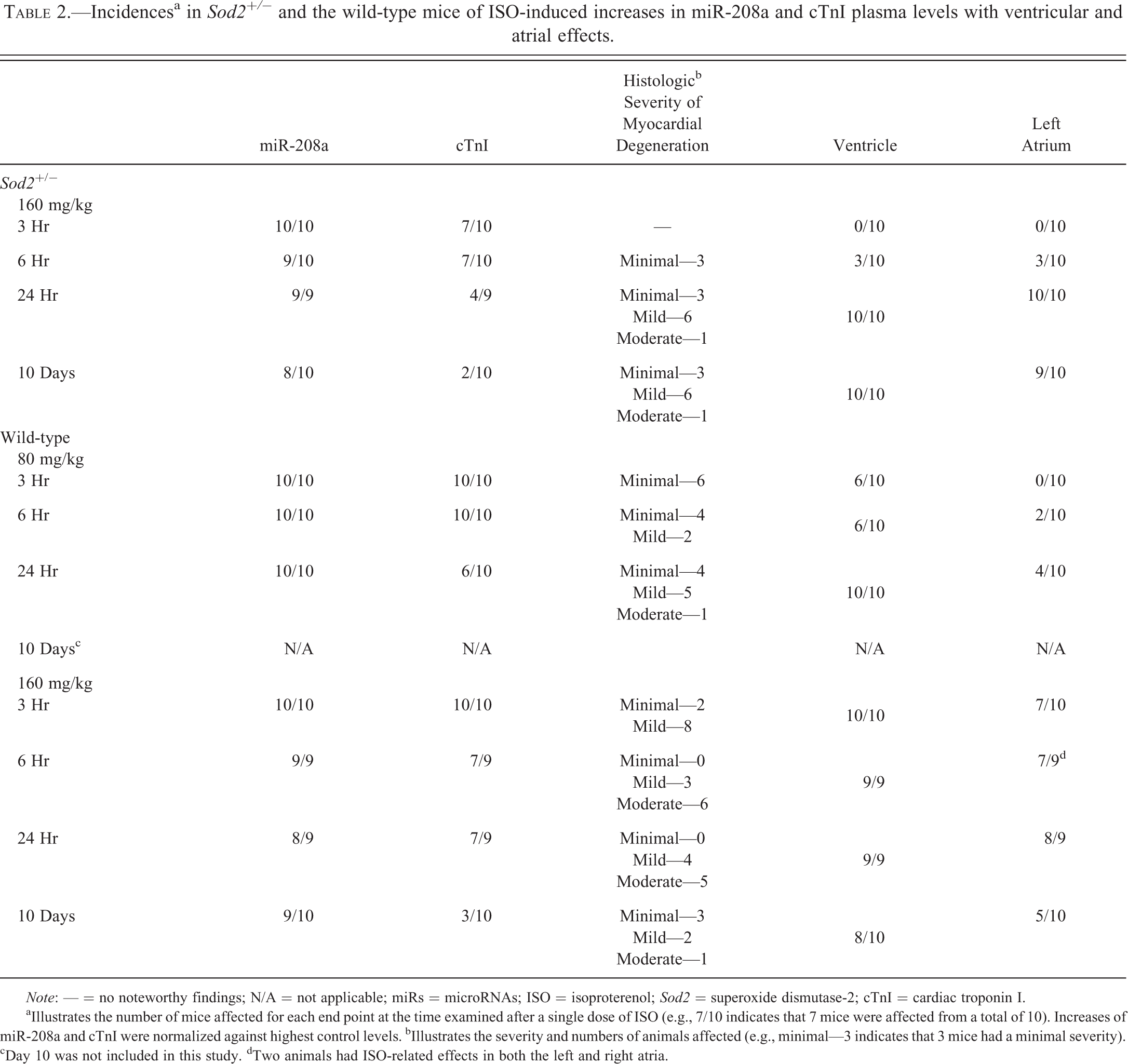
Note: — = no noteworthy findings; N/A = not applicable; miRs = microRNAs; ISO = isoproterenol; Sod2 = superoxide dismutase-2; cTnI = cardiac troponin I.
aIllustrates the number of mice affected for each end point at the time examined after a single dose of ISO (e.g., 7/10 indicates that 7 mice were affected from a total of 10). Increases of miR-208a and cTnI were normalized against highest control levels. bIllustrates the severity and numbers of animals affected (e.g., minimal—3 indicates that 3 mice had a minimal severity). cDay 10 was not included in this study. dTwo animals had ISO-related effects in both the left and right atria.
BNP, a marker for prognosis and risk stratification in the setting of heart failure in patients (Bhalla, Willis, and Maisel 2007), was also assessed in these studies. However, a single dose of ISO at 160 mg/kg did not lead to increases in BNP in either type of mouse at 3, 6, or 24 hr postdose (data not shown).
ISO given at similar systemic exposure (ISO at 80 mg/kg in wild-type mice; 160 mg/kg in Sod2+/− mice)
TK assessment of ISO in the wild-type mice demonstrated that ISO administered IP at 80 mg/kg resulted in similar systemic exposure to that achieved in Sod2+/− mice at 160 mg/kg (Table 3). Administration of ISO at 80 mg/kg in wild-type mice also led to increases in both cTnI and miR-208a, with mouse type-dependent differences in magnitude at the earlier time points (3 and 6 hr). The peak elevation of cTnI occurred at 6 hr, but that of miR-208a was similar at 3 and 6 hr after dosing (Figure 1C), and the magnitude of the miR-208a increase was lower than that of the parallel cTnI plasma concentration at 80 mg/kg in the wild-type mice. Interestingly, the magnitude of miR-208a was much greater than that of cTnI in Sod2+/− mice. Moreover, at 24 hr postdose, all wild-type mice had elevated miR-208a compared to 60% of the same mice with increases in cTnI (Table 2).
Summary of systemic exposure of ISO in both wild-type and Sod2+/− mice.a

Note: T max = time of maximum ISO plasma concentration; C max = maximum plasma concentration of ISO; AUC = area under the curve; ISO = isoproterenol; Sod2 = superoxide dismutase-2.
aBoth wild-type and Sod2 +/− mice (n = 4) were treated with a single dose of ISO. Plasma levels of ISO were determined at 1, 3, 6, and 24 hr postadministration. bThe C max and AUC (0–24) values are presented as the mean and standard deviation (SD).
In general at similar systemic exposure, there was notable differentiation between miR-208a and cTnI at 24 hr after dosing, which was depicted by a greater incidence of both wild-type and Sod2+/− mice with elevated miR-208a concentrations.
Tissue Expression in Atrium and/or Ventricle
In an effort to understand the specific cardiac tissue source of circulating miR-208a, atria and ventricles were collected from both wild-type and Sod2+/− mice 3 hr postdose (160 mg/kg) and miR-208a expression levels were measured by qRT-PCR. ISO administration led to significant decreases in miR-208a expression levels in the ventricles of both types of mice, compared to saline-treated mice (Figure 2B). By contrast, ISO did not cause such decrease in miR-208a levels in the atria of either type (Figure 2A). The observed decreases in the ventricle suggested that the ventricular tissue was the likely source of circulating miR-208a.
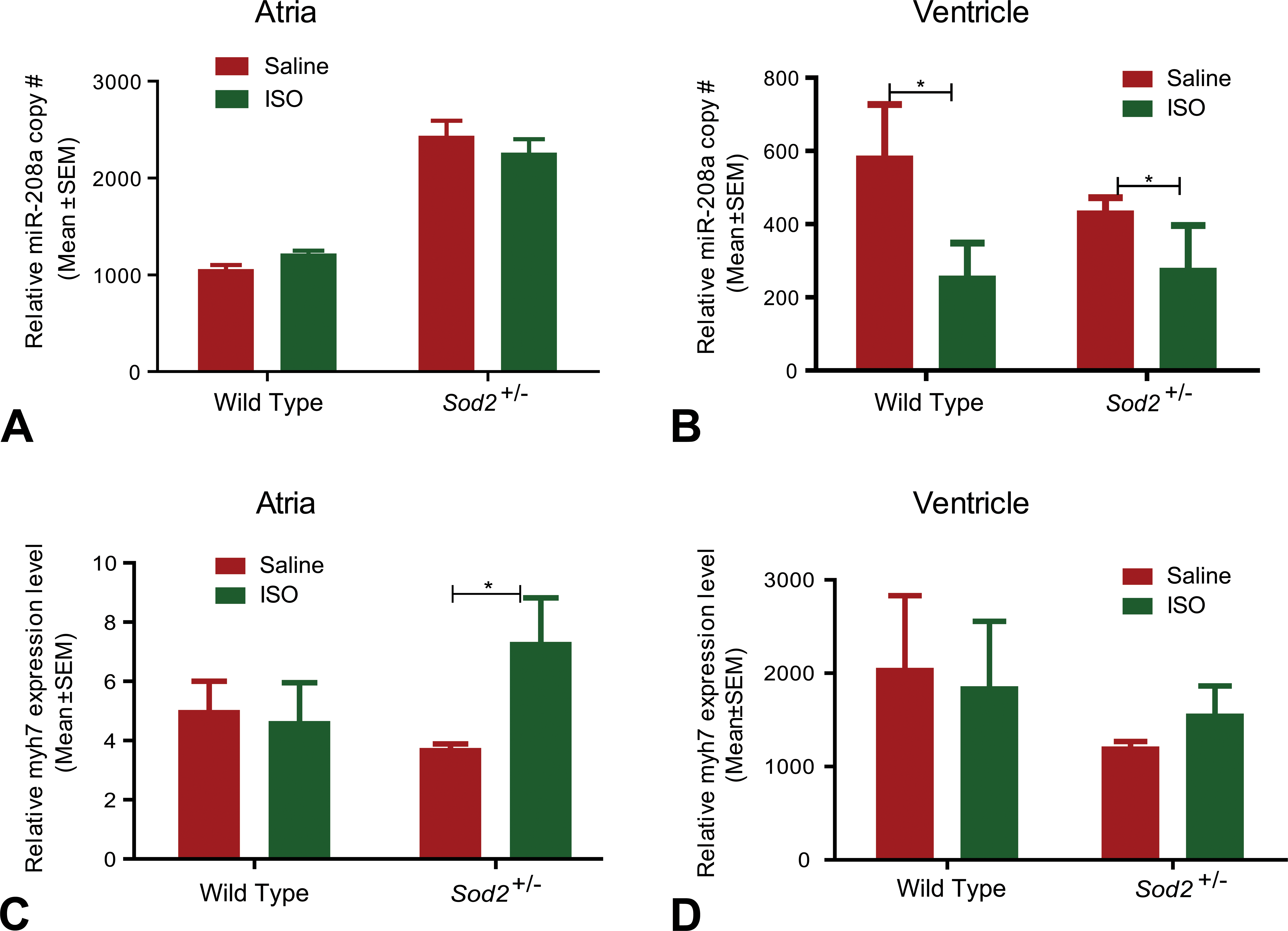
Expression levels of miR-208a and Myh7 in heart. Atrium (A) and ventricles (B) were collected from both wild-type and Sod2+/− mice at 3 hr after ISO administration at 160 mg/kg. miR-208a levels were measured by qRT-PCR. Relative copy numbers of miR-208a were calculated against total RNA loaded. Unpaired t-test was used to compare ISO-treated groups to saline-treated group. Atrium (C) and ventricles (D) were collected from both wild-type and Sod2+/− mice at 3 hr after ISO administration at 160 mg/kg. Myh7 levels were measured by qRT-PCR. Relative copy numbers of Myh7 were calculated against the total RNA loaded. Unpaired t-test was used to compare ISO-treated groups to saline-treated group. *p < .05. miR-208a = microRNA-208a; Myh7 = myosin heavy chain 7; Sod2 = superoxide dismutase-2; ISO = isoproterenol; qRT-PCR = quantitative real-time PCR.
We also examined the expression level of Myh7 by qRT-PCR to determine if concordant changes in Myh7 were present in treated mice. Three hours after ISO administration in the Sod2+/− mice, qRT-PCR analysis demonstrated a significant increase in Myh7 level in the atrium and a slight increase in the ventricle, compared to control, although the difference in the ventricle was not statistically significant (Figure 2C, D). However, there were no Myh7 changes observed in ISO-treated wild-type mice.
Histopathology Findings of Definitive Studies
Myocardial degeneration was evident in the wild-type and Sod2+/− mice at ≥3 hr and ≥6 hr, respectively, after the administration of ISO. Representative photomicrographs (Figures 3 –5) of the findings are provided herein, as there was no difference in the nature of the lesions between the 2 types of mouse.
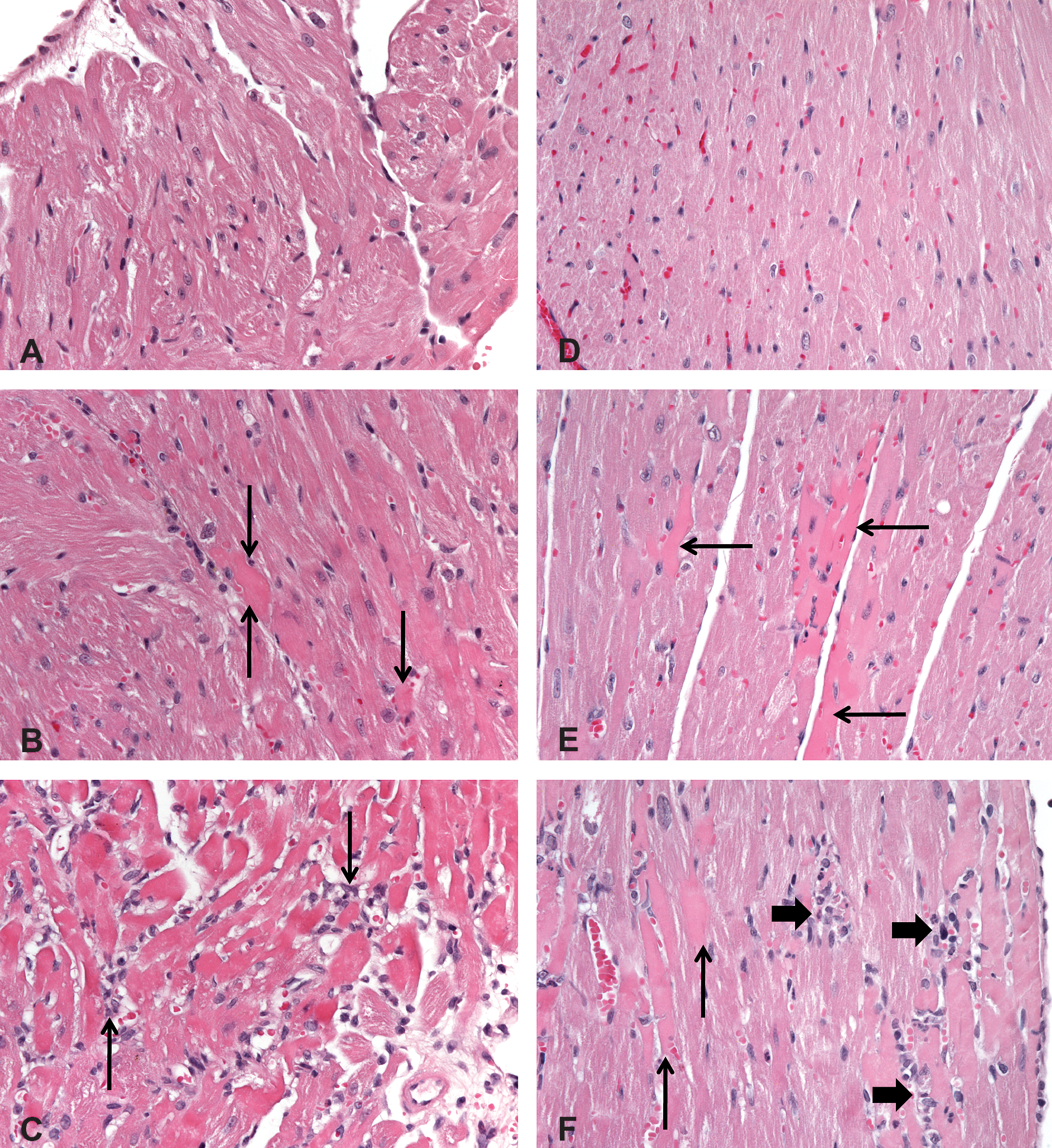
Histology of the atria and ventricles of the heart of normal and ISO-treated wild-type and Sod2+/− mice. Hematoxylin & eosin staining, 400×. A and D—Normal atrium (A) and ventricle (D) are evident from mouse treated with saline. B and E—Eosinophilia (arrows) of cardiomyocytes is evident in the atrium (B) and ventricle (E) at 3 hr after ISO treatment of a wild-type mouse. C and F—At 6 hr after ISO treatment, cardiomyocytes are loosely separated and infiltrated by macrophages (arrows) in the atrium (C) of a wild-type mouse and are homogenously eosinophilic (thin arrows) and infiltrated by neutrophils and macrophages (thick arrows) as shown close to the endocardium in the ventricle (F) of a Sod2+/− mouse. Sod2 = superoxide dismutase-2; ISO = isoproterenol.
In the wild-type mice given 80 or 160 mg/kg of ISO, myocardial degeneration was evident at the earliest time point examined (3 hr) after dosing and was generally minimal to mild (Figure 3B, E, Table 2). The severity of the changes generally was from mild to moderate at the later time points as the injury became more widespread and infiltrated by inflammatory cells (Figure 3C). In the ISO-treated wild-type mice, 6 of the 10 mice given 80 mg/kg and all mice (10 of the 10) given 160 mg/kg had myocardial degeneration at 3 hr postdose (Table 2).
Myocardial degeneration was not present at 3 hr postdose in Sod2+/− mice given 160 mg/kg and only occasionally evident (3 of the 10 mice) with minimal severity at 6 hr after dosing (Figure 3F, Table 2). However, at 24 hr after ISO dosing, all Sod2+/− mice had minimal to moderate changes. The myocardial degeneration was characterized by increased cytoplasmic eosinophilia, vacuolated or fragmented cytoplasm, and/or pyknotic nuclei of cardiomyocytes. At later time points (24 hr), there was mostly mononuclear cell inflammation with subsequent development of fibrous connective tissue by day 10 (Figures 4B and 5B). Interestingly, at similar systemic exposure, the wild-type mice had an early onset of histologic changes compared to Sod2+/− mice (Table 2). This suggests that Sod2+/− mice are not more sensitive to ISO-induced toxicity as hypothesized.
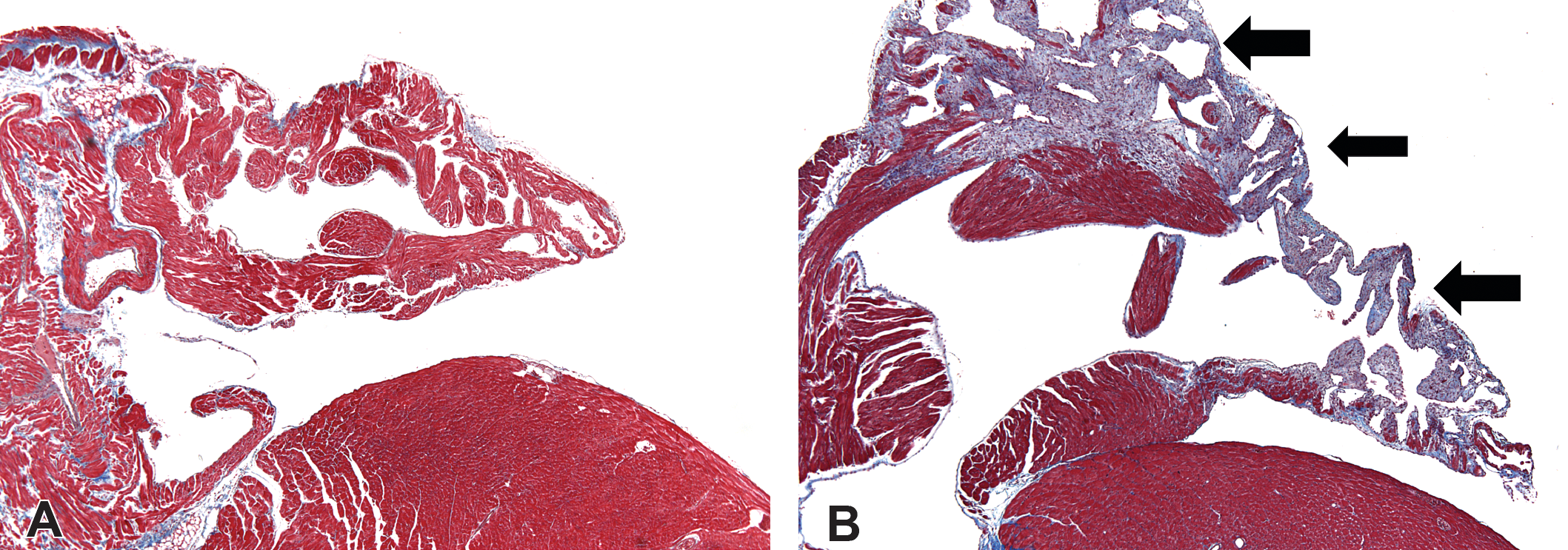
Masson’s trichrome staining of saline- and ISO-treated hearts (40×). (A) Normal atrium from a saline-treated mouse is shown. (B) Fibrosis (blue staining and arrows) is evident in the atrium of a Sod2+/− mouse on day 10 after ISO treatment. Sod2 = superoxide dismutase-2; ISO = isoproterenol.
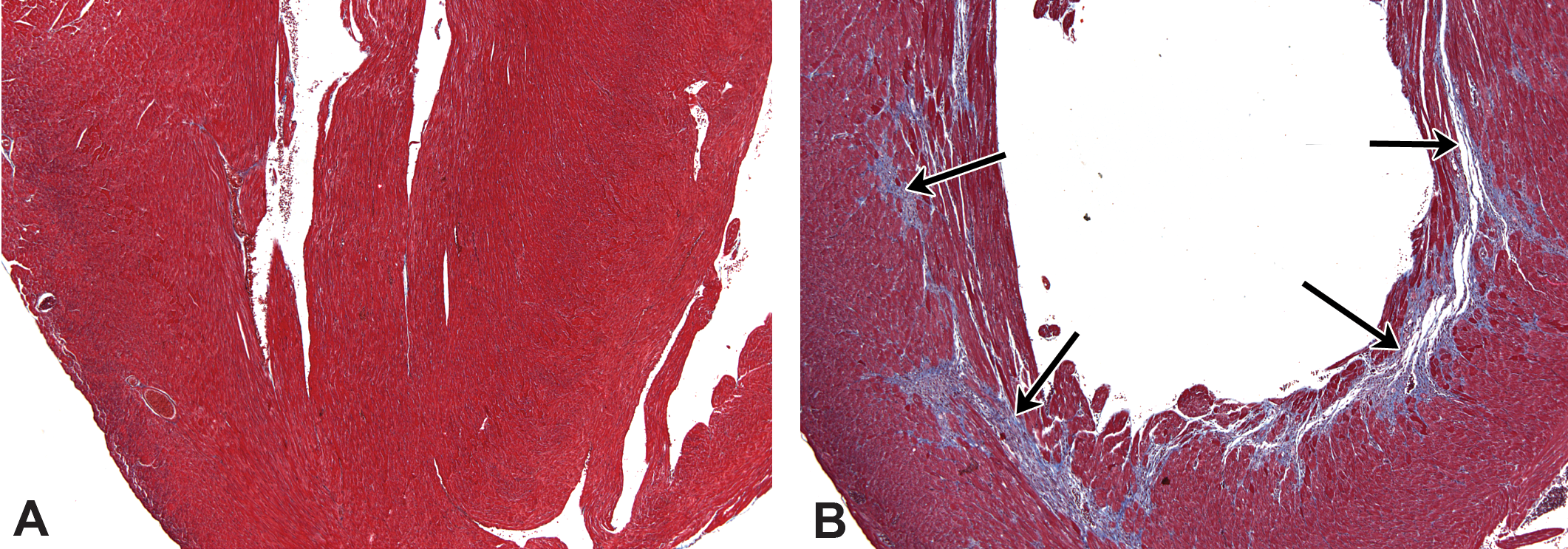
Masson’s trichrome staining of saline- and ISO-treated hearts (40×). (A) Normal ventricle from a saline-treated mouse is shown. (B) Fibrosis (blue staining and arrows) is evident in the inner contour of the left ventricle subendocardially, as shown in a Sod2+/− mouse on day 10 after ISO treatment. Sod2 = superoxide dismutase-2; ISO = isoproterenol.
In both the wild-type and Sod2+/− mice, myocardial degeneration was frequently present in the apex, the left ventricle wall, the interventricular septum, the right ventricular wall, the left atrium, and very occasionally in the right atrium. The patterns of the myocardial degeneration likely reflected areas that experienced the greatest oxidative stress. This pattern of effect followed the subendocardial inner contour particularly of the left ventricular wall (Figure 5B), as previously described for ISO-induced myocardial damage in mice (Engle et al. 2009; Faulx et al. 2005). In addition, the left atrium (Figure 4B, Table 2) was predominantly affected in both types of mouse, with occasional development of thrombosis. There was no general difference between the wild-type and Sod2+/− mice pertaining to the atrial effects, but the total number of mice exhibiting atrial lesions for both types totaled 51% (55 of the 108 mice examined). In both mouse types when the heart had ISO-induced changes, the ventricular wall was always affected and there were no instances of atrial effects without the ventricular wall being affected.
Caspase 3 Immunoreactivity
Caspase 3 immunoreactive staining was faintly present in cardiomyocytes of both mouse types in all ISO-treated mice at all time points. No differences were evident between the Sod2+/− and the wild-type mice in the nature or extent of caspase 3 immunoreactive staining. The staining was present in areas of the heart that overlapped with myocardial degeneration in the ventricles (Figure 6C) and atria (Figure 6B). No caspase 3 immunoreactivity was present in saline-treated mice of either type of mouse.
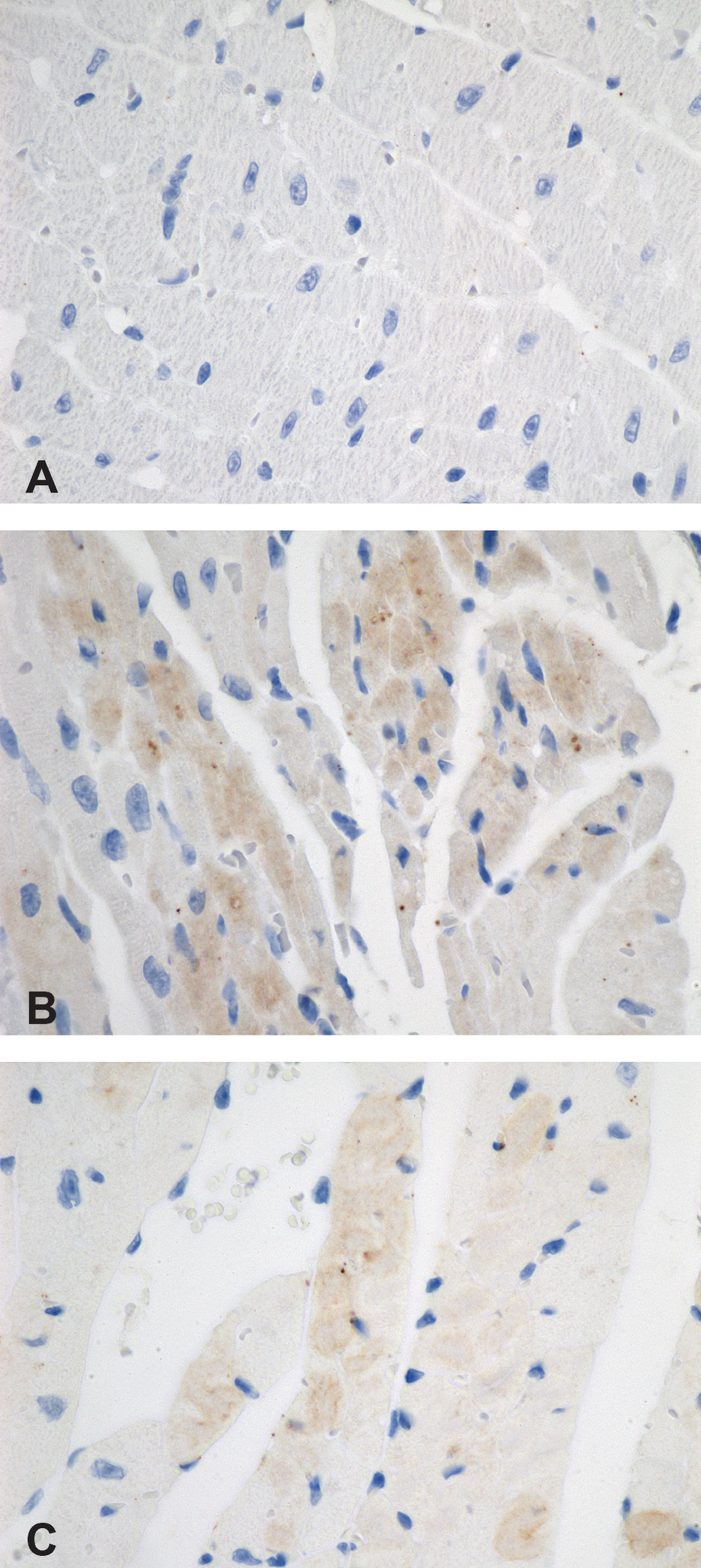
Caspase 3 immunohistochemical staining (A–C, 600×). A—Normal heart from a mouse treated with saline and devoid of caspase 3 immunoreactivity. B and C—Three hours after ISO treatment, caspase 3 immunoreactivity (brown staining) is present in the cytoplasm of cardiomyocytes in the left atrium (B) and ventricle (C) of a wild-type mouse. ISO = isoproterenol.
ISO TK Profiling
A TK study was conducted to measure the systemic exposure of ISO. Both the wild-type and Sod2+/− mice (n = 4) were treated with a single dose of ISO (160 mg/kg, IP), and plasma levels of ISO were determined at 1, 3, 6, and 24 hr postdose. The TK data revealed that both maximum plasma concentration of ISO (C max) and AUC (0–24) values in the wild-type mice were 3-fold higher than that in Sod2+/− mice after administration of 160 mg/kg (Table 3). C max values were 14,100 and 3,800 ng/ml in the wild-type and Sod2+/− mice, respectively; AUC (0–24) values were 34,100 and 10,700 ng. h/ml, respectively. In order to achieve similar systemic exposures in both types of mouse, a second TK study was conducted in the wild-type mice exploring doses of 20, 50, and 80 mg/kg. Data from the second TK study demonstrated that systemic exposure of ISO at 80 mg/kg in the wild-type mice was similar to that achieved at 160 mg/kg in Sod2+/− mice (Table 3). In the wild-type mice given 80 mg/kg and Sod2+/− mice given 160 mg/kg, C max values were 3,205 and 3,800 ng/ml, respectively; AUC (0–24) values were 10,390 and 10,700 ng. h/ml, respectively.
Discussion
This work investigated the utility of miR-208a as a biomarker of ISO-induced cardiac injury in both male wild-type and Sod2+/− mice. Significant increases in miR-208a were noted in this investigation with a concurrent profile to that of cTnI, similar to the work with the Sprague-Dawley rat after ISO administration (Ji et al. 2009). As a secondary objective, this work also tested the hypothesis that Sod2+/− mice would exhibit greater cardiac toxicity induced by ISO compared to wild-type mice. Because Sod2+/− mice exhibit increased oxidative stress and greater sensitivity to drug-induced hepatotoxicity compared to wild-type mice (Hsiao, Younis, and Boelsterli 2010), we anticipated this would also hold true for cardiac toxicity induced by ISO. Based on this study design, Sod2+/− mice do not appear to be more sensitive to ISO-induced toxicity compared to the wild-type mice at similar ISO exposures.
Findings from this investigation confirmed miR-208a as a potential biomarker for ISO-induced myocardial injury in wild-type and Sod2+/− mice. Significant increases in plasma miR-208a were noted in this investigation with a profile similar to that of cTnI. In Sprague-Dawley rats, miR-208a and concurrent increases in cTnI were also observed after ISO administration (Ji et al. 2009). However, in our study, there were notable differences between the wild-type and Sod2+/− mice in the plasma levels of miR-208a compared to cTnI. In the plasma of the wild-type and Sod2+/− mice, the copy numbers of miR-208a mirrored the profile of cTnI concentration, particularly at the earlier time points (3 and 6 hr), and there were no instances of above normal levels of cTnI without a concurrent abnormal level of miR-208a.
miR-208a may be a superior indicator of cardiac damage than cTnI after ISO-induced injury in mice. When intra-animal miR-208a and cTnI concentrations were examined in our investigations, miR-208a was consistently more predictive of myocardial damage compared to cTnI. This is supported by the higher (100%) incidence of miR-208a increases in Sod2+/− and the wild-type (80 mg/kg) mice at 24 hr postdose, compared to that (40% and 60%) of cTnI (Table 2). Similarly, a greater incidence (80% and 90%) of elevated miR-208a was present in both mouse types multiple days after dosing (day 10), compared to that (20% and 30%) of cTnI. At multiple days after the single dose of ISO in our studies, the histologic character of the lesion in the heart included mononuclear inflammatory cells and fibrosis, consistent with chronicity and attempts to repair the cardiac damage. It seems clear that despite the repair process, the plasma concentrations of miR-208a, and that of cTnI to a much lesser extent, were indicative of continuing myocardial damage. At this stage of the insult, however, miR-208a plasma levels were more indicative of myocardial cell injury status than that of cTnI, paralleling the incidence (≥80%) of histologic findings in the mice. This suggests differentiation between these biomarkers during the later stages of myocardial damage and that miR-208a is a better indicator of injury at ≥24 hr after ISO-induced cardiac injury in mice than that of cTnI. Wang et al. (2010) reported similar findings for AMI in patients as miR-208a was detectable in all the patients (100%), while parallel cTnI was only detected in 85% of patients.
There was a remarkable difference in the amount of miR-208a released in ISO-treated mice in this study with significantly greater levels measured in Sod2+/− mice compared to the wild-type (Figure 1). The reason for this unexpected finding in the Sod2+/− mice is unclear, but it is hypothesized that it may be related to the triggering of genes that work in concert during the development of cardiac hypertrophy. One of the hallmarks of cardiac hypertrophy in humans and in rodent models is an increase in β-Myosin heavy chain (MHC; Myh7) expression (Barry, Davidson, and Townsend 2008). Investigations in the regulation of cardiac hypertrophy and conduction in mice revealed that miR-208a was sufficient to induce cardiac remodeling and regulate the expression of hypertrophic components, including upregulation of the β-MHC gene (Myh7; Callis et al. 2009). In this study, Myh7 expression levels in ventricles were slightly elevated in the ISO-treated Sod2+/− mice but not in the wild-type mice (Figure 2D), suggesting that increased levels of miR-208a were induced in Sod2+/− mice and corresponded to the larger magnitude of miR-208a released into the circulation in Sod2+/− mice. In Sod2+/− mice, it has been reported that fractional shortening as a consequence of increased left ventricle dimensions occurs in instances of hypertrophy (Karakikes et al. 2013) or in dilated cardiomyopathy; however, this was not the case in the wild-type mice. Furthermore, Huang et al. (1998) reported that homozygous null mutations in Sod2−/− mice that died within 1 to 18 days was attributed to dilated cardiomyopathy, indicating a predisposition to this pathology during inadequate amounts of MnSOD. For reasons that remain currently unclear, these data suggest that Sod2+/− mice may be predisposed to cardiac effects or to a cardiac hypertrophic pathway response after cardiac insult.
Heart mitochondria isolated from Sod2+/− mice show a 50% reduction in MnSOD activity, which is associated with increased mitochondrial oxidative damage that results in increased mitochondrial cytochrome-c release and increased DNA fragmentation (Van Remmen et al. 2001). In addition, studies have shown increased death of cardiomyocytes isolated from Sod2+/− mice, compared with those from wild-type mice due to the difference in sensitivity to t-butylhydroperoxide treatment. Because mitochondrial cytochrome-c release and DNA fragmentation are well-established events in the induction of apoptosis, Sod2+/− mice were expected to demonstrate enhanced sensitivity to ISO-induced oxidative stress and cardiac injury. Surprisingly, there was no difference in cardiac damage in Sod2+/− mice when compared to the wild-type mice at similar ISO systemic exposures. We hypothesize that these incongruous results may be due in part to Sod2+/− mice not being as sensitive to oxidative stress because MnSOD contributes only 10 to 20% of the total SOD activity (Halliwell and Gutteridge 1995). In addition, in vivo compensation by other antioxidant defenses or differences in MnSOD expression may have contributed to this result. Unlike other antioxidant enzymes, MnSOD expression can be modified by a variety of physiological and environmental factors such as ionizing radiation, paraquat, thiol-modulating agents, and peroxynitrite (Van Remmen et al. 2001).
Mouse type-dependent differences were also noted in the onset of ISO-induced cardiac injury. Myocardial injury developed earlier in the wild-type mice than in Sod2+/− mice after ISO treatment (Table 2). Engle et al. (2009) reported similar strain differences where both plasma cTnI concentration and myocardial damage were greater in BALB/c mice than in CD-1 mice after ISO treatment. The reason for this difference was not investigated nor was systemic exposures of ISO measured in their study. The authors speculated that the difference was possibly due to a combination of desensitization of β-adrenergic receptors, strain-dependent receptor density inequality, cardiac capacity for aerobic metabolism, or simply adaptive responses. In this work, a single dose of ISO induced earlier cardiac injury in the wild-type mice compared to Sod2+/− mice, and the nature of the findings was consistent with those previously reported in other mouse strains and species (Faulx et al. 2005; Engle et al. 2009). Although it will require further investigation to characterize the mechanism by which the mouse types used in this work differed in the onset of cardiac injury, the increase in Myh7 expression levels in ISO-treated Sod2+/− mice suggests a different response to ISO (Figure 2C, D) in this type of mouse versus the wild-type.
Acute high doses of ISO have been reported to induce cardiomyocytes necrosis, apoptosis, interstitial fibrosis, and hypertrophy (Engle et al. 2009; Jimenez et al. 2011; Benjamin et al. 1989; Shizukuda et al. 1998; Teerlink, Pfeffer, and Pfeffer 1994), whereas chronic infusions at lower doses only result in ventricular hypertrophy (Faulx et al. 2005; Murad and Tucci 2000) and interstitial fibrosis (Zhang et al. 2008). A recent study by Zhang et al. (2008) using ISO at 125 to 500 μg/kg in Sprague-Dawley rats demonstrated a large number of apoptotic cardiomyocytes at 3 hr, which decreased over time and was rarely seen at the site of necrosis 24 hr postdose. To understand whether dual mechanisms of apoptosis and necrosis of cardiomyocytes or necrosis alone developed in ISO-induced cardiac injury, we preformed caspase 3 staining on selected heart sections for the detection of apoptosis. In the present work, a single dose of ISO administered at 80 (wild-type) or 160 (Sod2+/− ) mg/kg resulted in faint caspase 3 immunoreactivity at all time points (3, 6, 24 hr, and 10 days), similar to that reported for terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) staining in Sprague-Dawley rats by Zhang et al. (2008). The areas of caspase 3 staining colocalized with those of cardiomyocytes fragmentation and eventually with inflammatory cells and fibrosis, indicating a relationship among these processes. Although it is believed that necrosis is the main mechanism of cardiomyocyte death after infarction in humans and animals, there is evidence to suggest that apoptosis precedes necrosis and constitutes the prevailing form of cardiomyocyte death (Nadal-Ginard et al. 2003). Shortly after an ischemic event, it has been reported that apoptosis affects more than 80% and necrosis less than 20% of the cardiomyocytes in the ischemic zone, but with time, the 2 types of cell death overlap (Bardales et al. 1996). A more recent report by Dorn (2013) has suggested that the distinction between programmed apoptosis, programmed necrosis, and conventional necrosis (e.g., ischemia induced) requires careful attention because all 3 processes can increase caspase activity and TUNEL positivity. In our work, the presence of inflammatory cells in later time points ultimately characterized the cardiac lesions, indicating that necrosis played a primary role in the process of cardiomyocyte death.
Of particular interest in our study was the observation that atrium pathology was seen in both mouse types. The atrium has rarely been reported as a site of myocardial degeneration after ISO treatment in rodents despite the numerous reports of ISO-induced cardiac injury (Benjamin et al. 1989; Engle et al. 2009; Faulx et al. 2005; Jimenez et al. 2011; York et al. 2007; Zhang et al. 2008). The apex, ventricular walls, septum, and/or papillary muscles have been frequently reported as sites of ISO-induced myocardial degeneration (Zhang et al. 2008; Engle et al. 2009; Brady et al. 2010). In this investigation, these regions of the heart were affected as well as the atria; with the left atrium disproportionately more affected compared to the right atrium. This is consistent with the heavier workload of the left side of the heart. Thrombosis also developed occasionally but exclusively in the left atrium. Although the mechanism by which thrombosis developed was not investigated in this study, this change is usually initiated in the heart through endothelial damage or turbulent blood flow (Mosier 2007). The patterns of the myocardial degeneration reflected areas that most likely experienced the greatest oxidative stress in the heart and appeared to be characteristic of tachycardia-induced effects in rodents (Aragno et al. 2008; Zhang et al. 2008). In the ventricle, this pattern followed the inner contour particularly of the left ventricle and was the same as previously described for ISO-induced myocardial damage in rats (Engle et al. 2009). Although various areas in the ventricular wall were the most frequent sites of ISO-induced effects, the atria in these animals may not be as infrequently affected as suggested by the paucity of such reports in the literature because myocardial degeneration in our work was evident in the atrium of approximately a half of all (Sod2+/− and the wild-type) ISO-treated mice.
In summary, miR-208a was confirmed as a biomarker for the detection of myocardial injury in mice, and its release profile was comparable to cTnI. Importantly, increased levels of plasma miR-208a demonstrated an association with the development of cardiac lesions in both the wild-type and Sod2+/− mice and may be a superior biomarker of cardiac damage compared to cTnI at ≥24 hr after myocardial injury. Although this work indicated that miR-208a holds promise as an indicator of cardiac toxicity, further investigation of miR-208a and cTnI is required to determine which may be the superior biomarker of cardiac damage. Given the variations observed between the Sod2+/− and its wild-type, additional strain and species comparisons are warranted. Contrary to our expectation, Sod2+/− mice were not more sensitive to ISO toxicity compared to the wild-type after a single dose. However, given changes observed in Myh7 in this study, Sod2+/− mice may prove to be an interesting model to investigate cardiac hypertrophy through longer term treatment of ISO at lower doses.
Footnotes
Acknowledgments
Special thanks to individuals who directly contributed to this work: Aida Sacaan for review of the article and helpful discussions, Dingzhou Li for his assistance in the statistic analysis, Jessica Frey for her histology assistance, Mira Ko for her assistance on BNP assay, Michelle Hemkens for her assistance on the in vivo portion of the study, and Constance Benedict for the graphic arts assistance.
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: All studies were funded by Pfizer Drug Safety Research & Development. There were no external sources of funding.