
Abstract
Since its discovery in 1982, the global importance of Helicobacter pylori–induced disease, particularly in developing countries, remains high. The use of rodent models, particularly mice, and the unanticipated usefulness of the gerbil to study H. pylori pathogenesis have been used extensively to study the interactions of the host, the pathogen, and the environmental conditions influencing the outcome of persistent H. pylori infection. Dietary factors in humans are increasingly recognized as being important factors in modulating progression and severity of H. pylori–induced gastric cancer. Studies using rodent models to verify and help explain mechanisms whereby various dietary ingredients impact disease outcome should continue to be extremely productive.
Introduction
Helicobacter pylori, first isolated from inflamed gastric tissues of humans in 1982, is now firmly established as an important human pathogen. Prior to its discovery, despite earlier observations of gastric spiral organisms being present in human and animal gastric tissues (Freedberg and Barron 1940; Doenges 1938), an often stated principle, championed particularly by gastroenterologists, was the stomach for all practical purposes was considered to be sterile because of the stomach’s low pH. It is now known that H. pylori infection is persistent and is always associated with inflammation consisting of mononuclear cells and polymorphonuclear cells. When the infected individual is treated with antibiotics known to eradicate H. pylori, the gastritis regresses over time. However, if there is recrudescence of the organism due to ineffective antibiotic therapy or the patient becomes reinfected with another H. pylori strain, the gastritis reappears. It has been determined through rigorous clinical and epidemiologic studies that H. pylori is directly linked to peptic ulcer disease, and importantly, the World Health Organization (WHO) has listed H. pylori as a class I carcinogen (International Agency for Research on Cancer 1994). H. pylori is also considered to initiate and sustain low-grade gastric mucosa-associated lymphoma (Muller 2009). Prior to the discovery of H. pylori, peptic ulcer disease was linked to excessive gastric acid production and/or stress, whereas gastric cancer was linked to a variety of dietary factors documented by extensive epidemiologic surveys.
Although 50% of the world population is infected with H. pylori, only a small percentage (1–3%) of individuals have the gastric disease progress to gastric adenocarcinoma (GAC). GAC represents the most commonly diagnosed (∼95%) of gastric cancers and is the fourth and fifth most common cancer in males and females, respectively (Jemal et al. 2011). Worldwide, there were an estimated 989,000 cases of gastric cancer and 738,000 gastric cancer–associated deaths in 2008, making gastric cancer the fourth most common cancer and the second leading cause of cancer death, after that recorded for lung cancer (Ferlay et al. 2010).
A strong genetic predisposition to development of H. pylori–associated gastric cancer has been established. Gastric cancer risk is increased up to 3-fold in individuals with a first-degree relative with gastric cancer, and 10% of gastric cancer cases exhibit familial clustering. While it is well established that H. pylori can infect multiple family members, family history remains a risk factor even after controlling for H. pylori infection. However, only a small fraction of the familial clustering of gastric cancer is attributable to known family cancer syndromes. It was observed that H. pylori–infected patients who developed gastric cancer secreted lower levels of gastric acid compared to H. pylori–infected patients with duodenal ulcers. Thus, in a landmark study, the authors chose to study interleukin-beta (ILβ), a known proinflammatory cytokine with marked acid inhibitory properties in families with precancerous lesions attributable to H. pylori (El-Omar et al. 2000). They demonstrated that individuals with specific genetic polymorphisms in IL1β and IL1RN had a 2- to 3-fold increased risk of developing gastric atrophy and gastric cancer (El-Omar et al. 2000). These findings have been confirmed in some studies, but not others. In a meta-analysis of ILβ and Interleukin-1 receptor antagonist (ILR) antagonist gene polymorphisms and risk of gastric cancer, IL1β-511T and IL1RN*2 were associated with gastric cancer risk in Caucasians, but not in Asians. For IL1β-511T, the association in Caucasians was stronger when intestinal subtypes and noncardia gastric cancer cases were examined (Camargo et al. 2006). Further genetic polymorphisms in tumor necrosis factor-alpha (TNFα) and IL10, when combined with IL1B polymorphisms, result in higher risk genotype, conferring a 27-fold or greater risk of developing gastric cancer (El-Omar et al. 2003).
The mechanisms involved in the pathogenesis of GAC remains incompletely characterized but clearly the interactions of host, environmental variables, and H. pylori virulence factors, particularly the cag pathogenicity island (PAI) and vacuolating cytotoxin (VacA), play a role in cancer initiation and progression (Jones, Whitmire, and Merrell 2010). These virulence factors are polymorphic and affect a multitude of host cellular pathways and thereby contribute to differences in disease severity given that various cytotoxin-associated antigen (CagA) and VacA alleles differentially target selected pathways (Jones, Whitmire, and Merrell 2010). As reviewed by Jones, Whitmire, and Merrell (2010), cytotoxin-associated Gene A is encoded on the cag PAI, which is a horizontally acquired 40-kb DNA segment that encodes for a type-IV secretion system (T4SS; Jones, Whitmire, and Merrell 2010). cagA is the terminal gene on the cag PAI, and encodes for the 120- to 145-kDa immunodominant CagA protein (Covacci et al. 1993; Tummuru, Cover, and Blaser 1993). Once injected into host cells, CagA acts directly in an unphosphorylated state to influence cellular tight junction (Oliveira et al. 2009), cellular polarity (Zeaiter et al. 2008), cell proliferation and differentiation (Lee et al. 2010), cell scattering (Mimuro et al. 2002), and induction of the inflammatory response (Brandt et al. 2005). Upon entering the gastric epithelia, CagA localizes to the plasma membrane and can be phosphorylated by either Abl kinase or Src family kinases (Poppe et al. 2007; Tammer et al. 2007). These kinase phosphorylate tyrosine residues are located within the carboxy terminus of CagA. These repeats are based on the amino acid sequences found within the regions flanking the EPIYA sequence and are classified as 4 distinct EPIYA motifs: EPIYA-A, EPIYA-B, EPIYA-C, and EPIYA-D (Jones, Whitmire, and Merrell 2010). Once phosphorylated, CagA can form cellular interactions that influence cellular shape and motility (Tammer et al. 2007; Jones, Whitmire, and Merrell 2010; Brandt et al. 2007). The presence of CagA is associated with more severe disease. Cancer patients are at least twice as likely to be infected with an H. pylori strain that is cagA positive than one that is cagA negative (Gwack et al. 2006). The VacA is produced and secreted by the majority of H. pylori strains. VacA activity was first noted when H. pylori filtrates induced large host cell vacuoles (Leunk et al. 1988). VacA remains on the bacterial surface or is secreted as an approximately 88-kDa toxin (Cover and Blaser 1992). Exposure to alkaline or acidic conditions amplifies that activity of VacA, where the toxin induces large cytoplasmic vacuoles in host cells that contain the markers for late endosomes and lysosomes. VacA also causes apoptosis, which is dependent on interaction with the mitochondria (Foo et al. 2010). VacA also deregulates multiple cellular pathways as well as inducing inflammation. VacA intoxication of host cells induces production of a variety of inflammatory cytokines that include TNFα, IL-1β, IL-6, IL-10, and IL-13 (Supajatura et al. 2002).
Rodent Models for Helicobacter-associated Gastric Cancer
Animal models, particularly rodents, continue to play an invaluable role in dissecting interconnected mechanisms that determine gastric disease outcome.
Mice
Helicobacter felis Infection
Most of the major advances in our understanding of gastric cancer pathogenesis have been derived from studies with mice. While a number of other animal models have been developed for both gastric cancer (e.g., methylnitronitrosoguanidine [MNNG]-induced cancer in rats) and for Helicobacter-mediated gastric infection (ferret, gerbil, gnotobiotic pig, primate, etc.), the mouse model has clear advantages with respect to their small size, cost, ease of infection, reproducibility, and especially the power of genetic manipulation. The first study to ascertain whether gastric helicobacters would colonize the stomach of rodents utilized germfree Swiss Webster mice or Sprague-Dawley rats infected with H. felis (Fox et al. 1991; Lee et al. 1990). Although the rat is the preferred model for gastric carcinogenesis studies, long-term H. felis and H. pylori colonization studies in this species have not been performed. This model therefore requires further development before it can be used in carcinogenesis studies. In the first description of H. felis gastritis in the germfree mouse, a moderate degree of fundic glandular epithelial cell hyperplasia was evident at 4 weeks in portions of the gastric mucosa (Lee et al. 1990). This was characterized by an overall increase in thickness of the mucosa in areas of the inflamed tissue. The changes included lack of epithelial cell maturation and differentiation, increased basophilia, and increased nuclear to cytoplasmic ratio. Also of interest was the enlargement of individual glands and the presence of columnar type epithelium versus cuboidal epithelium. Occasional branching of glands was noted as well as luminal epithelium forming small pseudovillus projections. These lesions persisted for the duration of the 8-week study (Lee et al. 1990).
These studies have been extended by documentation of persistent active, chronic gastritis in germfree mice and specific pathogen free (SPF) mice, infected with H. felis for 1 year (Fox et al. 1993). Studies of 72- to 76-week colonization of conventional and SPF mice have provided additional clues on progression of the lesions in the stomach of infected mice. Studying the role of genetic factors in the pathogenesis of Helicobacter-associated gastritis by taking advantage of the wide range of inbred and other types of genetically defined mice was a logical next step in further exploration of Helicobacter pathogenesis using this model. Two different laboratories inoculated 3 inbred strains of mice with H. felis and tabulated the intensity of inflammation; in BALB/c mice inflammation was minimal, in C3H/He moderate, and was most severe in C57BL/6 when examined 2 to 11 weeks postinfection (Mohammadi et al. 1996; Sakagami, Shimoyama, and O’Rouke 1994). The authors also stated that C57BL/6 mice had mucus cell hyperplasia and parietal cell loss in gastric fundic glands (Mohammadi et al. 1996). Furthermore, the study demonstrated that by using congenic strains on the BALB/c and C57BL background, both major histocompatibility complex (MHC) and non-MHC genes contributed to H. felis–associated gastritis (Mohammadi et al. 1996). The H. felis–infected mouse model continues to be used frequently with considerable success in studying environmental and host factors centered on Helicobacter spp. pathogenesis (see Table 1 and below; Wang et al. 2000).
The Helicobacter-infected INS/GAS mouse model of Gastric Cancer.
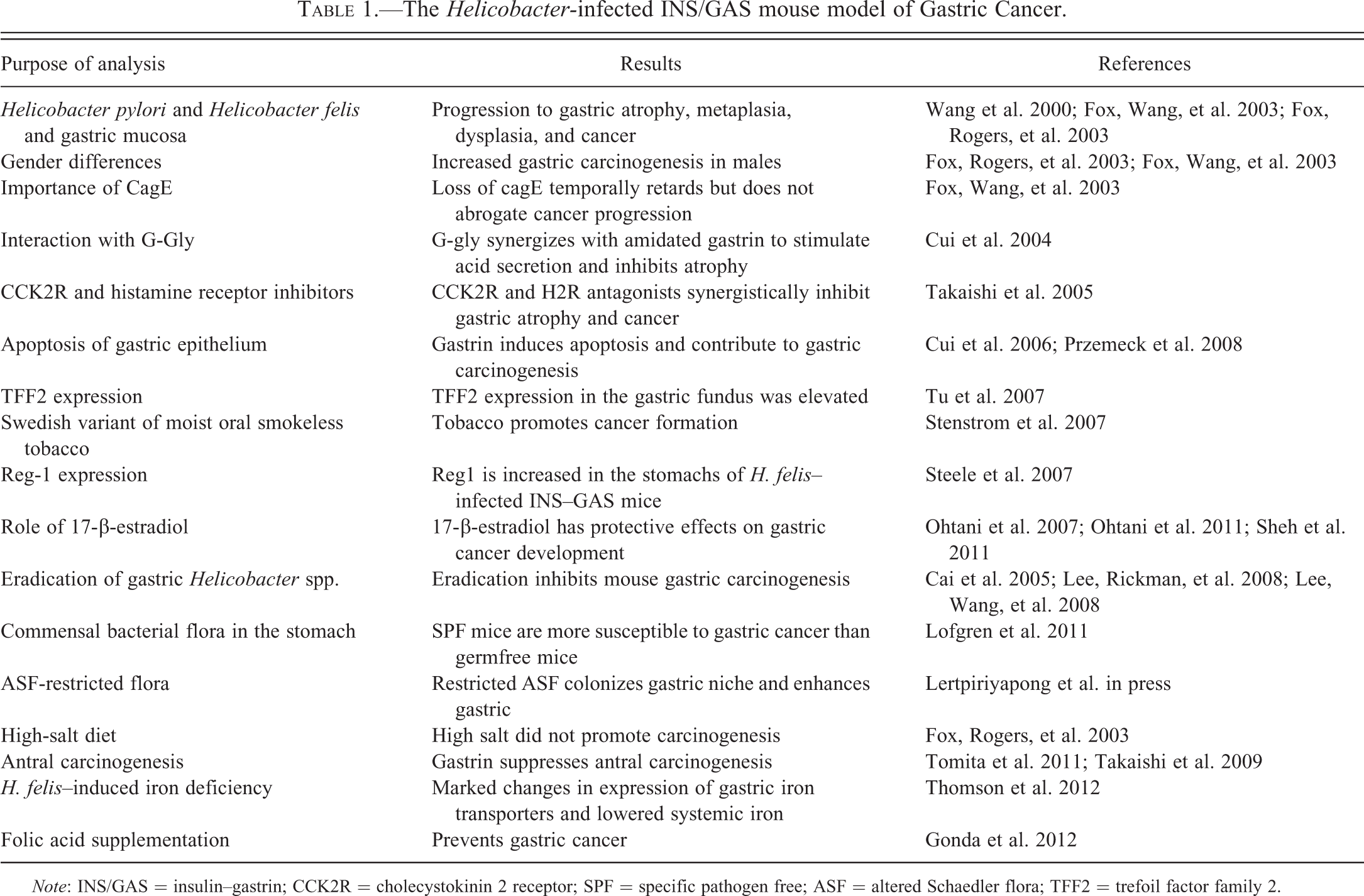
Note: INS/GAS = insulin–gastrin; CCK2R = cholecystokinin 2 receptor; SPF = specific pathogen free; ASF = altered Schaedler flora; TFF2 = trefoil factor family 2.
In the early investigations by a number of groups, it became clear from studies with inbred mice that progression to preneoplasia was largely determined by the host response. For example, the C57BL/6 inbred mouse strain responded to H. felis infection with a robust T helper (Th1) immune response, in contrast to the BALB/c inbred strain that showed a predominant Th2 response (Mohammadi et al. 1996; Sakagami, Shimoyama, and O’Rouke 1994; Wang and Fox 1998). Consequently, C57BL/6 mice showed rapid premalignant progression to atrophy and metaplasia with declining colonization levels, while BALB/c mice maintained high numbers of organisms with minimal epithelial injury or atrophy. Additional studies employing RAG and severe combined immune deficiency (SCID) mice and T-cell-deficient mice pointed to the importance of T-cell responses (Roth et al. 1999), while the use of cytokine knockout mice indicated that H. pylori–induced mucosal inflammation is Th1 mediated and exacerbated in IL-4 but not interferon-gamma (IFN-γ) gene-deficient mice (Smythies et al. 2000).
However, while genetic manipulation of the various cytokine genes clarified many aspects of the immunopathogenesis of the disease, the unmanipulated C57BL/6 mouse has proved surprisingly robust as a model for cancer development. Interestingly, many of the early H. felis infection studies included observation periods that extended up to 1 year but not beyond, and described changes of atrophy and metaplasia, but not more advanced pathology. The presence of dysplasia and invasive carcinoma in the H. felis–infected C57BL/6 mouse was first described by Fox and Wang (Fox et al. 2002), when the observation period was extended to 14 months. More recently, progression to antral carcinomas has been reported in mice in which the model has been extended out to 22 months (Cai et al. 2005).
The first Helicobacter-dependent mouse model of gastric cancer was reported by Wang et al. (2000) who described H. felis infection of insulin–gastrin (INS–GAS) transgenic mice in an FVB/N inbred background. The model has proved ideal as an accelerated model of carcinogenesis. A role for host and dietary factors also has been demonstrated using Helicobacter-infected INS/GAS mice (Table 1).
The gastric hormone gastrin stimulates gastric acid secretion and growth of the acid-secreting part of the stomach. H. pylori infection in humans results in a mild hypergastrinemia (1.5- to 2-fold elevation compared with uninfected subjects) that occurs early in the course of infection (Mulholland et al. 1993; Levi et al. 1989), precedes the development of atrophic gastritis, and often resolves after eradication of the infection. Chronic atrophic gastritis, a pathological condition characterized by decreased numbers of parietal cells and low levels of acid secretion and occurs as part of progression of Helicobacter spp. associated gastric lesions in the carcinogen cascade. The INS/GAS transgenic mice have a chimeric transgene in which the human gastrin gene is transcribed from the rat insulin I promoter (Wang et al. 1993). These mice express human heptadecapeptide gastrin (G-17) in the pancreatic β cells, which is then secreted into the circulation. Changes in gastrin secretion are divided into 2 distinct phases: an early phase (1–4 months) and a later phase (5 months and older). Gastrin levels increased modestly during the early phase, and approximately 2-fold compared with wild-type (WT) FVB/N mice (Wang et al. 1996). Slightly more than 50% of the serum gastrin (and all of the increase) in young INS–GAS mice is human gastrin resulting from pancreatic secretion. In contrast to the elevated plasma amidated gastrin levels, glycine-extended gastrin levels were all <20 pmol/L. Serum amidated gastrin levels are increased gradually beginning at 6 months, with levels peaking at 550 pmol/L by 20 months of age. Most of the increase in serum gastrin resulted from increased secretion of endogenous mouse amidated gastrin, with lesser changes in the level of human G-17, which increases 3-fold from 3 months to 20 months. During the late gastrin phase (5 months and older), INS/GAS mice have a gradual change in gastric function with a marked decline in the level of stimulated acid secretion. These decreases in acid production are mirrored by decreases in parietal and enterochromaffin-like (ECL) cell numbers (Wang et al. 2000).
In the INS/GAS mouse model, there is an increased expression of growth factors, heparin-binding epidermal growth factor, and TNFα. At 20 months of age, INS/GAS mice do not have increased ECL cell number, but rather exhibited gastric metaplasia, dysplasia, carcinoma in situ, and gastric cancer with vascular invasion. Experimental H. felis infection of INS–GAS mice led to accelerated (≤8 months) development of intramucosal carcinoma (85%), with submucosal invasion (54%) and intravascular invasion (46%; p ≤ .05; Wang et al. 2000). These findings support the conclusion that chronic hypergastrinemia in mice synergizes with H. felis and H. pylori infections and contributes to eventual parietal cell loss and progression to proximal gastric cancer. The model leads to accelerated gastric cancer in part due to the effect of elevated circulating levels of amidated gastrin that bind to cholecystokinin B (CCK-B) receptors in the gastric corpus to activate a histamine pathway, leading to altered growth and immune responses (Takaishi et al. 2005), and to relatively low levels of glycine-extended gastrin which may inhibit progression to preneoplasia (Cui et al. 2003). However, while hypergastrinemia clearly accelerates proximal gastric cancer, it also appears to protect against the more distal, antral gastric cancers (Takaishi et al. 2009; Tomita et al. 2011).
The INS–GAS mouse model has been used to demonstrate that male gender is important in Helicobacter-associated cancer progression (Fox, Rogers, et al. 2003; Fox, Wang, et al. 2003). Further studies demonstrated the protective effects of estrogen (Sheh et al. 2011; Ohtani et al. 2007). Thus, the INS/GAS model mimics the 2:1 ratio of male:female in gastric cancer development of humans. The INS/GAS model infected with either H. felis or H. pylori consistently reproduces the progression of gastric diseases that is recognized in humans infected with H. pylori, known as Correa’s model of gastric carcinogenesis (Figures 1 and 2; Fox and Wang 2007). However, the hypergastrinemic phenotype to date appears most prominent in INS–GAS mice in an FVB/N background, and is less prominent in a C57BL/6 background (Takaishi et al. 2009).
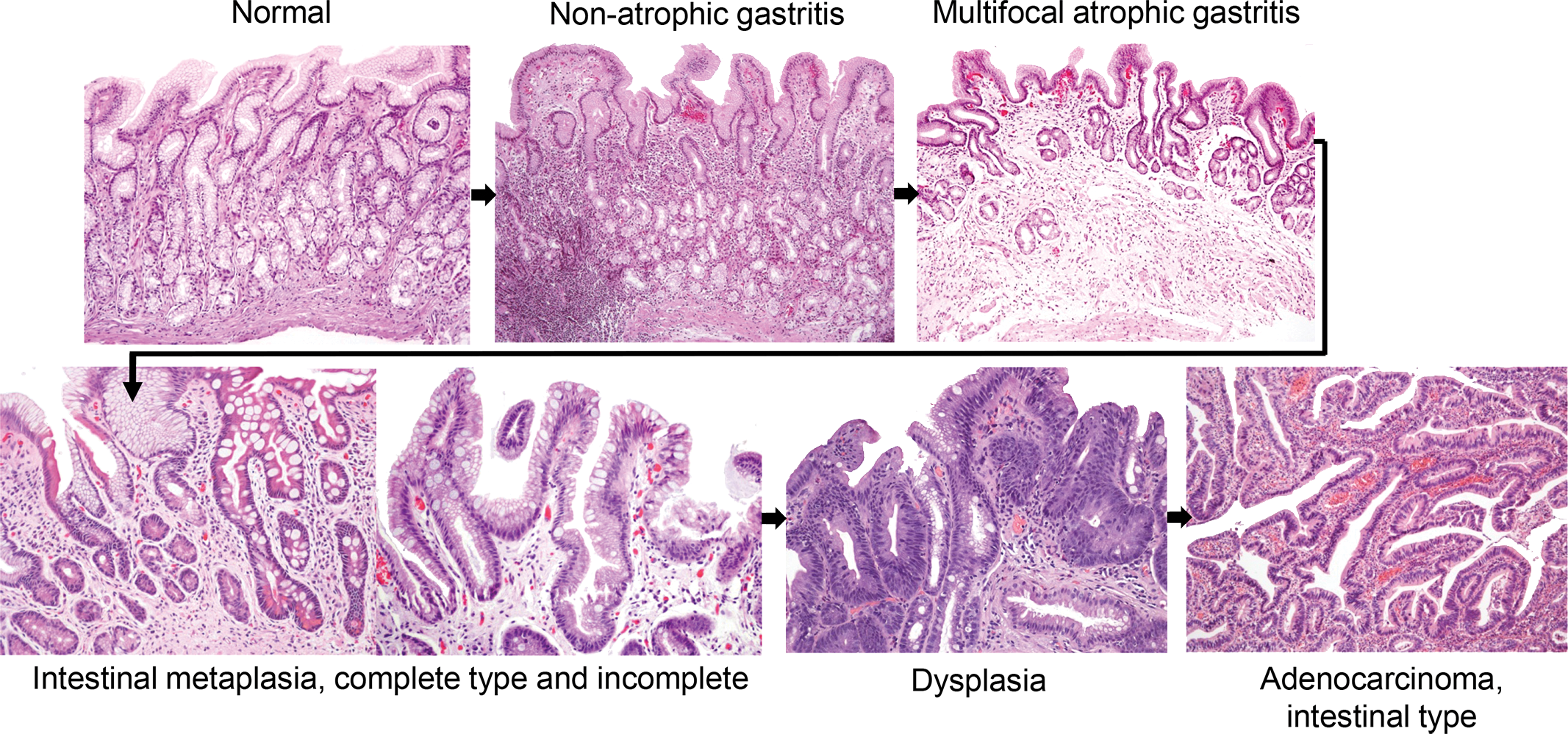
Correa’s cascade of gastric carcinogenesis. Courtesy of Pelayo Correa.
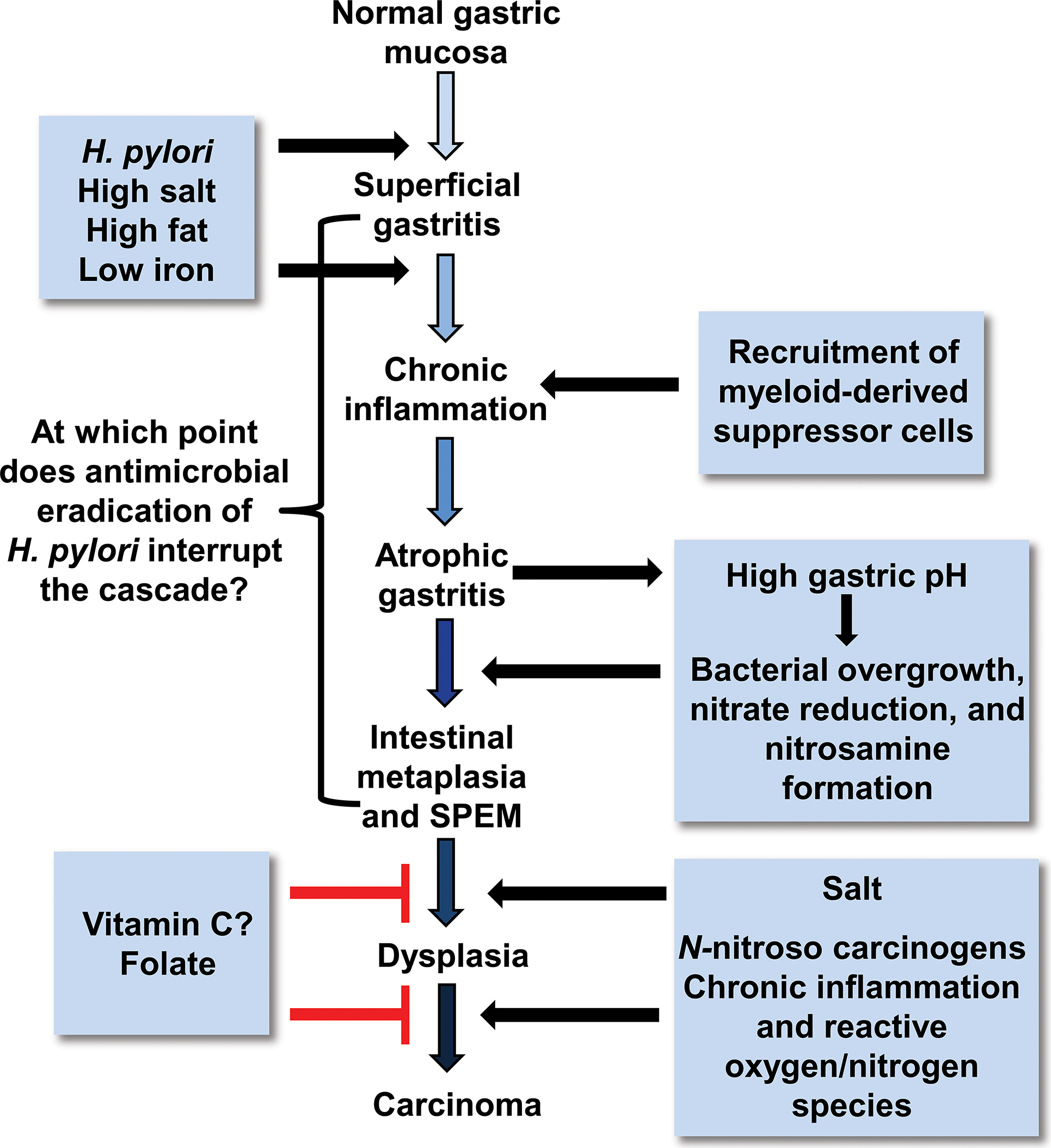
Proposed Correa pathway of pathological events and dietary influence on gastric cancer progression. In well-differentiated, intestinal-type gastric cancer, histopathological studies indicated that chronic Helicobacter pylori infection progresses over decades through stages of chronic gastritis, atrophy, intestinal metaplasia, dysplasia, and cancer (Correa 1991). The development of cancer has been attributed to alterations in DNA caused by chronic inflammation, recruitment, and engraftment of bone marrow–derived cells, an imbalance between epithelial cell proliferation and apoptosis, and, in a milieu of atrophy and achlorhydria, gastric colonization by enteric bacteria with nitrate reductase activity, which facilitates the formation of carcinogenic nitrosamines. Corpus-predominant atrophy, or the loss of specialized glandular cell types such as parietal and chief cells, appears to be the critical initiating step in the progression toward cancer (modified from Fox and Wang 2007).
H. pylori Infection
As described previously, the C57BL/6 mouse model of gastric H. felis infection has been very useful for the study for manipulation of the Th1–Th2 immune response. However, another factor is likely to be the particular H. pylori strain used to infect mice, and this question has been more difficult to address using the WT C57BL/6 mouse. A preliminary report published in 1991 indicated that fresh clinical isolates of H. pylori, but not stock culture isolates, colonized the stomach of athymic and euthymic mice for a short-time interval (Karita et al. 1991). The mouse model was further developed to test the ability of H. pylori strains that expressed VacA and the immunodominant CagA versus H. pylori strains that did not express the VacA and CagA antigens (Marchetti et al. 1995). The ability of H. pylori to colonize was enhanced (over that of clinical isolates) by serial passage of H. pylori in mice. Thus “mouseified” H. pylori strains used in the experiments assessing gastric pathology were isolated from mice 2 weeks after inoculation, expanded in vitro, and used in subsequent studies. The strains expressing the VacA and CagA experimentally produced gastritis for 8 weeks, whereas the strain lacking these antigens did not produce gastric pathology (Marchetti et al. 1995). In addition, the infected mice had a systemic antibody response demonstrated by immunoblot to several dominant antigens. Interestingly, the CagA antigen was not recognized, perhaps because of the early phase of infection in the mice (Marchetti et al. 1995) or expression of the functionality of CagA was lost after colonization (Rieder, Merchant, and Haas 2005). Also of considerable importance was the original description of the Sydney strain of H. pylori which colonizes mice up to 12 months after inoculation and, importantly, induces a significant gastritis, particularly in C57BL mice and B6129 mice (Lee et al. 1997; Rogers et al. 2005). However, most H. pylori strains—including the Sydney strain—while showing fairly consistent patterns of colonization, have proven less carcinogenic in C57BL/6 mice (Thompson et al. 2004), although a study published in 2005 demonstrated in situ carcinoma in C57BL/6 × 129SvEv (B6129) mice infected for 15 months with H. pylori SS1 (Rogers et al. 2005). It is now appreciated that use of H. pylori SS1, though successful in generating preneoplastic and neoplastic lesions in specified strains of mice, is somewhat limited due to its inability to express CagA in vivo. These results are compatible with H. felis mouse models of gastric cancer given that H. felis does not have Cag genes located on a PAI.
The cag PAI encodes an active T4SS, and after H. pylori epithelial cell adherence, the secretion system translocates the CagA protein into epithelial cells where the CagA protein undergoes tyrosine phosphorylation (CagAP-tyr; Jones, Whitmire, and Merrell 2010; Muller 2012). This process is associated with host cell morphological changes and phosphorylation of host cell proteins. Several genes within the cag PAI, but not cagA itself, are required for the induction of proinflammatory cytokines or chemokines, such as IL-8. The production of proinflammatory cytokines by H. pylori is dependent on the nuclear factor-κβ and nitrogen-activated protein kinase signal transduction cascades. Increased inflammation in cag+ H. pylori strains is also noted and importantly, human studies indicate that patients infected with CagA+ H. pylori are at a higher risk of developing gastric cancer than those infected with Cag− strains (Jones, Whitmire, and Merrell 2010; Muller 2012). Mice appear to be incompatible with cag+ H. pylori strains carrying functionally active T4SS. H. pylori strains rapidly switch off the cag PAI and downregulate proinflammatory responses. The widely used H. pylori SS1 strain has a complete cag PAI, but is unable to induce IL-8 or to deliver CagA into gastric epithelial cells in vitro. Using PMSS1 (H. pylori SS1 prior to serial mouse passage) with an intact functional CagA elicits more robust inflammation during its initial colonization in C57BL mice, but this feature is lost upon prolonged colonization, presumably due to functional loss of Cag, which recapitulates the in vivo behavior of SS1 (Arnold et al. 2011). Other mouse-adapted H. pylori strains used in different laboratories carry mutations or gene deletions in the cag-PAI and are unable to translocate and tyrosine phosphorylate CagA.
A number of other very useful murine models of gastric cancer, including genetically engineered mice (e.g., p27 [Kip1−/−]; IL1β), have been developed (Judd et al. 2004; Rogers and Fox 2004; Zavros et al. 2005; Tu et al. 2008). Indeed, in many of the models of gastric cancer, a long period of achlorhydria, associated with presumed bacterial overgrowth, appears to be the common denominator in mouse models of GAC (Zavros et al. 2005; Lertpiriyapong et al. in press; Lofgren et al. 2011). It is likely that various mouse models will continue to be developed and used to dissect the role of gastric Helicobacter infection and the associated immune response to precisely determine the cancer pathways most relevant to humans.
Gerbils
The Mongolian gerbil has been shown to have particular relevant features that have proven useful in addressing the potential of H. pylori to induce gastric cancer and the role cofactors play in the carcinogenic process. Mongolian gerbils were first reported as a model for experimental H. pylori infection in 1991 (Yokota et al. 1991). Japanese investigators noted premalignant lesions consisting of intestinal metaplasia, atrophy, and gastric ulcers in gerbils after experimental infection with H. pylori (Hirayama et al. 1996; Honda, Fujioka, Tokieda, Gotoh, et al. 1998). In one study, acute gastritis with erosions of the gastric mucosa occurred shortly after infection, whereas gastric ulcers, cystica profunda, and atrophy with intestinal metaplasia were observed at 3 to 6 months after H. pylori infection (Honda, Fujioka, Tokieda, Gotoh, et al. 1998). Following these findings, others in Japan have noted that gerbils infected with H. pylori from periods ranging from 15 to 18 months developed GAC (Honda, Fujioka, Tokieda, Gotoh, et al. 1998; Watanabe et al. 1998).
When infected with H. pylori for 62 weeks, 37% of the gerbils developed adenocarcinoma in the pyloric region, although vascular invasion and metastases were not observed (Watanabe et al. 1998). In another report, H. pylori induced GAC at 15 months postinoculation, but again the authors also did not record metastases or vascular invasion (Honda, Fujioka, Tokieda, Satoh, et al. 1998). Importantly, the histological progression in the gerbil closely resembled that observed in humans, in terms of the early appearance of intestinal metaplasia, well-differentiated histological patterns of the gastric malignancy, and location of the cancer in the antrum. Similar to findings in humans, the gerbils also had gastric ulcer disease in conjunction with gastric cancer (Hansson et al. 1996). The development of metaplasia with production of predominantly acid sialomucins was associated with tumor development. Whether the cancers arose directly from these metaplastic cells is unknown, but the tumors clearly originated deep in the gastric glands, in close proximity to these metaplastic cells (Watanabe et al. 1998).
Although most of the gastric tumors in this H. pylori gerbil model originated in the pyloric region of the stomach, significant changes in the oxyntic mucosa consistent with chronic atrophic gastritis were seen (Watanabe et al. 1998). Glandular tissue in the gastric body and fundus were atrophied and replaced by hyperplastic epithelium of the pseudopyloric type. The differences between this lesion and gastric atrophy in humans is that the gerbil corpus is not “thinner” consequent to the pseudopyloric hyperplasia (Wang et al. 1998; Watanabe et al. 1998). The diagnosis of atrophy in H. pylori–infected gerbils (as well as in mice) is not involved with the thickness of the mucosa but rather with the loss of oxyntic (parietal and chief) cell populations within the gastric glands. Accumulating data support that the parietal cell may regulate key differentiation decisions within the gastric glands, and ablation of parietal cells using transgenic technology (Li, Karam, and Gordon 1996) or their loss to infection with H. felis and H. pylori infection leads to altered glandular differentiation and neck cell proliferation; this corresponds to changes in gastric acid and gastrin physiology. The expansion of an aberrant neck cell (“regenerative hyperplasia” or “pseudopyloric hyperplasia”) in the gerbils is similar to that observed in the H. felis mouse model. This lineage has been shown to be spasmolytic polypeptide (SP) positive (Wang et al. 1998), and this SP-positive lineage also develops in H. pylori–associated gastric cancers in humans (Schmidt et al. 1998). H. pylori induced loss of oxyntic glandular tissue suggests that the gerbil becomes achlorhydric before the development of gastric cancer. Although the precise effects of chronic H. pylori infection on gastric acid secretion are not fully elucidated, serum gastrin levels, considered a risk factor in gastric cancer in the H. pylori–infected gerbil, are increased (Rieder, Merchant, and Haas 2005; Franco et al. 2005). Importantly, gerbils infected with H. pylori SS1 develop gastritis and dysplasia; however, these experiments published to date do not result in gastric cancer in the absence of carcinogens such as N-methyl-N-nitrosurea (MNU; Martin et al. 2010; Suzuki et al. 2002; Court et al. 2002).
Unlike mice, gerbils colonize efficiently with cag+ H. pylori strains and the importance in pathogenic potential of cagA in H. pylori strains has been shown in gerbils. Mongolian gerbils were infected for 32 weeks either with H. pylori cag+ strain B128 or with the isogenic mutant strain B128Δ cytotoxin-associated gene (cagY) or B128Δ cagA, defective in T4SS or in the production of its effector protein CagA, respectively (Rieder, Merchant, and Haas 2005). H. pylori B128-infected gerbils harbored high numbers of bacteria in the gastric antrum and corpus, whereas B128Δ cagY and B128Δ cagA colonized the antrum more densely than the corpus. All infected animals had robust antral inflammation and epithelial cell proliferation. B128-infected, but not mutant infected, gerbils had severe transmural inflammation with large lymph aggregates, increased proliferation, gastric atrophy, and mucous gland metaplasia in the corpus. Plasma gastrin levels and gastric pH values were significantly increased only in H. pylori WT B128-infected gerbils. In all infected gerbils, proinflammatory cytokines IL1β, IFNγ, and growth-regulated protein were increased in the antrum, but only in WT-infected animals was an increase seen in the corpus mucosa (Rieder, Merchant, and Haas 2005).
A recent study from Japan provided compelling evidence that the pattern of H. pylori colonization depends on the acid output and that this influences the long-term progression to neoplasia (Hagiwara et al. 2011). They examined the potential effect of proton pump inhibitor (PPI) therapy on progression of H. pylori–associated disease. The male gerbils were divided into 4 groups: H. pylori (ATCC43504) positives and negatives, with and without administration of a PPI (omeprazole 100 mg/kg body weight/day). At the end point of the 6-month experiment, the results provided support for previously published literature in humans. H. pylori–negative male gerbils did not develop gastritis, metaplasia, or cancer irrespective of PPI treatment; whereas all H. pylori–positive gerbils developed gastritis and most also developed metaplasia during the later stages of the disease. Treatment with omeprazole in H. pylori–positive animals accelerated progression to atrophy and enhanced hypergastrinemia, resulting in a significantly increased incidence of gastric cancer. The authors emphasized that hypergastrinemia might be promoting the development of H. pylori gastric cancer (Hagiwara et al. 2011). The findings support previous findings showing the increased risk factor of gastric atrophy in H. pylori–infected patients on PPI therapy (Kuipers 2006; Kuipers et al. 1996; Kuipers et al. 1995; Lundell et al. 2006). In humans, it remains to be shown whether chronic PPI therapy in H. pylori–infected individuals also increases the further progression to metaplasia, dysplasia, and neoplasia in the proximal stomach, and whether there are protective effects of hypergastrinemia on the gastric antrum (Tomita et al. 2011) are also observed with PPI therapy. These studies will require longer term follow-up of this subset of patients (Fox and Kuipers 2011).
The unusual susceptibility of this animal species to GAC using standard cag+ H. pylori strains underscores the overriding importance of host factors in determining the outcome from gastric Helicobacter spp. infection. It is important to note that many laboratories—particularly those outside of Japan—were unable to document the carcinogenic potential of H. pylori in the gerbil model. However, investigators in the United States using H. pylori strain B128, a Cag+ strain (7.13) serially passaged in gerbils, have now documented rapid onset GAC in gerbils. This study serves to highlight the importance of bacterial factors as well as host susceptibility in cancer induction (Franco et al. 2005).
Dietary Factors and Gastric Cancer
Fat
Epidemiological evidence supports the premise that both H. pylori and obesity are independent risk factors for gastric cancer (Ahmed and Sechi 2005; Chung and Leibel 2006). Obesity-induced inflammation underlies increasingly common diseases, such as type II diabetes and atherosclerosis (Kanneganti and Dixit 2012; James et al. 2012; Ferrante 2007; Zuniga et al. 2010). Importantly, several studies employing mouse models and focusing on the direct effect of adipose-derived factors on established tumors have shown that obesity promotes cancers (e.g., colorectal and pancreatic cancer) and implicate systemic inflammation as an important component of the tumorigenic process (Moon et al. 2013; Zyromski et al. 2009). Helicobacter-dependent gastric cancer in mice and humans results from a sustained gastric immune response involving both lymphoid- and myeloid-derived cells. The population of CD11b+Gr-1+, immature myeloid cells (IMC), increase in the gastric tissue, and circulation during the early stages of gastric Helicobacter spp. infection have been implicated in cancer initiation (Asfaha et al. 2013). IMCs are considered a heterogeneous cell population; myeloid-derived suppressor cells (MDSCs), a component of IMCs, suppress cytotoxic CD8 T cells and effectively prevent their antitumor activity and promote the survival of transformed epithelial cells (Pylayeva-Gupta et al. 2012; Ostrand-Rosenberg and Sinha 2009; Wang et al. 2013). Helicobacter-associated gastric cancer is also correlated with an increased Th1 and TH-17 immune response (Shi et al. 2010; Fox et al. 2000). Despite its exact role, an elevated Th17 has been positively associated with increased severity of gastritis, whereas its diminution is linked to decreased gastric dysplasia (Wang, Asfaha, et al. 2009). Cytokines and growth factors such as IL6 and granulocyte macrophage colony-stimulating factors (GM-CSF) also play a role in recruitment and activation of MDSCs (Pylayeva-Gupta et al. 2012; Ostrand-Rosenberg and Sinha 2009).
A recently completed study utilized H. felis–infected C57/BL male mice maintained on a high-fat diet (HFD), 45% of dietary calories from fat, to study the effect of obesity on immune modulation and the establishment of a protumorigenic gastric microenvironment (Ericksen et al. 2013). Mice on the HFD develop adipose inflammation, hyperglycemia, and insulin resistance (Collins et al. 2004; Zuniga et al. 2010). The authors reported that H. felis–infected mice in the HFD developed accelerated gastric cancer compared to infected mice fed a lean diet (13% fat). Obese mice had increased bone marrow–derived IMCs in both gastric tissue and blood, as well as increased numbers of CD4 T cells, IL17A, GM-CSF, and prosurvival gene expression in gastric tissue. Phosphorylated signal transducer and activator of transcription 3 (STAT3) was increased and likely played a role in promoting cellular transformation as well as regulating inflammatory gene expression. Analysis of adipose tissue in infected obese mice had increased accumulations of macrophages, increased IL6, C-C motif ligand 7, and leptin. Obesity and Helicobacter-induced gastric inflammation in concert also increased serum proinflammatory cytokines, including IL6, a cytokine known to promote tumor progression. The authors concluded that obesity accelerated Helicobacter-associated gastric cancer via cytokine-mediated cross talk between inflamed gastric and adipose tissue by augmenting immune responses at both tissue sites and contributing to a protumorigenic gastric microenvironment (Ericksen et al. 2013; Figure 3).
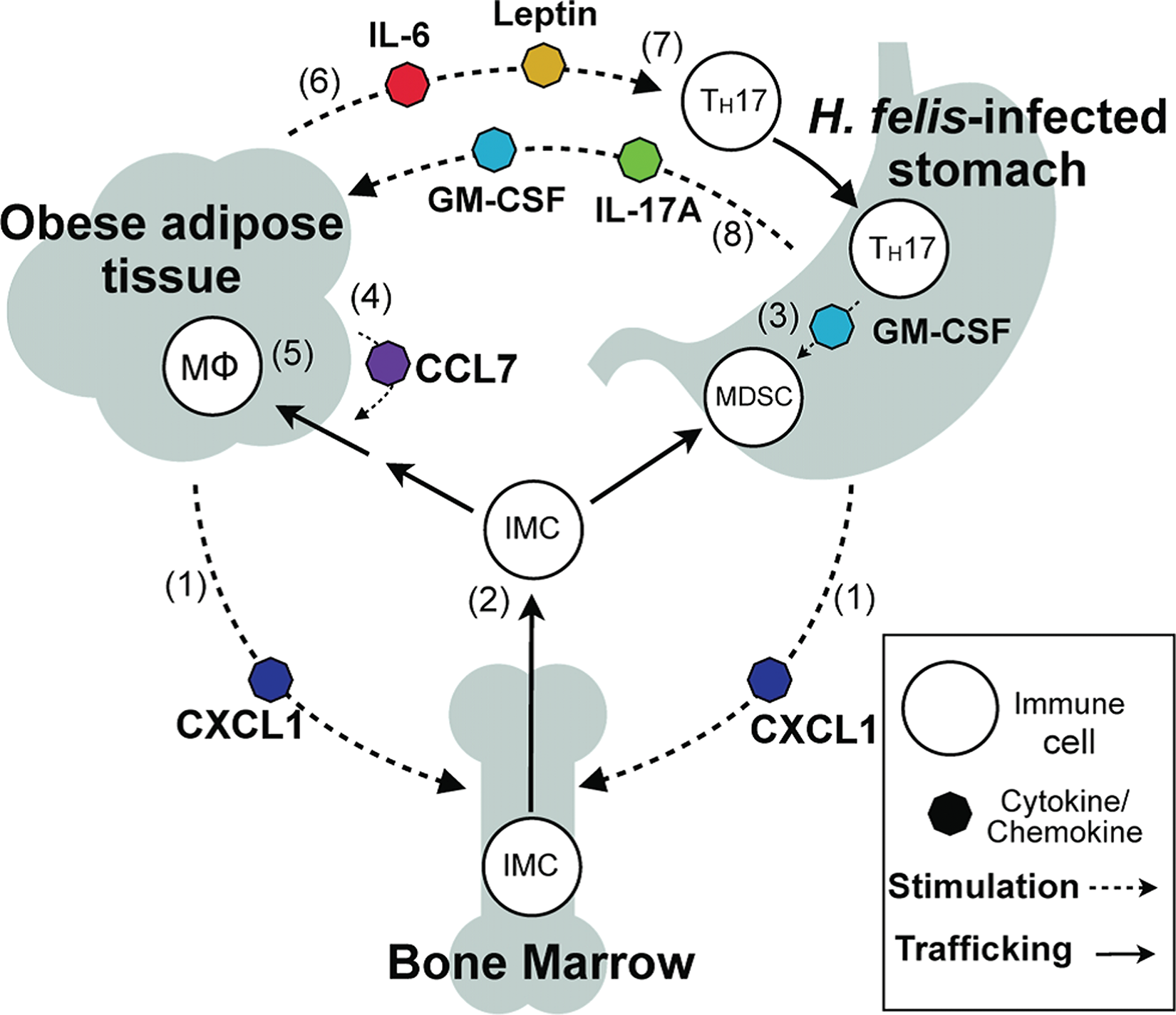
Schematic hypothesis of inflammatory cross talk during Helicobacter felis infection and diet-induced obesity. CXCL1 produced by adipose and gastric tissues (1) elevates circulating bone marrow–derived immature myeloid cells (IMCs); (2) IMCs recruited to gastric tissue by CXCL1 encounter TH17-associated cytokines, including GM-CSF, and develop into myeloid-derived suppressor cells; (3) adipose CCL7 secretion increases IMC recruitment to adipose (4), resulting in accumulation of adipose tissue macrophages (MΦ); (5) inflamed adipose produces IL-6 and leptin, (6) promoting TH17 differentiation, (7) TH17-associated cytokines IL-17A and GM-CSF produced in the stomach signal to adipose; (8) to increase CCL7 production and amplify adipose inflammation. CCL = C-C motif ligand; CXCL = Chemokine C-X-C motif ligand; GM-CSF = granulocyte macrophage colony-stimulating factor; IL = interleukin; TH17 = T helper 17 (reprinted with permission from Ericksen et al. 2013).
Folic Acid
Studies have supported the protective role of dietary folic acid (FA) against specified cancers. Results of these studies have varied and mechanisms responsible for its protective effect are not defined. Because a global reduction in DNA methylation is an early and consistent observation during carcinogenesis, it has been argued that reversal or protection against DNA hypomethylation provided by FA supplementation is a possible mechanism explaining FA chemopreventive effect (Kim 2004; Song, Sohn, et al. 2000; Song, Medline, et al. 2000). H. pylori infection and development of precancerous lesions have been associated with a loss of global DNA methylation (Cravo et al. 1996). Lower blood leukocyte (DNA methylation) levels were also observed in gastric cancer patients compared to matched controls (Hou et al. 2010). A recent study also found that high dietary folate increased survival rates in gastric cancer patients compared with low FA intake in advanced gastric cancer (Shitara et al. 2010). Interestingly, FA supplementation also reduced the incidence of N-ethyl-N-nitroguanidine (ENNG)-induced GAC in beagles (Xiao et al. 2002). Though not described in this report, commercially available beagles have a high incidence of gastric Helicobacter infection (Lee et al. 1992). Whether Helicobacter-promoted ENNG carcinogenesis in the dog is unresolved but should be considered.
In a study conducted in H. felis or control INS/GAS male mice, animals were fed a rodent diet containing 2 (control) or 8 (supplemented) mg FA/kg of diet (Research Diets, Newark, NJ). The diets were administered either at weaning or later at 8 and 16 weeks postinfection (Gonda et al. 2012). All mice were sacrificed at 28 weeks postinfection. The authors measured global DNA methylation in gastric tissue by H-deoxycytidine triphosphate (H-dCTP) incorporation assay and expressed data as a percentage of methyl-C content compared with uninfected nontransgenic FVB WT mice. For the first time, the authors found reduced global DNA methylation in the stomach of male H. felis–infected INS/GAS mice compared to WT uninfected mice; a significant effect was noted at 22 weeks postinfection, and became more severe at 28 weeks postinfection. There was a significant 18% reduction in folate content in infected INS/GAS mice, reflecting a deficiency of methyl donors in inflamed, dysplastic tissues. Serum homocysteine levels, an inverse measurement of methyl donor status, were also significantly elevated in H. felis–infected mice at 22 and 28 weeks postinfection. Importantly, FA supplementation prevented the loss of global methylation in both dysplastic epithelial and gastric stromal myofibroblasts, measured by 3 independent assays, and markedly reduced gastric dysplasia and mucosal inflammation. FA supplementation also had an anti-inflammatory effect as indicated by gene expression profiling and immunohistochemistry (IHC) for lymphocyte markers in gastric tissue (Gonda et al. 2012). However, other studies have demonstrated that treatment with demethylating agents also results in a reduction in preneoplastic lesions in H. pylori–infected gerbils (Niwa et al. 2013).
Iron
Nutritionally, iron deficiency remains a common global disorder affecting 500 to 600 million humans (Zimmermann and Hurrell 2007; World Health Organization/The United Nations Children’s Fund/United Nations University 2001), with children being particularly susceptible to iron deficiency anemia (IDA). A variety of clinical syndromes are recognized with IDA, including reduced growth, mental retardation, increased susceptibility to infectious microorganisms, and in extreme cases, mortality (Stoltzfus 2001). H. pylori has been linked to IDA, and interestingly, H. pylori–infected individuals are refractory to iron supplementation, but IDA can be reversed in these patients with H. pylori eradication (Barabino et al. 1999; Kurekci et al. 2005; Choe, Lee, and Kim 2000; Choe et al. 1999; Marignani et al. 1997). Hypochlorhydria, a marker for atrophic gastritis noted in chronic H. pylori infections, and sometimes observed with acute H. pylori infections, is considered a risk factor for IDA because under nonacidic conditions in the stomach there is an inability to reduce ferric iron to absorbable ferrous (Fe2+) iron (Windle, Kelleher, and Crabtree 2007; Charlton and Bothwell 1983; Annibale et al. 2003). Also under iron-restricted conditions, the virulence factors CagA and VacA in H. pylori are increased; iron release due to mucosal injury affords better access of H. pylori to the host’s iron stores and allows the organism to bind and utilize iron from hemoglobin, lactoferrin, and transferrin (Senkovich et al. 2010). In a recent report, investigators determined operable mechanisms of iron deficiency in H. felis–infected INS/GAS male mice over a 9-month period (Thomson et al. 2012). They noted that the infected mice, apparently on a standard mouse diet (although not stated), had parietal cell loss and hypochlorhydria accompanied by decreased serum iron and transferrin saturation, as well as hyperferritinemia and increased total iron-binding capacity (Thomson et al. 2012). The H. felis–infected INS/GAS mice at 9 months postinfection also had decreased gastric expression of iron metabolism regulators hepcidin (expressed in parietal cells and capable of regulatory acid secretion—(Schwarz et al. 2012), Bmp4, and Bmp6, but increased expression of the iron efflux protein, ferroportin, iron absorption genes and Lcn2, a siderophore-binding protein. The mice had low serum iron concentrations at 6 and 9 months postinfection, and at 9 months infected mice also had lowered transferrin, reflecting systemic iron deficiency. Iron storage capacity, measured by serum ferritin, was initially increased at 3 months in infected mice. The authors hypothesized that the H. felis–infected mice were attempting to sequester iron, making it unavailable for H. felis colonizing the stomach. However, by 6 and 9 months, H. felis–infected mice had significantly reduced serum ferritin, presumably due to overall paucity of the systemic iron (and thereby negating the need for iron storage; Thomson et al. 2012). Others have observed similar patterns of initial elevation in serum ferritin at 10 weeks post H. felis infection followed by a reduction at 30 weeks postinfection on WT mice maintained on an iron-restricted diet (Keenan et al. 2004). The authors further speculated that the marked changes in gastric iron transporters may also play a role in the rapid development of gastric cancer noted in gastric Helicobacter spp. infection in male, INS/GAS hypergastrinemic mice, known to be more susceptible to gastric cancer compared to their female counterparts (Fox, Rogers, et al. 2003; Fox, Wang, et al. 2003). The data generated with the H. felis model are Cag independent, given H. felis lacks a cag PAI. It will be interesting to ascertain if H. pylori SS1 or PMSS1-infected male INS/GAS mice will also have iron deficiency.
Importantly, iron deficiency has also been associated with stomach cancer along with other gastrointestinal tumors (Pra et al. 2009; Akiba et al. 1991; Harrison et al. 1997). H. pylori-associated gastric cancer has been recently used to explore the role of dietary iron as a cofactor in cag+ H. pylori (strain 7.13) associated gastric cancer in gerbils (Noto et al. 2013). The authors clearly demonstrated that iron depletion accelerated H. pylori–induced premalignant and malignant gastric lesions in a cagA dependent manner (Noto et al. 2013; Figure 4; Table 2). Importantly, irrespective of dietary iron status, infection with a cagA isogenic H. pylori mutant significantly attenuated inflammation, dysplasia, and GAC, highlighting the importance of cagA in gastric cancer development. Furthermore, in gerbils maintained on an iron-deficient diet, isogenic cagA mutants of H. pylori have impaired ability to colonize, suggesting that cagA plays a role in H. pylori iron acquisition (Tan et al. 2011). To determine whether the severity of gastric lesions under conditions of iron depletion were specific for H. pylori, gerbils either on iron-replete or iron-depleted diets were exposed to ethanol-induced inflammation. Proteomic analysis of in vivo adapted H. pylori harvested from gerbils on replete or deplete iron diets indicated that there was increased abundance of proteins that mediated bacterial–host interactions (Helicobacter sp. Q enolase) and cag T4SS function (CagA) in strains from the iron-depleted group (Noto et al. 2013).
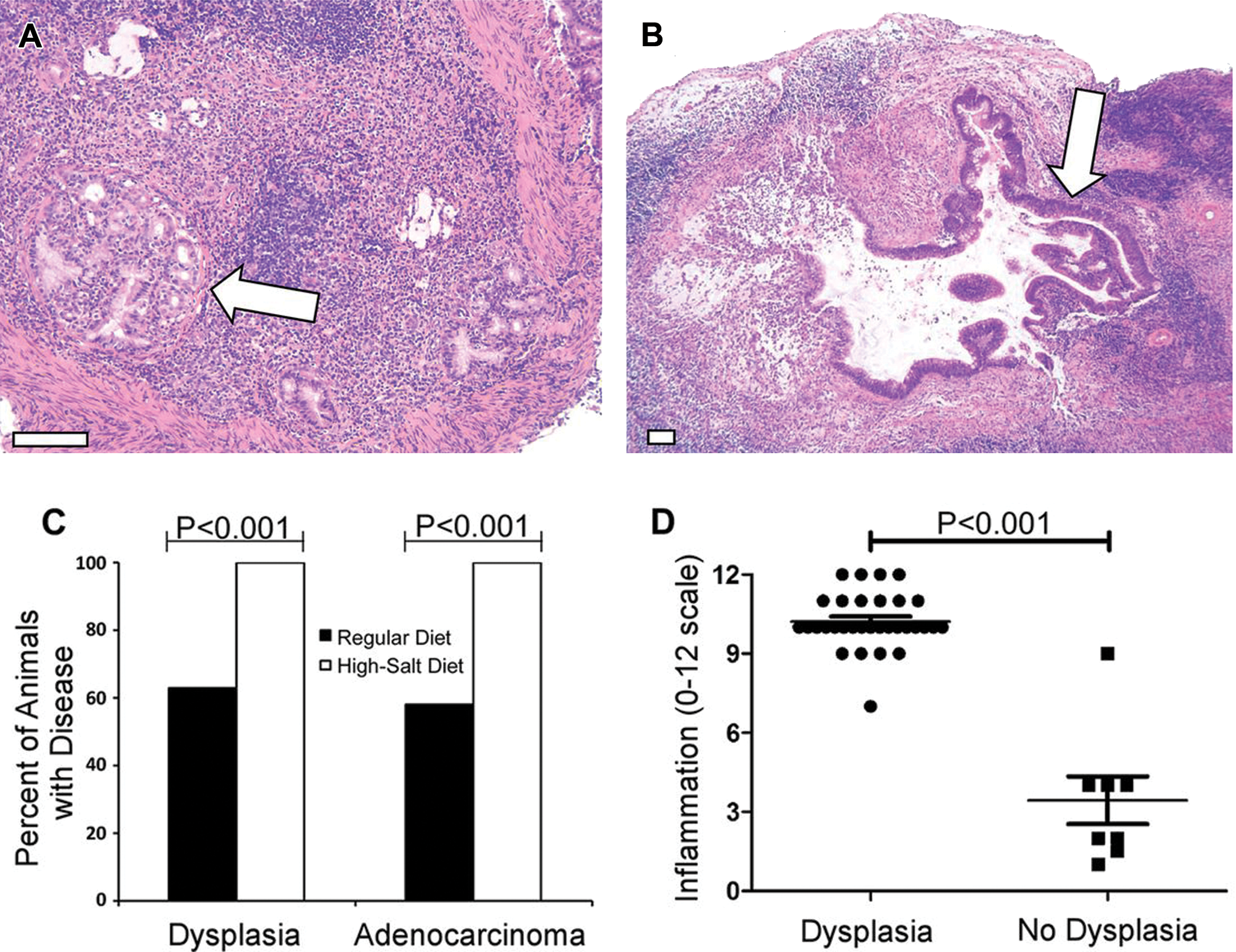
Analysis of gastric dysplasia and adenocarcinoma. Gerbils infected with WT Helicobacter pylori maintained on either a regular diet or a high-salt diet. (A, B) Representative micrographs of gastric tissue derived from WT-infected animals maintained on a high-salt diet (arrows indicate tumors). Magnification bars indicate 100 μm. (C) Gastric tissues were evaluated for dysplastic lesions and invasive adenocarcinoma. Only animals infected with the WT strain exhibited these abnormalities. Gastric cancer and dysplasia were detected significantly more frequently in the WT-infected high-salt diet group than in WT-infected regular diet counterparts (p < .0001). (D) WT-infected animals maintained on a regular diet and WT-infected animals maintained on a high-salt diet were analyzed. The total gastric inflammation score (scale of 0–12) was higher in WT-infected animals with gastric dysplasia than in WT-infected animals without dysplastic lesions. Horizontal bars indicate mean inflammation score + SEM for each group (modified with permission from Gaddy et al. 2013). WT = wild type; SEM = scanning electron microscope.
Iron depletion in gerbils increases the frequency and severity of gastric inflammation, dysplasia, and adenocarcinoma.

*Statistical significance (p ≤ .05) was determined by χ2 tests comparing iron-depleted gerbils to iron-replete gerbils. Reprinted with permissions from Noto et al. (2013).
Further, using scanning electron microscopy, the authors indicated that the H. pylori isolated from the iron-depleted gerbils and grown on human gastric epithelial cells had significantly more T4SS pili on their surface, compared to H. pylori isolated from gerbils on iron-replete diets. These data suggested that the increased assembly of the T4SS pili was attributable to H. pylori adaptation under iron depletion (Noto et al. 2013). This observation was tested using in vitro iron growth conditions with similar findings; the H. pylori grown in iron-deficient media had significantly more T4SS pili then the H. pylori grown in iron-replete media (Noto et al. 2013).
To compare data collected in the gerbil model, the authors compared H. pylori isolated from Colombian patients with the lowest serum ferritin levels to strains isolated with the highest ferritin levels. These H. pylori isolates from subjects with low serum ferritin levels induced significantly (p < .05) higher levels of IL8 compared with H. pylori strains isolated from patients with the highest ferritin levels (individuals with gastritis only or with intestinal metaplasia). The authors argued that the ability of H. pylori strains from a low ferritin serum level patient to induce more severe pathology was more likely to have accelerated the diseases early in the carcinogenic process. This hypothesis needs further longitudinal evaluations of H. pylori-infected populations on low dietary iron intake (Noto et al. 2013).
Salt
Historically, the role of salt in gastric cancer has received considerable attention in the literature (Geboers, Joossens, and Kesteloot 1985; Tuyns 1988). The deleterious effect of salt is based on epidemiological studies, biochemical analyses, and in vivo experimentation (Joossens and Geboers 1981; Takahashi and Hasegawa 1986). In a case control Belgium study, consisting of 1,087 cases (587 males, 500 females) of gastrointestinal cancer and 2,914 controls, the individuals were asked to respond to their taste for salt (Tuyns 1988). The question asked was “At the time you eat your food, do you add salt?” The possible responses were “never,” “sometimes,” or “always.” The relative risks of developing gastric cancer were adjusted for age, sex, and province where the patient lived. The relative risk for development of gastric cancer for individuals with high salt intake was 1.78 (confidence limits 1.19–2.75), which was statistically significant; however, the noted increase may be related to another causal factor, for example, H. pylori or other unknown factors of greater importance. The relative risk of 1.78 for gastric cancer in Belgian patients with high salt intake corresponds almost exactly to that noted by Correa studying black Americans in Louisiana (Correa 1985). A similar increased relative risk for gastric cancer with adding salt to their diet was noted in an Italian multicenter case control study (Buiatti et al. 1990). Other studies have incriminated increased risk of gastric cancer due to ingestion of preserved, often salty food (Tajima and Tominaga 1985), preference for salty foods (Tuyns 1988; You et al. 1988), or actual measurement of salt consumption (Correa et al. 1985; La Vecchia et al. 1987; Tsugane et al. 1991). In contrast, salt was not incriminated in a 1992 study (Friedman and Parsonnet 1992). In this smaller case–control study on 200 individuals residing in the United States who subsequently developed GAC and 200 age-matched control participants were asked “Do you usually salt food before tasting it?” Analysis of data indicated that there was no statistical evidence that routine salting of food led to increased risk of gastric cancer (Friedman and Parsonnet 1992). A joint WHO/Food and Agriculture Organization (FAO) report, given the association of dietary salt and increased risk of gastric cancer rarely raised the odds ratio above 2, concluded that high salt ingestion “probably increases the risk of stomach cancer” (World Health Organization 2003). Nevertheless, several more recent reports have analyzed the combinatory effects of high dietary salt and H. pylori infections in humans and have consistently shown increased rates of gastric cancer in these populations (Lee et al. 2003; Tsugane and Sasazuki 2007; Wang, Terry, and Yan 2009, D’Elia et al. 2012; Brenner, Rothenbacher, and Arndt 2009; Krejs 2010).
Given the late onset of gastric cancer in human populations, it is probable that both tumor initiators and tumor promoters play an important role in gastric carcinogenesis. In populations with a high risk for gastric cancer, dietary intake of fruits and vegetables is low and the diet often consists of high starch and low protein. Both low protein and high starch may favor acid-catalyzed nitrosation in the stomach and possible mutagenesis of gastric epithelia (Berretta et al. 2012; Tsugane and Sasazuki 2007). Because of the purported role of high-nitrate diets and subsequent nitrosamine formation in the stomach, numerous studies have been conducted in rodents administering various nitrosamines known to be potent carcinogens. In experiments with rats dosed with MNNG, NaCl was shown to have a promotional effect of induction of GAC when salt is given intragastrically weekly or in the diet during 12 to 20 weeks exposure to MNNG in drinking water (Newberne et al. 1987; Takahashi et al. 1983; Tatematsu et al. 1975). It is also known that salt causes damage to rat gastric surface epithelium followed by proliferation of gastric epithelium (Charnley and Tannenbaum 1985; Furihata et al. 1984; Ohgaki et al. 1989; Sorbye et al. 1988). In one study, 80% of rats given salt in the diet and MNNG in drinking water developed adenocarcinomas in the antrum, but not in the corpus (Takahashi et al. 1983). Also, ACI rats given a single initiation dose of MNNG (0.25 ml/10 g body weight MNNG in water [5 g/L]) by oral gavage and fed a 10% NaCl diet had significantly increased tumors in both forestomach and glandular stomach after being on the salt diet for 1 year (Watanabe et al. 1992).
Mice have also been used on a limited basis in studies addressing gastric damage due to salt intake. Mice (strain undesignated) fed a highly salted diet (salted codfish and yakuri sold for food consumption in Japan) developed acute gastric mucosal damage (Sato, Fukuyama, and Urata 1959). Swiss/ICR mice fed salted (10% w/w NaCl) rice diets for 3 to 12 months were noted to have developed hypertrophy of the forestomach and atrophy (considered a marker of premalignancy in humans) of the glandular stomach (Kodama et al. 1984). Parietal cell loss and statistically increased epithelial proliferation were also noted in B6 129 mice fed a high-salt diet (7.5%). A reduction in the parietal cells accounted for the atrophy observed in the fundus of the mice (Kodama et al. 1984). Although parietal cell loss has also been noted in C57BL and INS/GAS mice infected with H. felis or H. pylori (Fox et al. 2002; Mohammadi et al. 1996; Wang et al. 2000; Fox, Rogers, et al. 2003), these studies infer where uninfected mice receive salt in their diet that chronic mucosal damage per se may account for parietal cell loss in the fundus of mice.
More recent studies have incorporated NaCl in the diets of rodents infected with gastric helicobacters (Rogers et al. 2005; Fox et al. 1999). C57BL/6 mice infected with H. pylori SS1 for 4 months and maintained on a high-salt diet exhibited higher gastric urease activity, serum gastrin, bacterial colonization, and foveolar proliferation levels than did those on a basal diet; however, no meaningful effect of salt on gastric inflammation or oxyntic atrophy scores was observed (Fox et al. 1999). High salt likewise increased foveolar proliferation in uninfected but not H. pylori SS1-infected B6129 mice (Rogers et al. 2005). Thus, induction of proliferation in the absence of dysplasia does not seem to increase the risk of carcinogenesis in this model. In agreement with this concept, H. pylori SS1 infection was shown to induce DNA damage in Big Blue mice (containing a chromogen-inducible λ phage transgene for in vitro mutation quantitation), but no increase in mutation frequency was evident in infected animals fed a high-salt diet (Touati et al. 2003). B6129 mice fed a high-salt diet showed a shift in anti–H. pylori humoral immunity from a Th1 to a Th2 phenotype. This humoral immunity shift is not specific to B6129 mice. Others observed a similar increase in Th2-associated IgG1 relative to Th1-associated IgG2a in H. pylori–infected INS–GAS mice on a FVB strain background (Fox, Rogers, et al. 2003; Rogers et al. 2005). However, the salt-associated shift in humoral immunity to a Th2 pattern seemed to be H. pylori-specific. The authors found no increase in total IgG1 or IgE consistent with hypersensitivity nor did they detect antiparietal cell antibodies suggestive of autoimmune gastritis (Rogers et al. 2005).
Gerbils infected with H. pylori develop an antral gastritis/duodenitis that mimics the ulcerogenic form of the human disease (Ohkusa et al. 2003). Unlike mice, but similar to humans, the bacterial cag PAI seems to be a critical disease determinant in gerbils, particularly gastric cancer (Rieder, Merchant, and Haas 2005). The gerbil model has proved highly useful in addressing questions specifically related to cagA and its association with high dietary salt and other H. pylori virulence determinants in vivo. In earlier studies, authors have shown a synergistic promoting effect of H. pylori and high salt on MNU-initiated gastric cancer in the gerbil model (Nozaki et al. 2002). However, chemical initiation was required to induce cancer, as no tumors developed in gerbils untreated with MNU regardless of H. pylori infection status or diet. Gerbils fed a high-salt diet (2.5%) for 13 months were noted to have gastric atrophy and foveolar hyperplasia. However, the high-salt diet did not promote gastric cancer in gerbils infected with H. pylori SS1, which does not express cagA (Bergin, Sheppard, and Fox 2003). However, in a recent study, investigators have demonstrated that a high dietary intake (8.75%) in a Mongolian gerbil model enhances H. pylori–induced carcinogenesis (Gaddy et al. 2013). The high-salt diet used approximates the concentration of sodium chloride in dried fish, preserved in 3 to 20% salt, pickled foods containing up to 25% salt, and soy sauce which has 19% salt. The administration of high-salt diet to uninfected gerbils did not result in gastric cancer. The authors concluded that the effects of a high-salt diet on gastric cancer were dependent on the presence of H. pylori infection. Importantly, the high-salt diet increased incidence of gastric carcinoma in gerbils infected with WT cagA + H. pylori strain 7.13 but not in animals infected with a cagA mutant strain (Gaddy et al. 2013). This observation is consistent with several parts of the world with high rates of gastric cancer, also, a high prevalence of cagA + strains; a large percentage of these populations routinely have diets with a high salt concentration.
Others have reported that exposure to H. pylori to high-salt conditions in vitro leads to alterations in the expression of multiple H. pylori genes, included cagA and neutrophil activation protein (Liao et al. 2013; Loh et al. 2012). The authors also noted that cagA gene expression was elevated in the stomachs of WT-infected gerbils maintained on a high-salt diet compared to WT-infected animals maintained on a regular diet (Gaddy et al. 2013). Interestingly, both IL-1β transcription and inducible nitric oxide synthase (iNOS) transcription were significantly elevated in WT-infected gerbils maintained on a high-salt diet compared to the regular diet fed to gerbils infected with WT H. pylori. WT-infected animals maintained on a high-salt diet had markedly increased gastric pH compared to their regular diet WT-infected gerbil counterparts. This is consistent with the finding of atrophic gastritis and hypochlorhydria preceding the development of gastric cancer in humans. Parietal cells are reduced in the hypochlorhydric animals and likely accounts, at least in part, for the observed hypochlorhydria. They also hypothesized that hypochlorhydria could be due to perturbations in parietal cell function. To test this, the authors also measured hepcidin (produced in parietal cells), gastrin, and H, K-ATPase transcripts. The animals infected with WT H. pylori and maintained on a high-salt diet had decreased levels of all 3 transcripts compared to animals infected with WT H. pylori and maintained on a regular diet. They concluded that increased dietary salt consumption markedly alters the outcome of infected with cagA+ H. pylori strain 7.13 and proposed that high salt concentrations stimulate increased expression of cagA, and the actions of CagA lead to inflammation, hypochlorhydria, and enhanced carcinogenesis. They also raised the possibility that high salt concentrations lead to altered expression of multiple H. pylori genes (including cagA) as well as to the alterations in the host and that this constellation of alterations could enhance progression of carcinogenesis. Regardless of the mechanisms, their data argue that reduction in dietary salt could lower risk of gastric cancer in H. pylori-infected patients (Gaddy et al. 2013).
Vitamin C
Antioxidants, particularly vitamin C and β-carotene, have anticarcinogenic properties based on laboratory data as well as epidemiological evidence. Ascorbic acid scavenges nitrous oxide and makes the compound unavailable for nitrosation in the stomach. Furthermore, evidence indicates that many, but not all, patients with H. pylori gastritis have lowered ascorbic acid concentrations (Sobala, Pignatelli, and Schorah 1991; Sobala et al. 1993). Other investigators have shown that patients with precancerous lesions consisting of intestinal metaplasia and multifocal atrophic gastritis also have lowered ascorbate and elevated N-nitroso compound concentrations (Chen et al. 1990; Bartsch, Ohshima, and Pignatelli 1988). Supplementation of vitamin C has been associated with reduced gastric cancer risk in some human studies (Su et al. 2000; Correa et al. 1998). Despite initial promising results in a prospective trial in a very high-risk gastric cancer population supplemented with vitamin C at 2 g per day for 6 years (Correa et al. 2000), a follow-up study on this population indicated that vitamin C supplementation over a 12-year period did not provide any lasting protection against gastric cancer (Mera et al. 2005).
To test whether H. pylori does indeed impair ascorbic acid secretion into the stomach, animals which need dietary supplementation of vitamin C should ideally be used to mimic vitamin C metabolism in humans. Of the Helicobacter sp. infected animal models presently available, nonhuman primates fulfill this criterion, but to date have not been used in answering this important question. The guinea pig is the only other laboratory animal that has dietary requirements for vitamin C. Published data indicated that H. pylori–induced gastritis in this model (Shomer et al. 1998). Supplementation with vitamin C in the diet at 10-fold higher levels than a maintenance dose (3,102 mg/kg) for 6 weeks in guinea pigs, which lack L-gulono-γ-lactone oxidase similar to humans and gulo−/− mice, did not impact H. pylori colonization levels nor severity of gastritis (Taylor 2007).
In a later study using vitamin C–deficient B6.129P2—Gulo
tm1Umc
/mmcd (gulo−/−) mice lacking
Helicobacter-induced gastric disease in humans and mice is mediated by a Th1-predominant host immune response (Mohammadi et al. 1997; Fox and Wang 2007; D’Elios et al. 1997). Gulo−/− mice supplemented with high vitamin C had a similar Th1-promoted inflammatory response to H. pylori as infected WT mice. Low vitamin C-supplemented, H. pylori–infected gulo−/− mice had less severe gastric lesions associated with suppressed Th1 responses as evidenced by reduced levels of plasma IgG2c, reduced expression of gastric IFN-γ, and TNF-α mRNA, and increased H. pylori colonization levels. These data suggest that low dietary vitamin C impairs the host’s ability to sustain a proinflammatory response and conversely, that adequate vitamin C levels in the infected host may be important for maintaining a robust Th1 immune response to chronic H. pylori infection. Consistent with this report, others have reported attenuated Th1 immune responses in vitamin C–deprived gulo−/− mice. This was manifested by less severe pneumonia and suppressed pulmonary expression of proinflammatory IL-1β and TNF-α mRNA in vitamin C–deprived gulo−/− mice during the first few days of acute viral influenza (Li, Maeda, and Beck 2006). In hosts with low or no vitamin C intake, attenuated Th1 responses to other types of infection, such as tuberculosis in humans or Klebsiella pneumoniae sepsis in gulo−/− mice, may also be important in predicting survival (Gaut et al. 2006; Hemila et al. 1999).
Using a mouse model of intestinal parasitic infection that causes a Th2 immune response, the effect of modulating the Th1-associated response to H. felis infection has been analyzed (Fox et al. 2000). In C57BL/6 mice coinfected with intestinal helminths and H. felis, reduced systemic Th1 immune responses and lower levels of Th1-mediated gastric cytokines were associated with increased H. felis colonization levels (a marker of less severe gastric pathology) and less severe premalignant lesions (Fox et al. 2000). In another series of mouse experiments, the authors demonstrated that lower bowel colonization with enteric helicobacters accentuated the severity of H. pylori gastritis. Colonization with these helicobacters led to a lower gastric Th1 immune response, apparently triggered by regulatory T cells sensitized by shared antigens between enteric Helicobacter spp. and H. pylori (Ge et al. 2011; Lemke et al. 2009). This may have direct relevance to the immune status of the African population where enteric helicobacters are commonly isolated from diarrheic children (Lastovica and le Roux 2000). These results may in part explain the “African enigma” where helminth and enteric Helicobacter infections are common and the incidence of gastric cancer is low, despite a high prevalence of H. pylori infection (Holcombe 1992). The H. pylori-infected, low vitamin C-supplemented gulo−/− mouse model may offer another possible explanation to the African enigma. In Gambia, due to the impact of drought on the food supply, mean daily intake of vitamin C approaches zero for 7 months of the year and is accompanied by low plasma vitamin C levels (Bates, Prentice, and Paul 1994). It is tempting to speculate based on studies of the H. pylori–infected gulo−/− mice on low vitamin C, that minimal vitamin C intake during H. pylori infection may be one of the factors contributing to a lower reported incidence of gastric cancer in some populations in Africa because of attenuated gastric immune responses resulting in less severe premalignant lesions. Although this hypothesis is consistent with a report of diminished mitogen responses of peripheral blood mononuclear cells from pigs affected by heritable vitamin C deficiency (Schwager and Schulze 1998), the biological significance of these responses requires further studies. The authors concluded that high vitamin C supplementation in this model, similar to previous epidemiological studies in humans (Mera et al. 2005; Waring et al. 1996; Phull et al. 1999; Bjelakovic et al. 2004), did not prevent progression of H. pylori–induced gastritis and development of premalignant gastric lesions. In contrast, low dietary vitamin C resulted in less severe gastric disease by downregulating gastric and systemic Th1 immune responses to H. pylori infection.
Footnotes
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
The author(s) received the following financial support for the research, authorship, and/or publication of this article: NIH support: P01CA028842 (JGF), P01CA026731 (JGF), P30ES002109 (JGF), and R01CA093405 (TCW).