
Abstract
Inflammatory bowel diseases (IBD) such as ulcerative colitis and Crohn’s disease lead to altered gastrointestinal (GI) function as a consequence of the effects of inflammation on the tissues that comprise the GI tract. Among these tissues are several types of neurons that detect the state of the GI tract, transmit pain, and regulate functions such as motility, secretion, and blood flow. This review article describes the structure and function of the enteric nervous system, which is embedded within the gut wall, the sympathetic motor innervation of the colon and the extrinsic afferent innervation of the colon, and considers the evidence that colitis alters these important sensory and motor systems. These alterations may contribute to the pain and altered bowel habits that accompany IBD.
Introduction
The functions of the gastrointestinal (GI) tract are subject to complex neural regulation. The enteric nervous system (ENS), which is located within the wall of the gut, contains reflex circuits capable of modulating gut functions independently of connections with the central nervous system (CNS). Notwithstanding this, there are multiple afferent and efferent connections with the CNS. Extrinsic afferent neurons convey information to the CNS, while parasympathetic and sympathetic motor pathways interact with the ENS to modify gut activity. This review will use the example of colonic inflammation to illustrate the plasticity of the innervation of the gut and its potential functional consequences.
Structure and Function of the ENS
The ganglia of the ENS are located in 2 plexuses: the myenteric plexus located between the circular and longitudinal smooth muscle layers and the submucosal plexus, which is located within the connective tissue beneath the mucosa (Furness 2012). The submucosal plexus of large mammals, including humans, exists in 2 connected but separate layers, whereas smaller mammals, including rodents, have only 1 (Furness 2006). Enteric ganglia contain the cell bodies and neurites of intrinsic primary afferent neurons (IPANs), interneurons, and motor neurons, which together form microcircuits involved in the reflex regulation of motility, blood flow, and secretion (Brookes 2001; Furness 2006; Gershon, Kirchgessner, and Wade 1994; Wood et al. 1999).
Enteric reflexes are initiated by the activation of IPANs. IPANs innervate one another, interneurons, and motor neurons to regulate gut functions (Furness et al. 2004). IPANs are located in both plexuses and respond to changes in the chemical and contractile state of the organ by the discharge of action potentials (APs) and the release of neurotransmitters. AP discharge can be either in direct response to sensory stimuli, for example, distension, or via the release of substances, including serotonin, from the basolateral surface of enteroendocrine cells (Bertrand et al. 2000). IPANs typically exhibit Dogiel type II morphology—a smooth cell body with multiple axons—and are electrophysiologically classified as afterhyperpolarizing (AH) neurons because of a characteristic long-lasting afterhyperpolarizing potential (AHP) following an AP (Bornstein, Furness, and Kunze 1994; Furness et al. 2004). Ascending and descending interneurons form connections to neurons within the myenteric or submucosal plexus of neighboring ganglia and are innervated by IPANs and other interneurons. Motor neurons are innervated by interneurons or IPANs and in turn release neurotransmitters onto effector cells within the smooth muscle, the vasculature, or the mucosal epithelium. Motor neurons and interneurons are electrophysiologically classified as S neurons as they exhibit fast excitatory postsynaptic potentials and generally lack long-lasting AHPs (Bornstein, Furness, and Kunze 1994; Furness et al. 2003). These neurons commonly exhibit Dogiel type I, uniaxonal morphology.
Motility is regulated by the innervation of the smooth muscle layers by myenteric neurons, superimposed upon the pacemaker activity of interstitial cells of Cajal (Huizinga and Lammers 2009; Sanders et al. 2012). The motor neurons that innervate the circular and longitudinal muscle layers release a number of excitatory and inhibitory neurotransmitters. Acetylcholine (ACh) and substance P primarily mediate contraction, whereas vasoactive intestinal peptide (VIP), nitric oxide (NO), and purines elicit relaxation (Furness 2006; Sanders 1998). Motor neurons from the submucosal plexus, along with a small minority of myenteric neurons, innervate the mucosal epithelium release transmitters that cause Cl− and H2O secretion (Furness 2006). In doing so, enteric secretomotor neurons play an important role in fluid homeostasis by partially counteracting the ongoing electrolyte absorption. Enteric vasodilator reflexes within the submucosal plexus enable the GI tract to meet the metabolic and nutrient absorption requirements during digestion (Lundgren 1984; Sjovall et al. 1983a; Vanner and Macnaughton 2004; Vanner and Surprenant 1996). The activity of the ENS can be modulated by endocannabinoids, lipid mediators that act primarily via cannabinoid receptor type 1 (CB1) and cannabinoid receptor type 2 (CB2) receptors located on enteric neurons (Hons et al. 2012). CB1 and/or CB2 receptor activation has been shown to reduce GI motility, secretion, and possibly inflammation (Izzo and Camilleri 2008, 2009; Izzo and Sharkey 2010). Interestingly, axons from myenteric and submucosal neurons innervate the lamina propria and are found in close proximity to lymphoid cells and follicles in mice (Crivellato et al. 2002; Defaweux et al. 2005), which suggests a potential role for enteric neurons in immunomodulation. However, the physiological and pathophysiological significance of the proximity of enteric varicosities to immune cells has not yet been fully elucidated.
Effects of Colitis on the ENS
The effects of colitis on the ENS are summarized in Table 1. A common finding from studies of the inflamed colon is a reduction in the number of enteric neurons that does not appear to be restricted to specific neural populations. This has been observed in the myenteric plexus of mouse and guinea pig (Boyer et al. 2005; Linden et al. 2005) and the submucosal plexus of rats (Sanovic et al. 1999) with trinitrobenzene sulfonic acid (TNBS)–induced colitis, as well as the myenteric plexus of the human ulcerative colitis (UC) patients (Bernardini et al. 2012; Geboes and Collins 1998). Enteric neuron density remains diminished following the resolution of inflammation (Linden et al. 2005). The loss of enteric neurons appears to occur downstream of the activation of caspase-dependent apoptosis. Enhanced activation of the P2X7 receptor, pannexin-1-signaling complex (Gulbransen et al. 2012), and neutrophil infiltration of enteric ganglia (Boyer et al. 2005) have each been proposed as causes of neuronal apoptosis in the inflamed bowel.
Summary of the effects of colitis on the ENS.
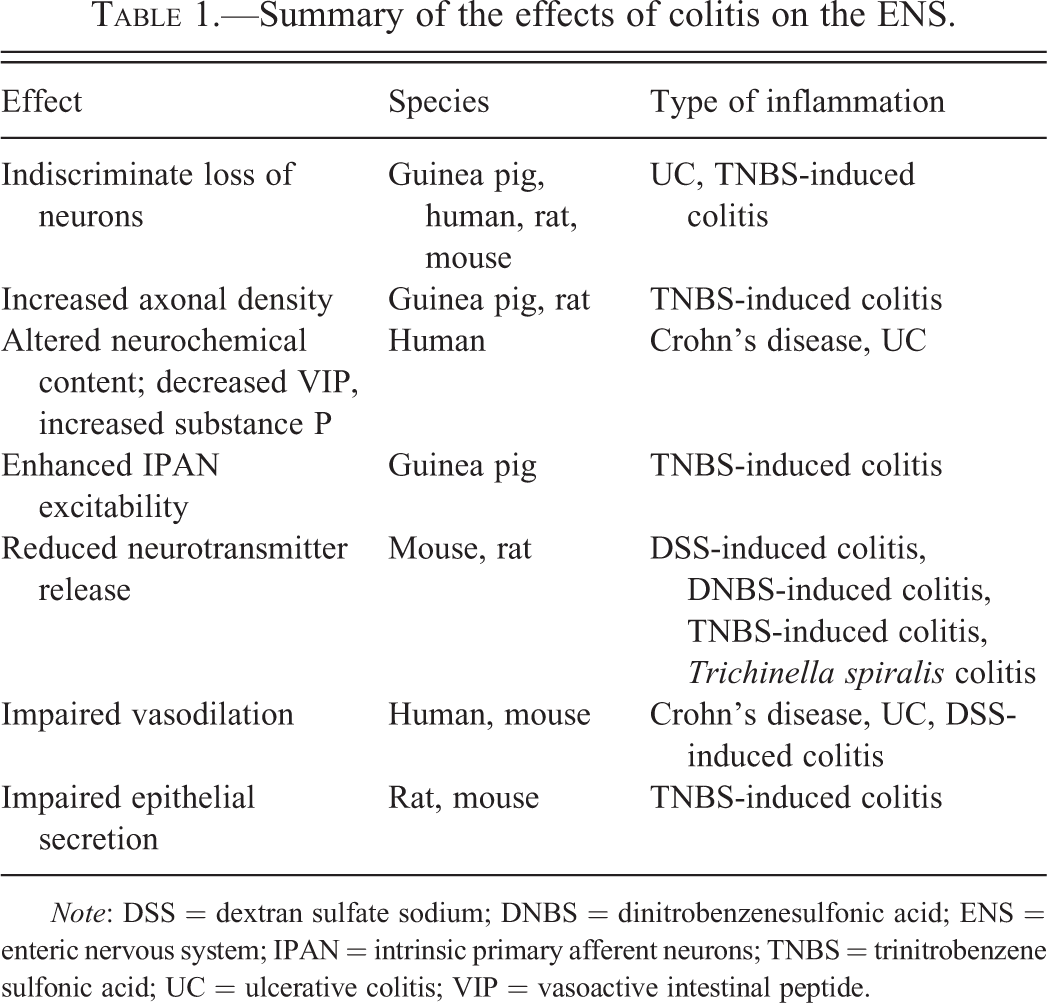
Note: DSS = dextran sulfate sodium; DNBS = dinitrobenzenesulfonic acid; ENS = enteric nervous system; IPAN = intrinsic primary afferent neurons; TNBS = trinitrobenzene sulfonic acid; UC = ulcerative colitis; VIP = vasoactive intestinal peptide.
Despite a loss of enteric neurons, several studies have demonstrated that there is enhanced neurite outgrowth following colitis. During the acute inflammatory phase of TNBS-induced colitis, there is initial damage to enteric axons followed by axon sprouting (Lourenssen et al. 2005). Following TNBS-induced inflammation there is an overall increase in axon density within the muscle (Lourenssen et al. 2005) and mucosa (Nurgali et al. 2011) of inflamed gut from rats and guinea pigs, respectively. The neurochemical content of enteric neurons also changes during inflammation. Immunoreactivity for VIP decreases and substance P increases in tissues from colon of inflammatory bowel disease (IBD) patients compared to controls (Mazumdar and Das 1992; Neunlist et al. 2003; Vento et al. 2001). Additionally, immunoreactivity for VIP, NO synthase, and neuropeptide Y (NPY) increases in tissues from the ileum of Crohn’s disease patients compared to controls (Belai et al. 1997).
In addition to structural alterations to the ENS, numerous studies have identified substantially altered electrophysiological properties of enteric neurons from the inflamed and post-inflamed gut. At rest, IPANs are relatively inexcitable because of their prolonged AHP, which limits AP firing (Furness et al. 2004). Hyperexcitability of IPANs from inflamed regions of the GI tract has been well documented in guinea pig TNBS-induced colitis during (Linden, Sharkey, and Mawe 2003; Lomax, Mawe, and Sharkey 2005) and following the resolution of inflammation (Krauter et al. 2007; Lomax, O’Hara, et al. 2007). The increased excitability of these neurons has been attributed to a reduction in the AHP (Lomax, Fernandez, and Sharkey 2005; Lomax et al. 2006). Prostaglandin E2, which is elevated during colitis, reduces the AHP and has been identified as a possible mediator of the increased enteric neuron excitability (Linden et al. 2004; Manning et al. 2002).
Evidence from several studies suggests that a reduction in intracellular Ca2+ signaling may underlie the increased activity of these neurons (Mawe et al. 2009). The generation of the long-lasting AHP is mediated by a Ca2+-activated K+ conductance (gKCa; Hirst, Johnson, and van Helden 1985; Vogalis et al. 2001), and reduced intracellular [Ca2+] lowers the activation of the gKCa, which would subsequently reduce the AHP. However, there is also evidence that an increase in current through hyperpolarization-activated cation channels may contribute to IPAN hyperexcitability (Linden, Sharkey, and Mawe 2003).
Another important effect of colitis is that it alters enteric neurotransmitter release (Collins et al. 1992) as well as effector tissue responsiveness to these neurotransmitters. Purinergic neuromuscular transmission to the vascular and circular smooth muscle is reduced during experimental colitis in mice and rats (Antonioli et al. 2010; Lomax et al. 2007; Neshat et al. 2009; Roberts et al. 2012). ACh released from submucosal vasomotor neurons binds to endothelial muscarinic receptors, which stimulates endothelial NO synthase. NO produced from endothelial cells diffuses into the adjacent vascular smooth muscle cells where it activates soluble guanylyl cyclase and causes smooth muscle relaxation and vasodilation. Studies of arterioles from IBD patients and mice with colitis have demonstrated that inflammation impairs endothelium-dependent vasodilation due to free radical damage to endothelial cells (Hatoum et al. 2003; Mori et al. 2005). Similarly, the secretory response of the epithelium to enteric neurotransmitters is inhibited during colitis due to free radical effects on epithelial cells (Asfaha et al. 1999).
Structure and Function of the Sympathetic Innervation of the GI Tract
The sympathetic nervous system (SNS) is comprised of preganglionic sympathetic neurons, postganglionic sympathetic neurons (PGSNs), and adrenal chromaffin cells (ACCs). Axons of preganglionic neurons extend out from the intermediolateral cell column of the thoracolumbar spinal cord and form synapses with PGSNs within the paravertebral and prevertebral ganglia, as well as the ACCs of the adrenal medulla (Janig 1988). Prevertebral sympathetic ganglia lie ventral to the abdominal aorta and include the celiac, superior mesenteric, and inferior mesenteric ganglia. Axons from PGSNs synapse with their targets where they primarily release norepinephrine and cotransmitters, including ATP and NPY (Szurszewski et al. 2002; Szurszewski and Miller 1994), while ACCs are neuroendocrine cells that release catecholamines and cotransmitters into systemic circulation. The overall effect of the sympathetic innervation of the GI tract is a reduction in activity achieved by lowering epithelial electrolyte secretion, blood flow, and gut motility (Lomax et al. 2010). There is also evidence that the SNS interacts with the immune system to modulate GI inflammation (McCafferty et al. 1997; Straub et al. 2008; Straub et al. 2006) by actions on cells of the innate and adaptive immune systems (Elenkov et al. 2000; Sanders 2012).
The axons of GI-related PGSNs follow the mesenteric arteries to the GI tract. These axons pass through the serosa and innervate the microvasculature and enteric neurons within the myenteric and submucosal plexus (Lomax et al. 2010). Rather than innervating the smooth muscle of the gut directly, PGSNs cause a presynaptic inhibition of neurotransmission from enteric neurons (Hirst and McKirdy 1974). This is in contrast to sympathetic innervation of the sphincters, where the smooth muscle is directly innervated and has a small tonic activation (Lomax et al. 2010). The PGSNs that supply the submucosal ganglia reduce secretion by an indirect inhibitory effect on secretomotor neurons and are tonically active (Sjovall et al. 1983b). There is also dense innervation of the GI microvasculature, mesenteric veins, and the portal vein (Sjovall et al. 1983a; Vanner and Surprenant 1996). Increased sympathetic activity decreases the share of cardiac output received by the gut, thus making it available to other organ systems.
Effects of Colitis on the SNS
The effects of colitis on the SNS are summarized in Table 2. There is a large body of evidence suggesting that GI inflammation affects the SNS as there are marked changes in SNS excitability (Dong et al. 2008), neurotransmitter release (Blandizzi et al. 2003; Swain et al. 1991), and structure (Dvorak et al. 1980; Dvorak and Silen 1985; Magro et al. 2002; Straub et al. 2008). Patients with active UC display increased SNS activity at rest (Furlan et al. 2006; Ganguli et al. 2007; Maule et al. 2007), while Crohn’s disease patients and animal models typically have decreased SNS activity (Swain et al. 1991). Nerve dysfunction may continue after clinical symptoms are no longer observed in the patient as there is often increased SNS activity in IBD patients while in clinical remission (Sharma et al. 2009). Further, nerve dysfunction is not restricted to the inflamed region demonstrated by decreased norepinephrine in both the inflamed colon and the uninflamed small intestine during TNBS-induced colitis in rats (Blandizzi 2007; Jacobson, McHugh, and Collins 1995; Motagally, Neshat, and Lomax 2009; Swain et al. 1991) and dextran sulfate sodium (DSS)–induced colitis in mice (Motagally, Neshat, and Lomax 2009).
Summary of the effects of colitis on the sympathetic innervation of the colon.
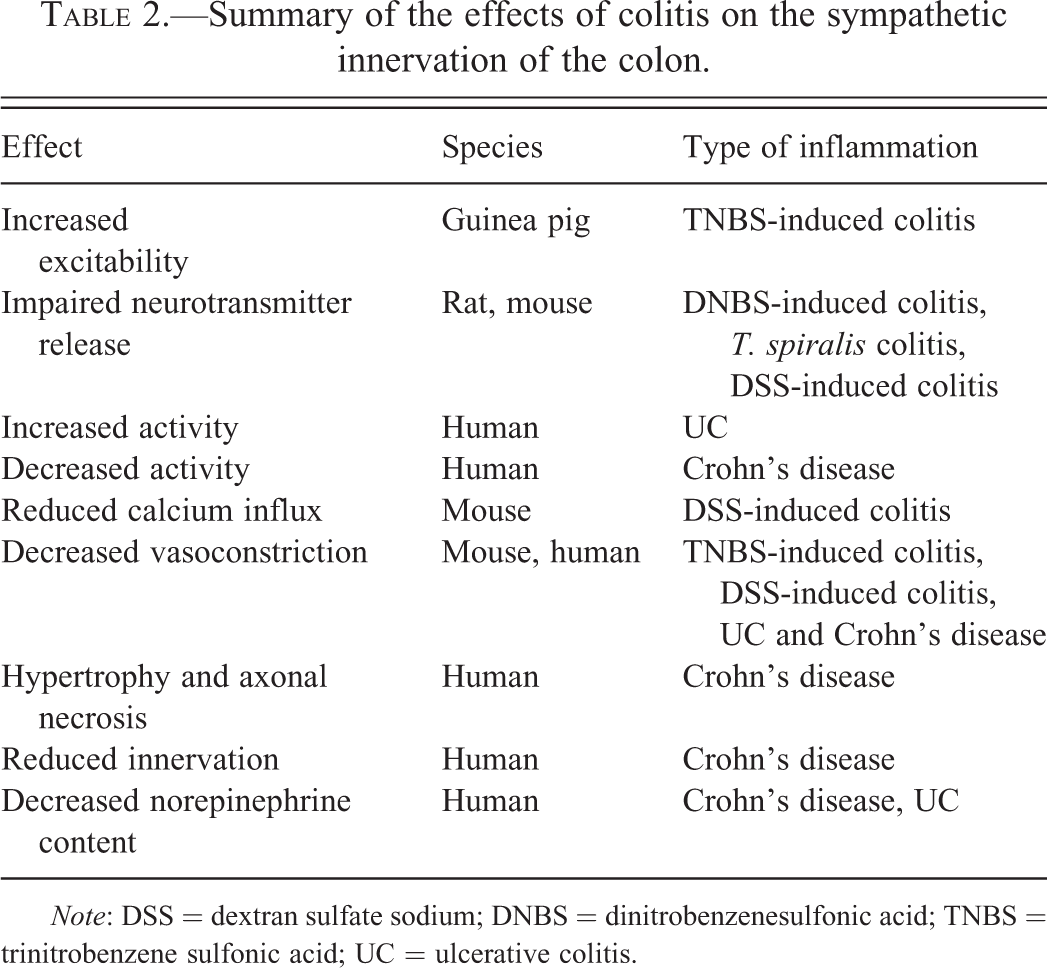
Note: DSS = dextran sulfate sodium; DNBS = dinitrobenzenesulfonic acid; TNBS = trinitrobenzene sulfonic acid; UC = ulcerative colitis.
Recent research has made progress toward understanding the potential mechanisms underlying decreased noradrenaline release during colitis. DSS-induced colitis in mice reduced N-type voltage-gated Ca2+ current in sympathetic neurons (Motagally, Neshat, and Lomax 2009); this effect was recapitulated by exposure of naive sympathetic neurons to the proinflammatory cytokine tumor necrosis factor-α (TNF-α; Motagally et al. 2009). It has long been known that presynaptic voltage-gated Ca2+ channels are responsible for coupling APs to neurotransmitter release, where a decrease in Ca2+ current can lead to a decrease in neurotransmitter release. In a separate study, dinitrobenzene sulfonic acid–induced colitis in rats upregulated presynaptic α2-adrenoceptors on sympathetic nerve terminals, which inhibit noradrenaline release, at both inflamed and uninflamed sites (Blandizzi et al. 2003). Sympathetic vasoconstriction of enteric blood vessels is due to the postjunctional effects of noradrenaline and ATP, or a related purine, on vascular smooth muscle (Evans and Surprenant 1992; Vanner and Surprenant 1996). Decreased sympathetic constriction of submucosal arterioles (Birch et al. 2008), which has been observed during IBD, has been linked to increased expression of CD39, a protein on the plasma membrane of vascular endothelium which breaks down extracellular ATP (Lomax, O'Reilly, et al. 2007; Neshat et al. 2009).
The structure of the SNS within the GI tract also appears to be altered during IBD. Electron microscopic studies of ileum from Crohn’s disease patients have reported both hypertrophy of autonomic axons and severe axonal necrosis (Dvorak et al. 1980; Dvorak and Silen 1985). Immunohistochemical techniques later demonstrated reduced sympathetic innervation (Straub et al. 2008), an observation supported by decreased colonic mucosal norepinephrine concentrations in Crohn’s disease patients (Magro et al. 2002). Contrary to these findings, other investigators used similar techniques and found that Crohn’s disease patients had increased sympathetic innervation (Birch et al. 2008; Kyosola, Penttila, and Salaspuro 1977). Collectively, these findings indicate that the structure of the SNS is affected during IBD; it appears that the density of sympathetic axons can increase or decrease, perhaps depending on which region of the tissue is analyzed and whether the inflammation is active or in remission.
Structure and Function of Extrinsic Afferent Innervation of the GI Tract
There are 2 populations of extrinsic afferent neurons that innervate the GI tract: those that synapse in the brain stem and those that synapse in the spinal cord (Brookes et al. 2013; Furness 2012). The axons of the neurons that synapse in the brain stem are contained in the vagus nerve; thus the neurons are called vagal afferent neurons. Their cell bodies are contained in the nodose and jugular ganglia, while their peripheral axons project to a variety of visceral organs and their central nerve terminals innervate neurons in the nucleus of the solitary tract (Grundy 2006). Vagal afferent neurons are particularly important in monitoring the state of the upper GI tract and their activation is thought to contribute to the feeling of satiety. Since vagal afferent neurons do not substantially innervate the colon of most species, the effects of colitis on their structure and function will not be considered further.
Spinal afferent neurons that innervate the gut are pseudounipolar neurons with cell bodies in thoracolumbar and lumbosacral dorsal root ganglia (DRG), peripheral axons in the gut, and central projections that terminate in the dorsal horn of the spinal cord (Brookes et al. 2013). A subset of spinal afferent neurons responds to noxious stimuli and give rise to the sensation of visceral pain. The remaining spinal afferent neurons convey information to the CNS on the contractile and chemical state of GI organs. The nerve terminals of spinal afferent neurons are located in the muscle layers, the mucosa, and along blood vessels (Brookes et al. 2013). The mucosal endings can be excited by the release of enteroendocrine substances, such as serotonin released in response to luminal stimuli, or by direct mechanical stimulation. Different types of mechanosensitive endings have been classified either by their responsiveness to specific mechanical stimuli or by their morphology (Brierley et al. 2004; Brunsden et al. 2007; Lynn et al. 2005; Lynn and Blackshaw 1999; Lynn and Brookes 2011).
In addition to the role of spinal afferent neurons in exciting second-order neurons in the dorsal horn of the spinal cord, their peripheral axons can release substance P and calcitonin gene–related protein to cause vasodilation. There is also a unique group of afferent terminals in the rectum termed rectal intraganglionic laminar endings (Lynn et al. 2005). As the name suggests, these endings terminate in and around myenteric ganglia, and their function is thought to be the detection of low-threshold mechanical stimuli. Therefore, rectal intraganglionic laminar endings may be involved in physiological reflexes of the distal colon, such as defecation. Viscerofugal neurons are a specialized class of enteric neuron whose axons project out of the gut and synapse on PGSNs as a component of the afferent limb of intestinointestinal reflexes (Lomax et al. 2000; Szurszewski et al. 2002).
Effects of Colitis on Extrinsic Afferent Pathways
The changes in extrinsic afferent pathways during colitis have received a lot of attention due to their potential importance in the generation of visceral pain, which often accompanies IBD (Table 3). There are a number of complex electrophysiological changes that occur during inflammation, which increase the excitability of colonic afferent neurons. In addition, a number of pronociceptive substances (e.g., TNFα, bradykinin, and eicosanoids) are released during inflammation that can directly excite colonic afferent neurons. Recording from afferent nerves directly innervating the inflamed colon have fairly consistently shown that responses to chemical and mechanical stimuli are enhanced (Feng et al. 2012; Hughes et al. 2009). This is thought to be due in part to upregulation or sensitization of receptors for sensory mediators (Coldwell et al. 2007; Matsumoto et al. 2012; Wynn et al. 2004) and mechanosensitive ion channels (De Schepper et al. 2008). Superimposed upon these changes is an increased cellular excitability, which lowers the threshold for neuronal AP discharge in response to a given stimulus. Changes in the function of voltage-dependent Na+ (Beyak et al. 2004) and K+ channels (Stewart et al. 2003), as well as leak K+ channels (La and Gebhart 2011), which together contribute to the generation and central propagation of APs, underlie the increased excitability of spinal afferent neurons during colitis.
Summary of the effects of colitis on colonic extrinsic afferent innervation.
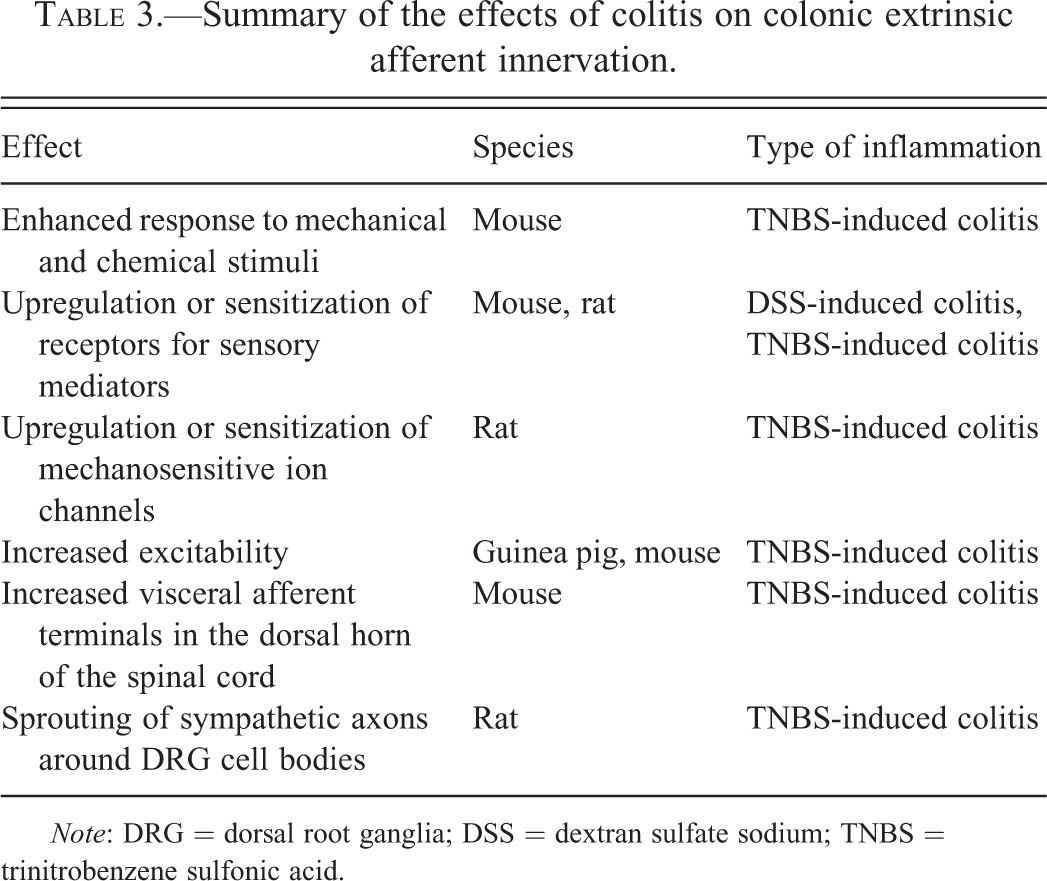
Note: DRG = dorsal root ganglia; DSS = dextran sulfate sodium; TNBS = trinitrobenzene sulfonic acid.
The electrophysiological evidence of altered afferent signaling during colitis is complemented by findings of neurochemical and neuroanatomical changes in visceral pain pathways during colitis. For example, the expression of calcitonin gene–related protein is enhanced in the DRG of mice, and a retrograde tracer study demonstrated an increase in the terminals of visceral afferent neurons in the dorsal horn of the spinal cord following TNBS-induced colitis in mice (Harrington et al. 2012). In addition, sympathetic axons appear to sprout axon collaterals that form baskets around the cell bodies of DRG neurons (Xia et al. 2011). In nerve injury models of neuropathic pain, this sympathetic sprouting is thought to contribute to nociception (Gibbs et al. 2008). Further studies are needed to determine the contribution of neuroanatomical remodeling of afferent pathways to the pain that accompanies IBD. Taken together, these studies point to significant changes in afferent function induced by colitis, with most leading ultimately to increased afferent traffic to the CNS. These changes are likely to contribute to the pathogenesis of visceral pain.
Conclusion
The innervation of the GI tract is complex and capable of impressive plasticity of form and function. Using the example of colitis, each of the three branches of the innervation of the colon undergo changes in structure, excitability, and neurotransmitter release that would contribute to the functional consequences of colitis. Given the myriad roles of the enteric innervation in modulating gut function and conveying visceral sensations, it is possible that some of the off-target effects of compounds on the GI tract are due to interactions with its innervation.
Footnotes
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: Crohn’s and Colitis Foundation of Canada and the Canadian Institutes of Health Research.