
Abstract
(+)-Usnic acid (UA) has been known to be a strong uncoupler, and mitochondrial and endoplasmic reticulum (ER)–related stresses are suggested to be involved in the mechanism of hepatotoxicity. However, it has not been clarified whether UA causes toxicity in other mitochondria-rich organs such as the heart. We elucidated whether UA induces cardiotoxicity and its mechanism. UA was orally administered to rats for 14 days, and laboratory and histopathological examinations were performed in conjunction with toxicogenomic analysis. As a result, there was no alteration in blood chemistry, whereas cytoplasmic rarefaction of myocardium was observed microscopically. This finding corresponded to the swollen mitochondria observed ultrastructurally. Immunohistochemically, expression of prohibitin, indicating mitochondrial imbalance, increased in the sarcoplasmic area. Toxicogenomic analysis highlighted the upregulation of gene groups consisting of oxidative stress, ER stress, and amino acid limitation. Interestingly, the number of upregulated genes was larger in the amino acid limitation-related gene group than that in other groups, implying that amino acid limitation might be one of the sources of oxidative stress, not only mitochondria and ER-originated stresses. In conclusion, the heart was manifested to be one of the target organs of UA. Mitochondrial imbalance with complex stresses may be involved in the toxic mechanism.
Keywords
Introduction
(+)-Usnic acid (UA), 2,6-diacetyl-7,9-dihydroxy-8,9b-dimethyl-1,3(2H,9bH)-dibenzofurandione, is a naturally occurring dibenzofuran derivative found in several lichen species. It has been used for various chronic diseases, such as pulmonary tuberculosis, pain, fever, wounds, athlete’s foot, and other dermal lesions (Cocchietto et al. 2002; Ingolfsdottir 2002). UA agents have been marketed as dietary supplements in the United States to help weight loss, because they increase fat metabolism and raise the basal metabolic rate (Guo et al. 2008). The pharmacological effect of UA is explained by the uncoupling of oxidative phosphorylation in mitochondria, characterized as stimulated oxygen consumption and induction of maximal stimulation of respiration (Johnson, Feldott, and Lardy 1950; Pramyothin et al. 2004). However, severe hepatitis provoked by UA agents, Lipokinetix (Syntrax, Cape Girardeau, Missouri) or uncoupling protein-1 (BDC Nutrition, Richmond, Kentucky), was reported in humans, and the Food and Drug Administration issued a warning letter titled “FDA Warns Consumers Not to Use the Dietary Supplement Lipokinetix” in 2001 (Guo et al. 2008).
Subsequent elucidations about this pivotal toxicity have revealed that UA induces cytotoxicity (cell membrane damage) in hepatocytes in vitro, and reactive oxygen species production in mitochondria by the inhibition of the respiratory chain is considered critical (Han et al. 2004; Pramyothin et al. 2004). Several in vivo studies have also revealed that mitochondrial uncoupling and functional inhibition lead to generate oxidative stress and cell death (Liu et al. 2012; Lu et al. 2011). Additionally, it has been reported that mitochondrial swelling in the hepatocyte is induced by UA treatment in rats (Pramyothin et al. 2004). These studies support that mitochondrial dysfunction/damage is one of the main sources of this “oxidative stress” in the hepatocyte.
On the other hand, endoplasmic reticulum (ER) has been suggested as a primary target of UA. Downregulation of ERp29, one of the secretory processing proteins that expresses ubiquitously in ER, has been detected in proteomic analysis in UA-treated rat livers (Liu et al. 2012). ER stress is also considered an integral constituent of UA-induced hepatotoxicity.
As described above, UA-induced toxicity has been studied mainly in the liver. On the other hand, toxic effects by UA against other organs are not well known, whereas necrosis of skeletal muscle has been reported in animals (Cook et al. 2007; Dailey et al. 2008). Especially, organs that contain rich mitochondria, including the heart, have not been investigated. In the present study, we tested UA-induced myocardial toxicity in rats. Detailed histopathology was performed in conjunction with toxicogenomic analysis to elucidate the presence of lesion and toxic mechanisms.
Material and Method
Animals and Experimental Design
A total of 27 female F344/DuCrlCrlj rats aged 7 weeks were purchased from Charles River Japan, Inc. (Kanagawa, Japan). The animals were housed under controlled conditions (24 ± 2°C temperature, 40–70% relative humidity, and a 12:12 light–dark cycle) and fed a standard laboratory diet and water ad libitum during the experimental period. Animals were randomly divided into 3 groups of 9 animals each and treated orally with 30 and 100 mg/kg of UA (Wako Pure Chemical Industries, Ltd., Osaka, Japan) or vehicle (0.5% methylcellulose, Wako Pure Chemical Industries, Ltd.) once daily for 14 consecutive days. The higher dose level was set based on preliminary data where animals became moribund or died after 4 days’ treatment with UA at 300 mg/kg (data not shown), and the lower dose was set to find dose dependency. The first day of administration was defined as day 1. The animals in each group were further subdivided into 2 groups for laboratory examinations and histopathology (5 animals) and electron microscopy and toxicogenomic analysis (4 animals). Each group was defined as toxicity and satellite groups, respectively. All of the animals were observed for general conditions once daily throughout the experimental period. Body weights were measured on days 1, 4, 8, 11, and 15. Food consumption was measured weekly per each cage and was divided by the number of animals in the cage to express the group mean value. On the day after the final administration (day 15), blood was collected from the abdominal aortas of rats under isoflurane anesthesia. Using the serum obtained, creatine kinase (CK), lactate dehydrogenase (LDH), aspartate aminotransferase (AST), and alanine aminotransferase (ALT) were determined with an automatic analyzer (H7180, Hitachi High Tech, Tokyo, Japan), and troponin-T was measured by quantitative rapid immunoassay with a coding chip (Cardiac Reader, Roche Diagnostics, Tokyo, Japan). All animals were euthanized by exsanguination under isoflurane anesthesia. The experimental protocol was approved by the Ethics Review Committee for Animal Experimental of Daiichi Sankyo Co., Ltd.
Light Microscopy and Immunohistochemistry
The hearts of the toxicity group were excised, macroscopically examined, and weighed. As a relative heart weight, heart-to-body weight was calculated. The tissue was fixed in 10% neutral buffered formalin. The tissue was longitudinally trimmed, embedded in paraffin, sectioned at a thickness of 4 μm, and stained with hematoxylin and eosin. Immunohistochemistry for prohibitin, to detect the imbalanced function of mitochondria, was performed using a 2-step peroxidase 3,3′-diaminobenzidine staining technique with a DAKO Envision+ Kit (DAKO Japan, Tokyo, Japan) according to the manufacturer’s instructions. Mouse monoclonal anti-prohibitin (Clone II-14-10, DAKO Japan) was used at 1:75 dilution. The sections were counterstained with hematoxylin to visualize nuclear stainability of myocardium.
Electron Microscopy
The left ventricles of the hearts in all animals of the satellite group were excised and cut into a few small pieces for electron microscopy (approximately 1 × 1 × 1 mm in size) immediately after exsanguination (within 3 min). The tissues in the central portion of the left ventricle (in the middle of the apex and base and the middle layer of the myocardium, between the endocardium and epicardium) were minced, fixed in 2.5% glutaraldehyde in 0.1 M sodium phosphate buffer, postfixed in 0.1 M sodium phosphate buffered 2% osmium tetroxide, dehydrated through a graded ethanol series, and embedded in epoxy resin. Sections at approximately 1-μm thickness were prepared from epoxy resin tissue-bearing blocks, stained with toluidine blue to identify the target area. Ultrathin sections were cut from each of the two selected blocks of each animal, stained with uranyl acetate and lead citrate, and examined by an electron microscope.
RNA Extraction
An aliquot of the sample (approximately 100 mg) for microarray analysis was obtained from the left ventricle tissue remaining after the sampling for electron microscopy in the satellite group. The tissues were collected immediately after exsanguination (within 5 min) and wiped softly to reduce blood contamination. The samples were immediately frozen in liquid nitrogen and stored at −80°C until analysis. Heart samples were homogenized with the TRIzol® Reagent (Life Technologies, Carlsbad, California), and the total RNA was isolated according to the manufacturer’s instructions. The total RNA was treated with deoxyribonuclease I (Takara, Shiga, Japan) in the manufacturer’s buffer supplemented with 10 mM dithiothreitol, 0.5 mM deoxyribonucleoside triphosphates, and 40U RNase inhibitor (Toyobo Life Science, Tokyo, Japan).
DNA Microarray Analysis
Microarray analysis was performed using a GeneChip® IVT Express kit (Affymetrix Inc., Santa Clara, California) for complementary DNA (cDNA) synthesis, purification and synthesis of biotin-labeled cDNA according to the manufacturer’s instructions. Every biotin-labeled complementary RNA target sample (approximately 10 μg) was individually hybridized to a GeneChip® Rat Genome 230 2.0 Array (Affymetrix, Inc.) at 45°C for 16 hr followed by washing and staining with streptavidin–phycoerythrin using Fluidics Station 400 (Affymetrix, Inc.). The scanned image was analyzed with a Microarray Suite 5 (MAS5) algorithm using genechip operating software (Affymetrix, Inc.). All the MAS5-analyzed data were scaled by global normalization.
The UA-induced up- and downregulated genes were selected by the following criteria: (1) the mean signal intensity values were at least 1.5-fold higher/0.67-fold lower than the concurrent control value and (2) statistical significance from the concurrent control with a p value less than .05 (Student’s t-test). Gene information (molecular activity and biological function) was extracted from the Gene Ontology website (http://www.geneontology.org). The Entrez IDs and the Gene symbols of each gene were retrieved from the website of the National Center for Biotechnology Information (http://www.ncbi.nlm.nih.gov/).
Real-time Quantitative Reverse Transcriptase–Polymerase Chain Reaction (RT-PCR) Analysis
Of the upregulated genes in microarray analysis, 20 genes were selected to be analyzed with a real-time quantitative RT-PCR. They were classified into 3 groups based on their functions: oxidative stress, ER stress, and amino acid limitation–related genes including several kinds of amino acyl tRNA synthetase (aaRS). TaqMan Gene Expression Assays were used as probes and primers and were summarized in Supplementary Table 1. The quantitative polymerase chain reactions (qPCR) were performed using qPCR Mastermix Plus (Eurogentee, Philadelphia, Pennsylvania). The reactions were performed at 50°C for 2 min and 95°C for 10 min, with amplification in 40 cycles of 95°C for 15 s and 6°C for 1 min. Glyceraldehyde-3-phosphate dehydrogenase (Applied Biosystems, Foster city, California) was used as an endogenous control.
Statistical Analysis
All the quantitative data except for food consumption were expressed as the group mean ± standard deviation (SD). Values for body weights, heart weights, and real-time quantitative RT-PCR were employed with Dunnett’s multiple comparison tests. In microarray analysis, the data were analyzed by an F-test to evaluate the homogeneity of variance. If the variance was homogeneous, a Student’s t-test was applied. If the variance was heterogeneous, an Aspin–Welch’s t-test was performed. A p value less than .05 (two-tailed) was considered significant.
Results
Laboratory Examinations
No abnormal clinical signs were observed during the experimental period. There was no significant difference in body weights between UA-treated and the control groups, whereas increased tendency in food consumption was seen at 100-mg/kg/day group on days 7 to 8 and 14 to 15 (Table 1). CK and LDH decreased at 30 and 100 mg/kg/day; however, they were not considered toxicologically significant changes (data not shown). Heart weight, AST, ALT, and troponin-T did not show significant changes.
Body weight and food consumption of female rats receiving 14-day oral administration of (+)-usnic acid (UA).

Note. aMean ± SD. bMean.
Pathological Examination and Immunohistochemistry
Macroscopically, no abnormal findings were found in the heart and other thoracic and abdominal organs. Under the microscope, myocardial cytoplasm showed diffuse rarefaction in the septum and ventricle in the all animals of the UA-treated group at 100 mg/kg/day. Morphologically, the lesion was characterized as a scattered myofibrils and extended sarcoplasmic area in the cytoplasm (Figure 1). Immunohistochemistry for prohibitin revealed an increased staining intensity in the myocyte of this group. The change was seen diffusely in the myocardium, although was more apparent in the middle layer of the left ventricle (Figure 2A and B). Increased expression of prohibitin was detected as an increased number of positive granules and corresponded to the sarcoplasmic area in myocytes (Figure 2C and D). In electron microscopy, swollen mitochondria were noted in the myocyte of all the animals in the UA-treated group at 100 mg/kg/day. An electron-lucent mitochondrial matrix and discrete cristae were also observed (Figure 3A and B). Neither the number of mitochondria nor ER showed apparent change compared to that in the control group.
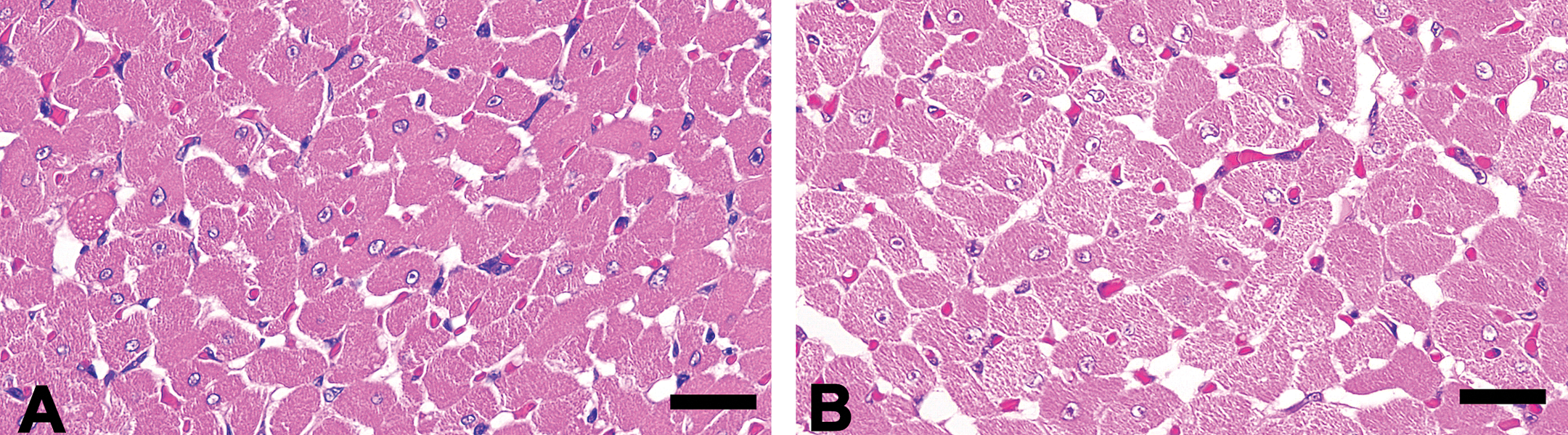
Histological appearance of the left ventricular myocardium. The control animal (A). The animal in the 100-mg/kg/day group. The cytoplasmic rarefaction is observed in the myocardiocyte (B). Hematoxylin and eosin staining. Bars: 25 μm.
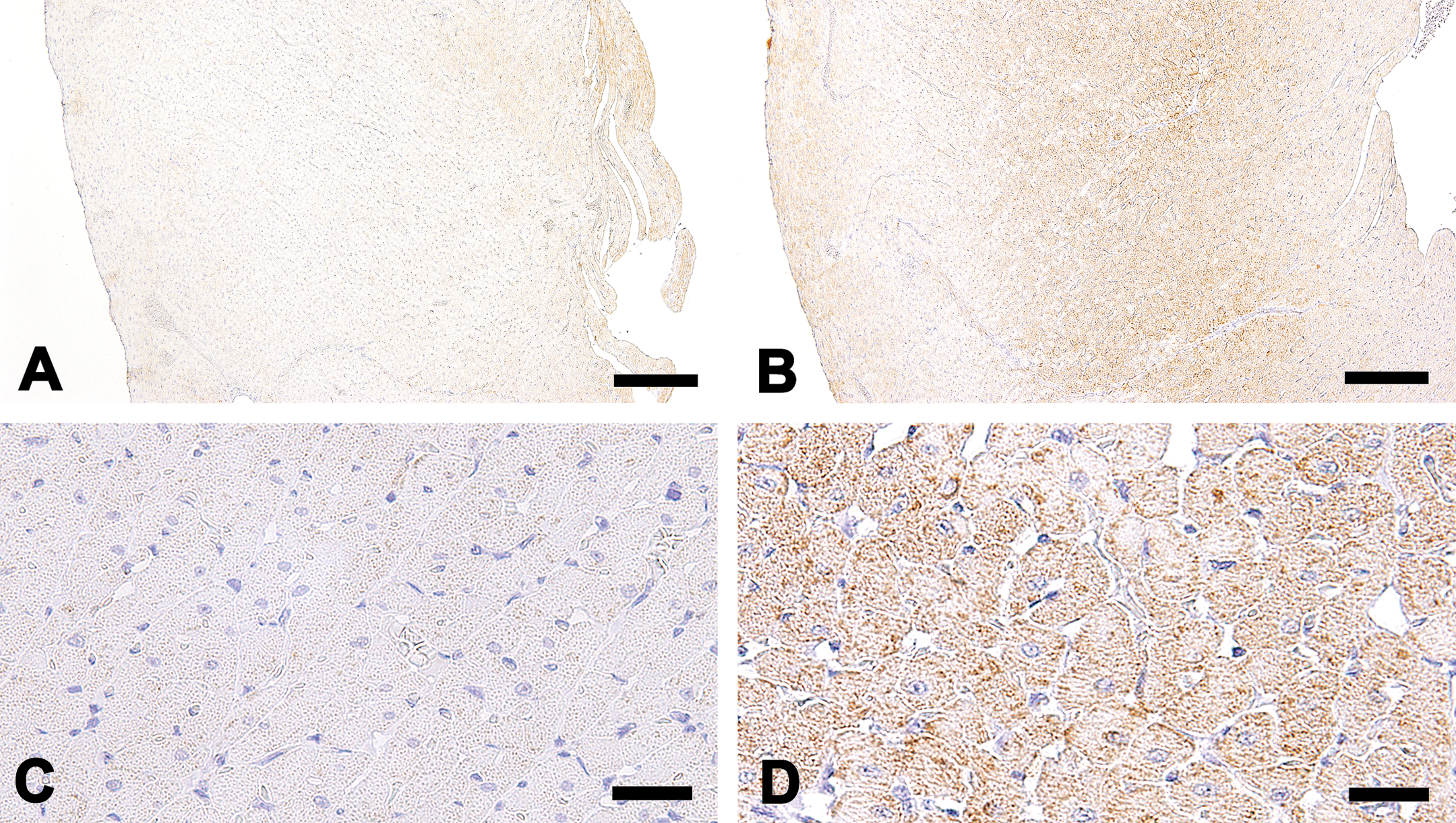
Immunohistochemistry for prohibitin in the left ventricular myocardium. The control animal (A). The animal in the 100-mg/kg/day group. Increased intensity in the myocardium is observed especially in the middle layer of left ventricle (B). The control animal (C). The animal in the 100-mg/kg/day group. Increased intensity and number of positive granules are localized in the sarcoplasmic area (D). Bars: A and B (200 μm) and C and D (25 μm).
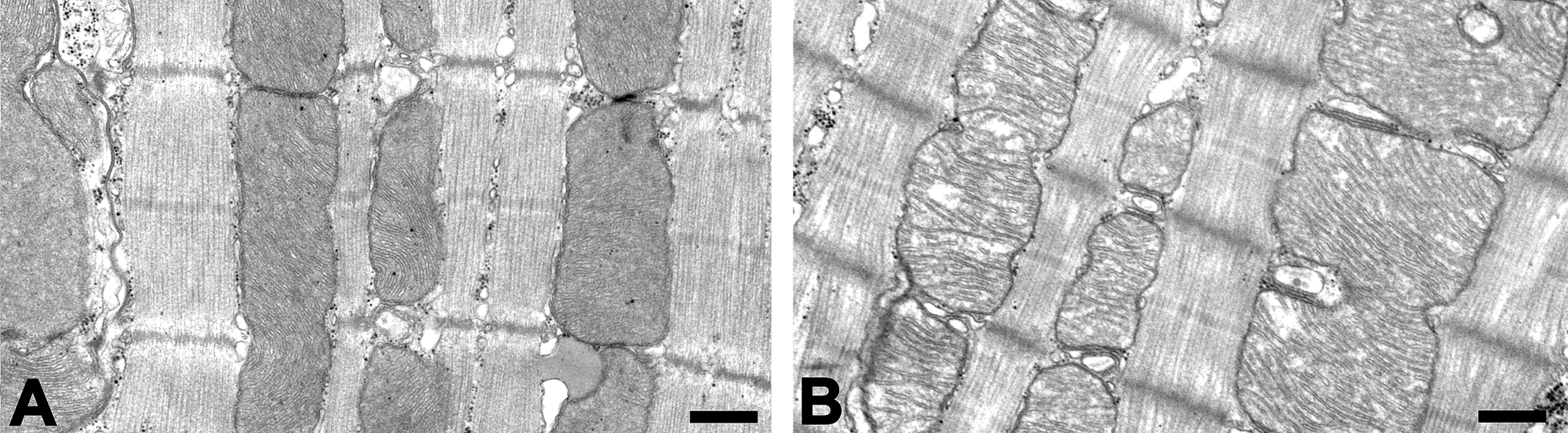
Representative ultrastructural appearance of the left ventricular myocardium. The control animal (A). The animal in the 100-mg/kg/day group. Swollen mitochondria, electron-lucent mitochondrial matrix, and discrete cristae are observed (B). Bars: 500 nm.
Analysis of UA-induced Gene Expressions
In DNA microarray analysis, 222 genes were upregulated more than 1.5-fold and 385 genes were downregulated less than 0.67-fold in the UA-treated groups. All results of DNA microarray analysis are attached as supplemental data (Supplementary Tables 2 and 3). Based on the results of microarray analysis and the previous reports of UA-induced hepatotoxicity (Das et al. 2011; Hitomi et al. 2004; Liang et al. 2013; Ohoka et al. 2005; Wang et al. 1996), upregulated and representative genes (NAD(P)H dehydrogenase, quinone 1 [Nqo1] and thioredoxin reductase 1 [Txnrd1] for oxidative stress; tribbles homolog 3 (Drosophila) [Trib3], caspase 4, apoptosis-related cysteine peptidase [Casp4], and DNA damage inducible transcript 3 [Ddit3] for ER stress) were selected for measurement by real-time RT-PCR. Additionally, three genes (methylenetetrahydrofolate dehydrogenase (NADP+ dependent) 2 [Mthfd2], asparagine synthetase [Asns], and activating transcription factor 5 [Atf5]), which showed high increasing rates and 12 of the upregulated genes of aaRS were added to find a broad spectrum of amino acid limitation. The selected genes are shown in Table 2 (5 genes were redundant). The fluctuations of these genes corresponded to those in the microarray analysis, showing increases in the UA-treated group at 100 mg/kg/day (Figures 4, 5, and 6A and B).
List of genes upregulated in DNA microarray in the heart of female rats receiving 14-day oral administration of (+)-usnic acid.
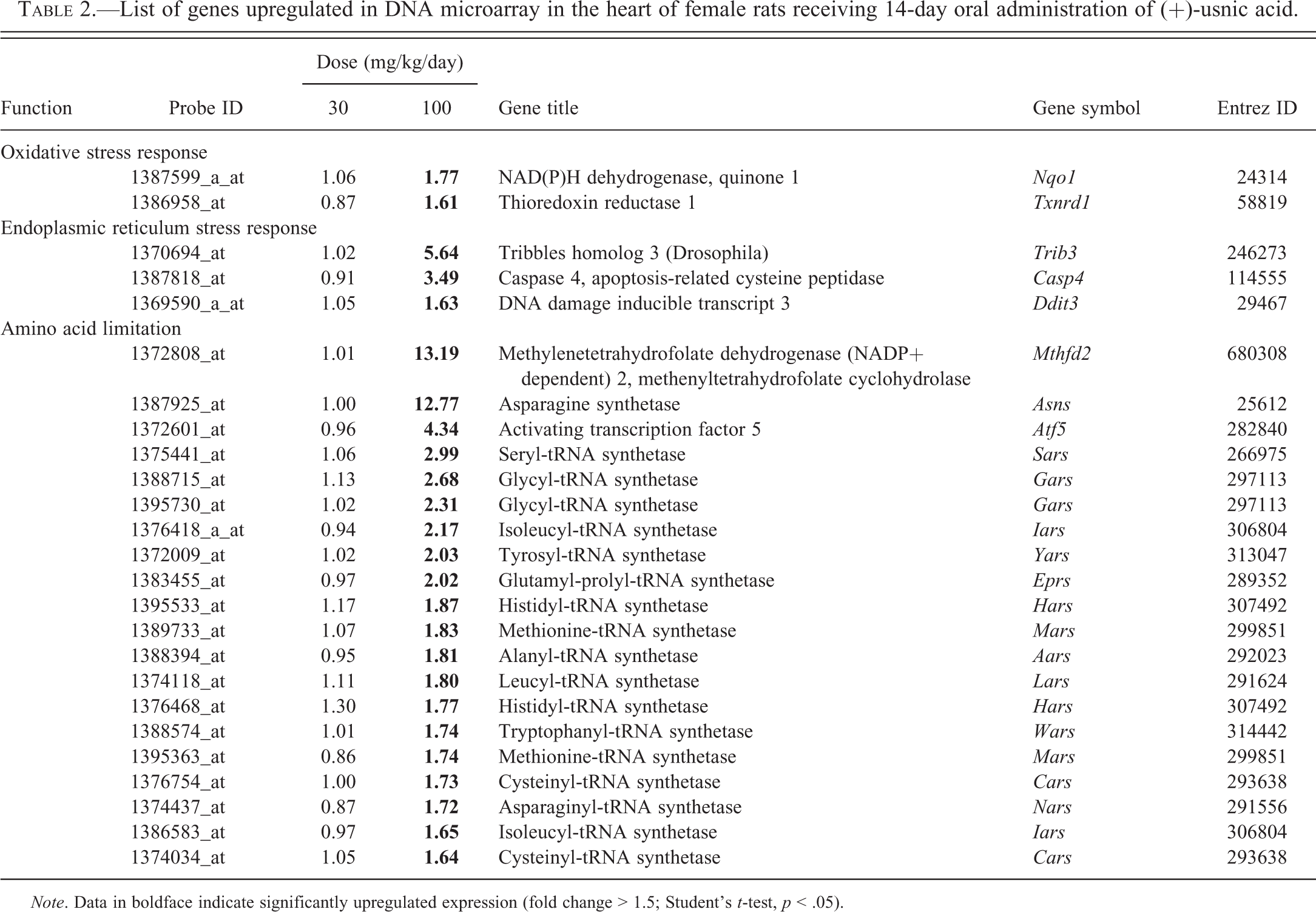
Note. Data in boldface indicate significantly upregulated expression (fold change > 1.5; Student’s t-test, p < .05).
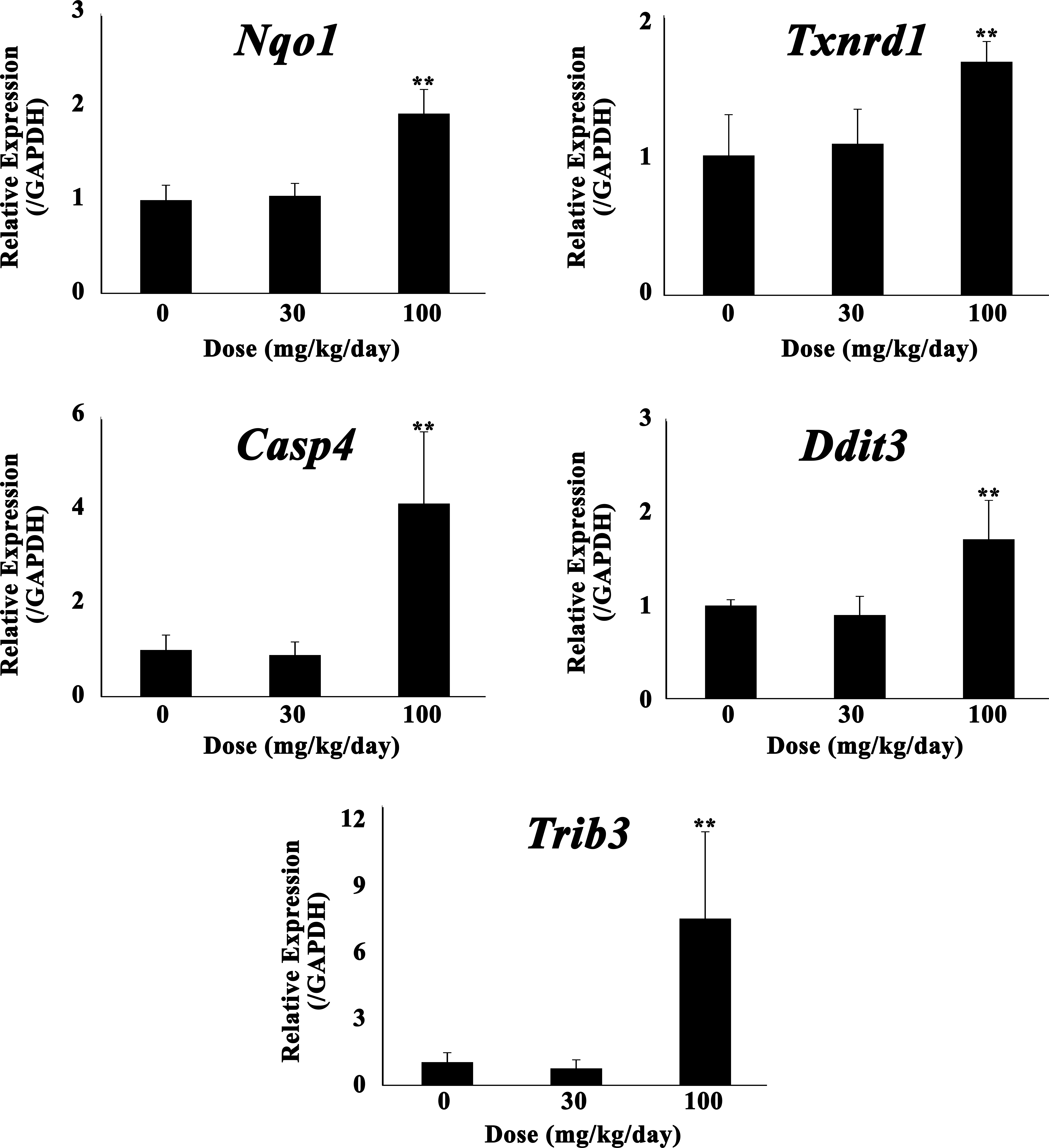
Myocardial gene expression changes of oxidative stress (Nqo1 and Txnrd1) and endoplasmic reticulum stress (Casp4, Ddit3, and Trib3)-related genes by quantitative real-time RT-PCR after 14-day oral administration of UA. Each column represents the mean ± SD of the increased mRNA expression levels in the hearts of 5 rats given 0, 30, or 100 mg/kg/day of UA. *p < .05. **p < .01: significantly different from the control group (Dunnett’s multiple comparison test). mRNA = messenger RNA; RT-PCR = reverse transcriptase–polymerase chain reaction; SD = standard deviation; UA = (+)-usnic acid.
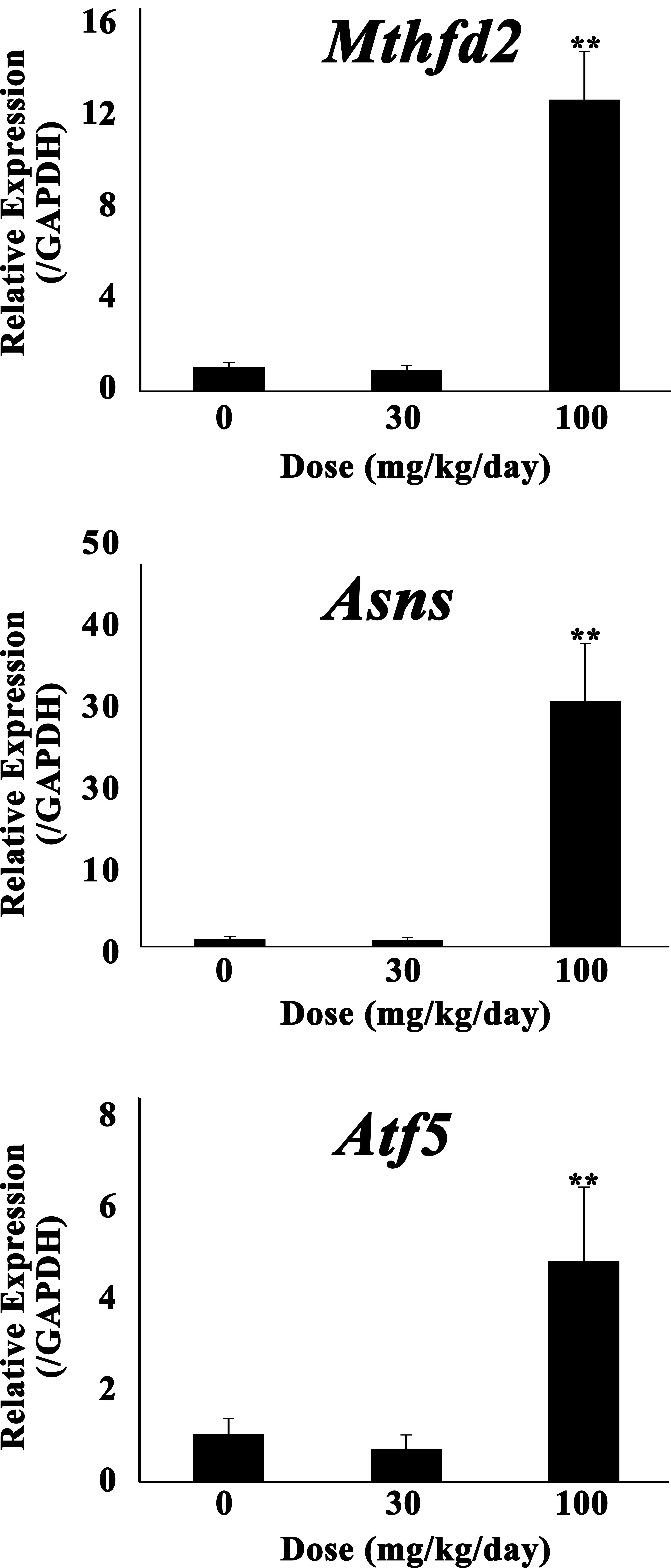
Myocardial gene expression changes of amino acid limitation–related genes (Mthfd2, Asns, and Atf5) by quantitative real-time RT-PCR after 14-day oral administration of UA. Each column represents the mean ± SD of the increased mRNA expression levels in the hearts of 5 rats given 0, 30, or 100 mg/kg/day of UA. *p < .05. **p < .01: significantly different from the control group (Dunnett’s multiple comparison test). mRNA = messenger RNA; RT-PCR = reverse transcriptase–polymerase chain reaction; SD = standard deviation; UA = (+)-usnic acid.
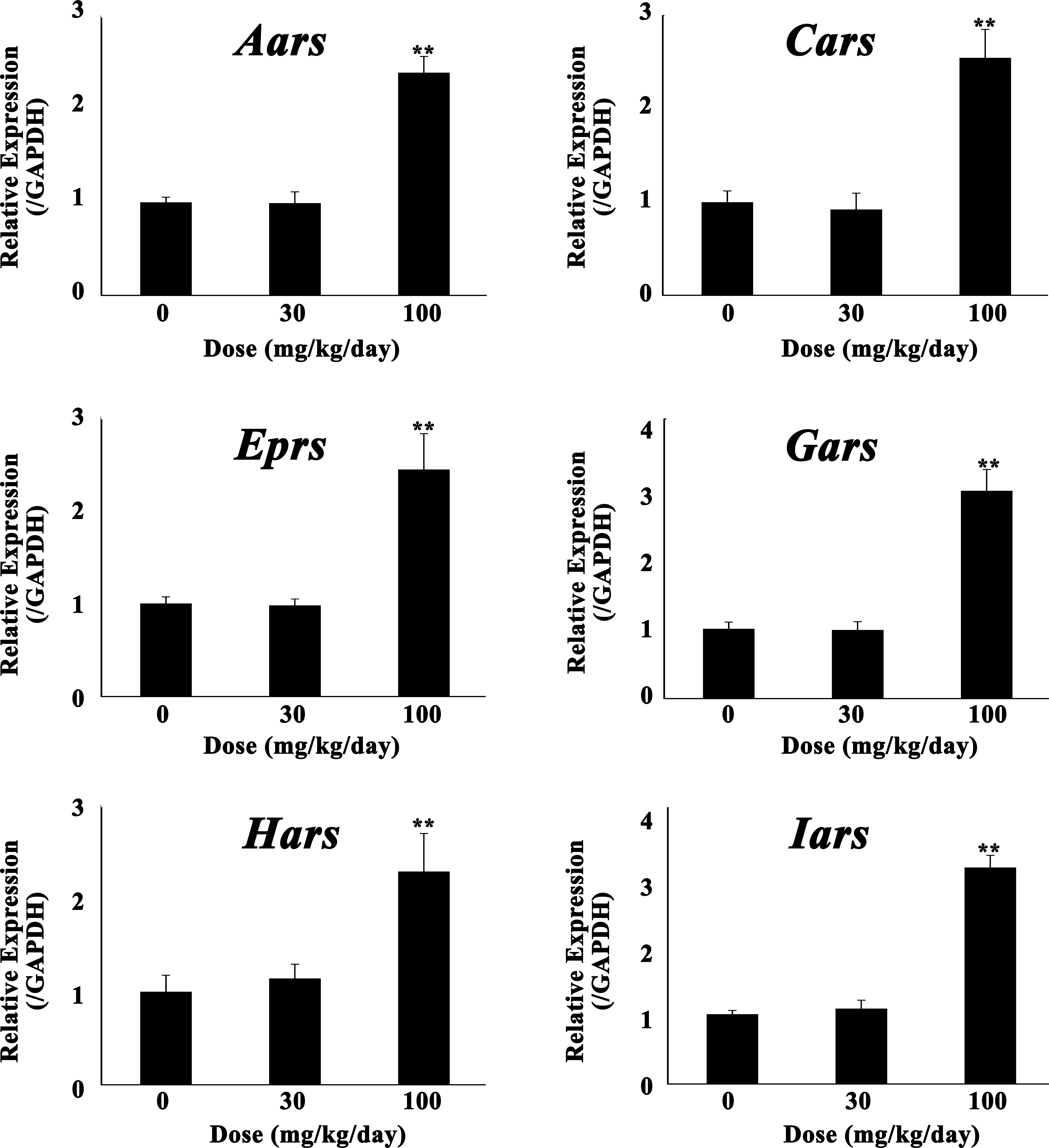
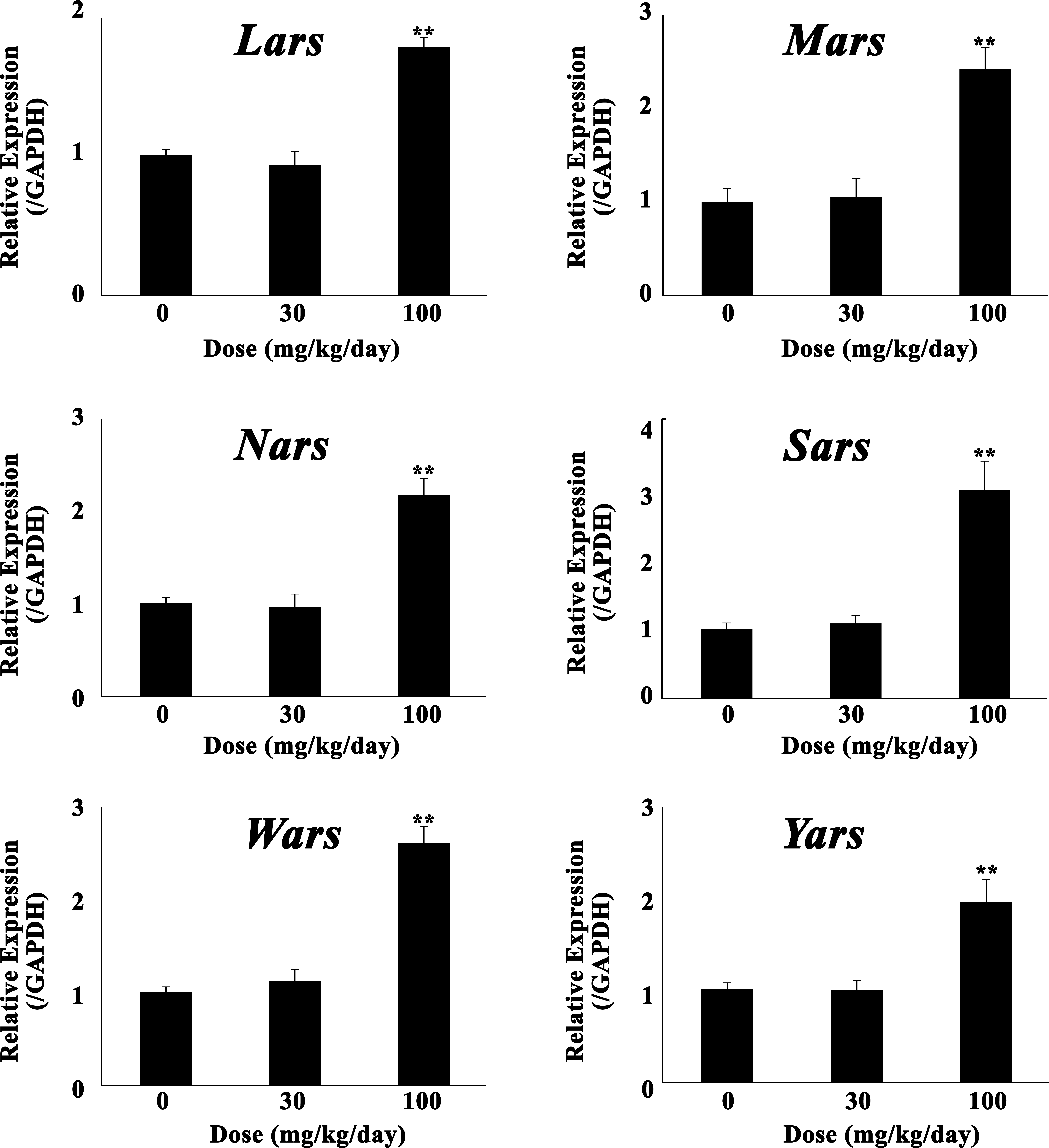
Myocardial gene expression changes of amino acyl tRNA synthetases ([A] Aars, Cars, Eprs, Gars, Hars, and Iars and [B] Lars, Mars, Nars, Sars, Wars, and Yars) by quantitative real-time RT-PCR after 14-day oral administration of UA. Each column represents the mean ± SD of the increased mRNA expression levels in the hearts of 5 rats given 0, 30, or 100 mg/kg/day of UA. *p < .05. **p < .01: significantly different from the control group (Dunnett’s multiple comparison test). mRNA = messenger RNA; RT-PCR = reverse transcriptase–polymerase chain reaction; SD = standard deviation; UA = (+)-usnic acid.
Discussion
In the present study, 14-day administration of UA provoked swelling of mitochondria in the myocardiocyte at 100 mg/kg/day. It has been reported that UA-induced hepatocellular mitochondrial swelling is not accompanied by increased transaminases (Pramyothin et al. 2004). Since there were no changes in AST, ALT, and troponin-T in the present study, the toxic effect of UA was considered to be less or comparable with that in the liver reported previously, whereas pathological examination was not performed in the liver. The present study revealed the heart was one of the target organs of UA, and the morphological characteristic, swelling of mitochondria, was similar to the liver. Although a high concentration has been detected in the lung and liver, tissue distribution of UA in the heart was relatively low in rats (Krishna, Ramana, and Mamidi 1995). The present result possibly implies the heart is susceptible to UA-induced toxicity. On the contrary, no remarkable alterations were observed in any examinations including toxicogenomics in the 30-mg/kg/day group. Systemic exposure of UA was not obtained in the present study, and it was unclear whether dose-dependent and/or enough exposure was achieved at 30 mg/kg/day. Although no toxicokinetic information has been available, it would be worthy to explore to identify UA’s toxicity as a whole picture.
Immunohistochemically, increased intensity and the number of positive granules for prohibitin were observed in the myocardium. Prohibitin is an intracellular protein, predominantly localized in the mitochondrial inner membrane, and has been known to regulate the membrane and cristae structure, protein degradation, protein/lipid scaffolds, and cellular proliferation (Artal-Sanz and Tavernarakis 2009; Merkwirth and Langer 2009; Nijtmans et al. 2002; Osman, Merkwirth, Langer 2009; Shibanuma et al. 2011; Zhou et al. 2012). Imbalanced oxidative phosphorylation and excessive formation of reactive oxygen species lead to an increased expression in prohibitin (Artal-Sanz and Tavernarakis 2009; Lee, Arnouk, et al. 2010). For example, prohibitin expression increases in pancreatic β cells under an ethanol-induced oxidative stress condition (Lee, Nguyen, et al. 2010). Prohibitin has been proposed as an oxidative stress biomarker in the retina of an aged or diabetic mice model (Lee, Arnouk, et al. 2010). The information strongly supports the speculation that increased expression of prohibitin was a reactive response to an oxidative stress generated from mitochondria.
In the previous reports, there has been no detailed information about food consumption in UA-treated animals. However, in humans who take UA, decreased body weight is known, and it is explained by UA’s pharmacological effect as an uncoupler. UA reduces the production of adenosine triphosphate and promotes thermogenesis with increased calorie consumption by an uncoupling effect of oxidative phosphorylation (Hsu et al. 2005). The increased tendency of food consumption was considered a compensatory response to sustain their body weight gain against the weight loss effect. With regard to the decrease of CK and LDH, the reason was not explained. There has been no report showing the relationship between UA treatment and alterations in CK and LDH mechanistically.
Based on DNA microarray and quantitative real-time RT-PCR analyses, oxidative stress was proven to be involved in this cardiotoxicity, same as the liver. Both Nqo1 and Txnrd1 are well known as antioxidant genes that can be induced under an oxidative stress condition. Upregulation of these was consistent with the morphological and immunohistochemical findings in the present study. On the other hand, Nqo1 has been reported as an ER stress preventer in several tissues, such as benzo[a]pyrene-treated enterocytes and aminochrome-treated neurons (Bock 2012). Nqo1 might represent a part of ER stress in addition to oxidative stress.
While an ER-related histological/ultrastructural abnormality was not obvious, typical ER stress–related genes such as Casp4, Ddit3, and Trib3 were upregulated in our work. Casp4 is one of the ER stress-specific caspase in humans and is suggested to be involved in the pathogenesis of Alzheimer’s disease (Hitomi et al. 2004). Upregulated Ddit3 and Trib3 (a putative target gene of the Ddit3 transcription factor) have also been reported under various ER stress conditions (Ohoka et al. 2005; Wang et al. 1996). In addition, Ddit3 is upregulated when oxidative and/or mitochondrial stresses are evoked (Ariyama et al. 2008; Zhang, Yang, and Cohen 1999; Zhao et al. 2002). Moreover, Casp4 and Trib3 have been suggested to be related to apoptosis of the cell. In humans, it has been reported that ER stress-induced activation of an unfolded protein response mediates Casp4 and caspase-3 activation and apoptosis in the human brain of infantile neuronal ceroid lipofuscinosis (Kim et al. 2006). Additionally, induction of Trib3 is regulated by the PERK-ATF4-CHOP pathway and mediates apoptosis (Hegedus, Czibula, and Kiss-Toth 2007; Ohoka et al. 2005). Genetic fluctuations in the present study possibly indicate that myocardiocyte was confronted with a pre-apoptotic status and that ER stress was involved in UA-induced toxicity in the heart to some extent. In the liver, dilated ER in the hepatocyte has been reported in rats (Pramyothin et al. 2004). This finding is seen only at a high dose level accompanying severe mitochondrial swelling, implying that mitochondria have higher susceptibility than ER against UA-induced toxicity. Our results where ER showed no apparent histological change might support the above hypothesis; however, the present results are not sufficient to determine the reason for the lack of changes occurring in ER. Further investigations in different durations and/or dose levels will be required.
Of the large number of upregulated genes, 15 genes (Mthfd2, Asns, Atf5, alanyl-tRNA synthetase [Aars], cysteinyl-tRNA synthetase [Cars], glutamyl-prolyl-tRNA synthetase [Eprs], seryl-tRNA synthetase [Gars], histidyl-tRNA synthetase [Hars], isoleucyl-tRNA synthetase [Iars], leucyl-tRNA synthetase [Lars], methionine-tRNA synthetase [Mars], asparaginyl-tRNA synthetase [Nars], glycyl-tRNA synthetase [Sars], tryptophanyl-tRNA synthetase [Wars], and tyrosyl-tRNA synthetase [Yars]) were classified into amino acid regulators or their catalyzers and were shown to increase quantitatively. Mthfd2 catalyzes the conversion of folic acid intermediates and is considered necessary for the synthesis of purines to support growth during embryogenesis (Di Pietro, Wang, and MacKenzie 2004). It is known as a typical starvation reactive gene: Mthfd2 is induced by starvation in cultured myotubes (Stevenson et al. 2005). Asns is induced in total amino acid deprivation or depletion of a single essential amino acid (Hutson and Kilberg 1994). Atf5, regulated by the alternative 5′-untranslated regions of its messenger RNA (mRNA), is a transcription factor in the cyclic adenosine monophosphate response element binding protein/activating transcription factor family, and its mRNA expression is regulated by amino acid limitation during the post-transcription level (Watatani et al. 2007). The other 12 genes are categorized as aaRS. AaRS consists of 2 groups (classes I and II) and catalyzes esterification of a specific amino acid or its precursor. Upregulated genes belong to both of the classes. Furthermore, Ddit3 is also induced by amino acid limitation both at transcriptional and at post-transcriptional levels (Bruhat et al. 1997). It was suggested that the metabolism or absorption in a variety of amino acids were inhibited.
This phenomenon is considered due to the uncoupling effect of UA. Disturbance of amino acid metabolism by high energy demand (as uncoupler) for increasing gluconeogenesis has been reported in the metabolomics analysis of UA-treated livers in rats (Lu et al. 2011). Moreover, the upregulation of skeletal muscular Mthfd2 and Asns has been reported in a mouse mitochondrial myopathy model (Tyynismaa et al. 2010). Respiratory chain deficiency induces a mitochondrial stress response with gene fluctuation mimicking starvation; however, it is still possible to consider that UA directly inhibits cellular transport of amino acids. Bacterial amino acid transport is known to be inhibited by uncouplers in yeast (Nicholas and Ordal 1978). In addition, starvation of essential amino acids has been known to induce oxidative stress and apoptosis in vitro (Eisler, Frohlich, and Heidenreich 2004). While the magnitude of the contribution of amino acid limitation to oxidative stress response was undetermined, amino acid limitation in myocardiocyte was considered to contribute to a part of oxidative stress. Sequential detection of intracardiac and serum amino acid concentrations combined with metabolomic analysis may provide detailed information.
In a different compound that has a membrane uncoupling effect, similar mitochondrial findings have been demonstrated. Bupivacaine, a local anesthetic agent, has been reported to induce mitochondrial swelling ultrastructurally with reversible PR interval and QRS prolongation in Langendorff-perfused guinea pig model (Hiller et al. 2013). Moreover, an in vitro study indicates the involvement of ER stress and mitochondrial dysfunction to bupivacaine-induced apoptosis in neuroblastoma-derived SH-SY5Y cell (Li et al. 2013). The information seems to be consistent with the present study and there might be common mechanisms in uncouplers. In addition, the UA-induced functional effect on the heart is a matter of interest. In conclusion, the heart was revealed to be one of the primary targets of UA in the present study. Histopathological features were mitochondrial swelling and increased intensity and the number of positive granules for prohibitin in the myocardiocyte. By means of the findings and results in the toxicogenomic analysis, the imbalanced mitochondrial function and oxidative stress were considered to be involved in the toxic mechanism. Additionally, ER stress and amino acid limitation-related genes fluctuated, suggesting their pivotal role in UA-induced cardiotoxicity. Further studies are necessary to clarify detailed mechanisms of toxicity including an initial and developed status.
Footnotes
Supplementary Material
Supplemental Table S1. List of TaqMan® Gene Expression Assays for real-time quantitative RT-PCR analysis.
Supplemental Table S2. Upregulated genes in female rats received 14-day oral administration of (+)-usnic acid.
Supplemental Table S3. Downregulated genes in female rats received 14-day oral administration of (+)-usnic Acid.
Acknowledgment
The authors are grateful to Keiko Okado, Daiichi-Sankyo RD NOVARE Co., Ltd., for her technical assistance in the slide preparation for electron microscopy and to Mai Takahashi, Daiichi-Sankyo Co., Ltd., for her technical assistance in the animal experiment.
The author(s) declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
The author(s) received no financial support for the research, authorship, and/or publication of this article.