
Abstract
Mitochondria, endoplasmic reticulum (ER), cytoplasmic lipid droplets (CLD), and Golgi vesicles use cross talk to control hepatocyte metabolism, growth, and stress. Interpretation of ultrastructural change requires knowledge of how cross talk pathways function, how differential activation of hepatocellular signals influences organelle structure, and how organelles position themselves to become central hubs for stress responses. Mitochondria, by coupling energy production to pathways for protection, form critical platforms for innate signaling. Mitochondrial outer and inner membranes activate channels and signals to translocate peptides that drive oxidative phosphorylation, β-oxidation of fatty acids, and calcium ion (Ca2+) flux. In cell stress, mitochondrial signals initiate fusion and fission, reactive oxygen species (ROS) control, autophagy, apoptosis, and senescence. Specialized tethering proteins tie mitochondria to ER to support translocation of metabolites. For Ca2+ translocation, ER pores are connected to mitochondrial voltage-dependent anion channels, and for mitochondrial fission, unique membrane proteins pull ER to mitochondria. In toxic injury, cytosolic cytokines translocate to alter metabolism. Toxic effects on ER lipid synthesis lead to Golgi vesicle reduplication and transport of perilipin and other protein cargos into CLDs. How cellular proteostasis, oxidative homeostasis, and ion balance are maintained depend upon the effectiveness of mitochondrial ROS defense responses, unfolded protein responses in mitochondria and ER, and other organelle defenses.
Introduction
Hepatotoxic injury typically leads to marked ultrastructural alterations in mitochondria, endoplasmic reticulum (ER), cytoplasmic lipid droplets (LD), and endomembrane systems. Structural and functional changes in these organelles depend upon specific, and often overlapping, cross talk pathways that control hepatocyte metabolism, growth, stress, and repair. Interpretation of ultrastructural change requires knowledge of how cross-talk pathways function, how differential activation of hepatocellular signals influences organelle structure, and how organelles position themselves to become central hubs for stress responses. From the elucidation of pathways for cross talk, we are beginning to understand the global consequences of dysfunctional mitochondria. The capacity for adaptation, autophagy, or apoptosis serves to make the mitochondria–ER–cytoplasmic lipid droplets (CLD) axis a central hub for responses to cell stress. To cause disease, many toxins disrupt these pathways, and viruses and bacteria corrupt signal pathways and ubiquitylation machinery to regulate virulence.
Mitochondria: The Master Organelles
Mitochondria provide the enzymes and structural framework for generation of adenosine triphosphate (ATP) by the citric acid cycle and by the coupled reactions of electron transport and oxidative phosphorylation (OXPHOS). Because of the global need for ATP and the hazards arising from its production, there is a powerful mandate for mitochondria to control many major cellular metabolic systems. They do this by providing platforms from which innate signaling occurs. They are positioned in the cell for maximum interaction with other organelles (Figure 1). Repositioning of mitochondria in the cytoplasm, as with most other organelles, is effected by the cytoplasmic cytoskeleton. Microtubules and microfilaments control organelle movement both in the cytoplasm and in the nucleus since the two are connected by specific linker proteins embedded in nuclear envelope.
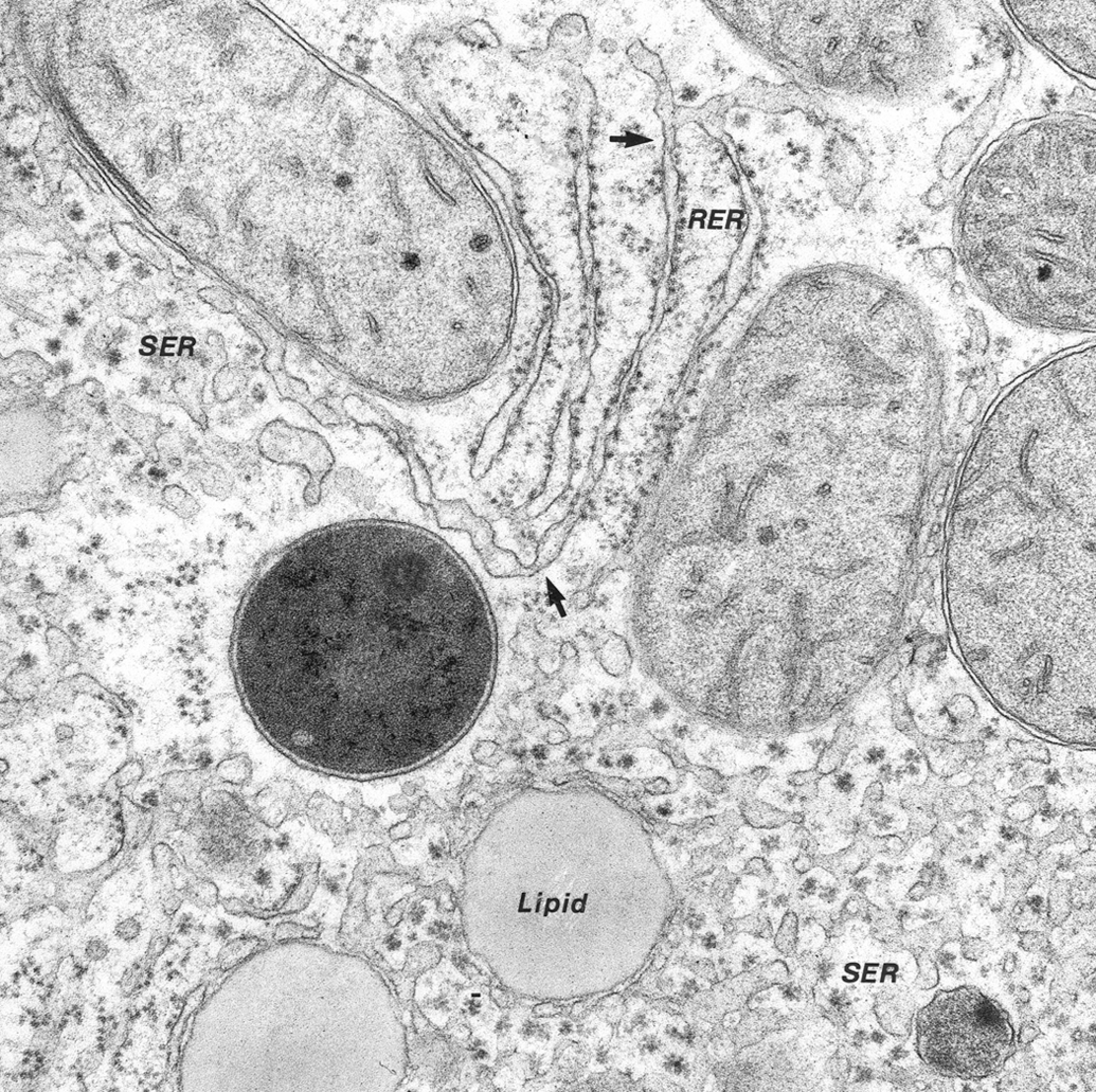
Cytoplasm: normal hepatocyte, dog. Relationships of rough endoplasmic reticulum (RER), smooth endoplasmic reticulum (SER), peroxisomes (lower right), mitochondria, and lipids. Cisternae of SER and RER communicate (arrows). Glycogen rosettes and lipid globules are associated with SER. Dense calcium-sequestering bodies are present in mitochondria (From Cheville 2009).
Mitochondrial DNA (mtDNA), which bears the genome for core OXPHOS proteins, is dispersed throughout the matrix packaged in histone-free mitochondrial nucleoids, DNA-protein complexes of ∼100 nm diameter. Super-resolution microscopy reveals mammalian nucleoids to be uniform in size and to contain single copies of mtDNA or clusters of genomes (Kukat et al. 2011). Mitochondrial transcription factor A is a major component of the mitochondrial nucleoid. Peptides produced by mitochondrial genes include at least 13 polypeptides that form the core of the OXPHOS complexes; ultrastructurally, these polypeptides have been localized to mitochondrial ribosomes in the matrix. Mutations in genes for OXPHOS complexes typically lead to abnormal mitochondria with whorled cristae (Cameron et al. 2011). Mitochondrial translation is specialized in expressing membrane proteins and couples protein synthesis to peptide insertion on the MIM (Perreault et al. 2009). Mitochondria also have regulatory RNA interference components similar to microRNAs, the small noncoding RNAs that associate with argonaute proteins to regulate gene expression at the posttranscriptional level in the cytoplasm (Bandiera et al. 2011).
Mitochondrial membranes are unique in their spectrum of receptors, pores, channels, and phospholipids. The membrane phospholipid makeup directs many transport functions. Movement of cholesterol into and out of the membrane bilayers can control many activities; for example, cholesterol accumulation in the MIM can reduce the efficiency of the respiratory chain, activation of OXPHOS, overproduction of ROS, and suppression of intrinsic antioxidant defenses. Caveolins, particularly CAV-1, bind cholesterol to regulate cholesterol flux in the membrane (Mastrodonato et al. 2012).
Cardiolipin is the signature membrane phospholipid molecule, and defects in cardiolipin lead to markedly abnormal mitochondria. Taffazin (Taz1p), a cardiolipin translocase, is required for normal cardiolipin function. Human Barth syndrome is an X-linked mitochondrial disorder arising from mutation in the TAZ1 gene for tafazzin; the disease has been duplicated in a tafazzin KO mouse model. Children with the TAZ1 mutation suffer dilated cardiomyopathy and delayed growth: ultrastructurally, their abnormal mitochondria have focal lesions in the MIM, linear zones of adhesions, often with large tubular structures that are derivatives of the adhesion zones (Acehan et al. 2007).
The MIM is impermeable to ions and most small molecules (except where a path is provided by membrane transport protein). The MIM is closely linked to the MOM. The noncristae part of the membrane bears integral membrane proteins that are dedicated to mitochondrial–cytosol transport. Osmotic balance is maintained by water transporting aquaporins. In hepatocytes, the aquaporin-8 channel is permeable to water and ammonia, and is regulated by the thyroid hormone T3.
The MOM is porous—freely permeable to ions; it maintains mitochondrial shape and structure. The MOM contains many porins that form wide aqueous channels through the lipid bilayer and is permeable to molecules <5,000 daltons (including small proteins). Within the lipid bilayer, lipid rafts, clusters of lipoprotein receptors, are critical for signaling events with the cytosol (Table 1). Voltage-dependent anion channels (VDAC) in the MOM are the common pathway for exchange of metabolites with cytosol; it is responsible for at least 4 major systems: ATP rationing, oxidative stress protection, calcium homeostasis, and apoptosis regulation. Electron microscopic data show that VDAC-1 is a cylindrical channel with a diameter of 2.6 to 3 nm; it contains β-barrel proteins for cargo transport and signaling (Hiller et al. 2008). Too small for passage of cytochromes, the formation of heterologomers of VDAC and Bax/Bak results in large pores and loss of cytochrome c (Cyt c). VDAC is an important component of the protein complex that transports and distributes cholesterol in mitochondrial membranes.
Mitochondrial membrane pores and signal molecules.

Note: MIM = mitochondrial inner membrane; MOM = mitochondrial outer membrane; VDAC, voltage-dependent anion channels.
MOM hormone receptors include those for thyroxin, steroid hormones, glucocorticoids, estrogens, and androgens; all have been identified by immunogold EM (Cheville 2009). Receptors signal the mitochondrial genome to produce the hormone response elements that regulate OXPHOS and ATP synthesis; for example, steroid hormones regulate energy production, inducing nuclear and mitochondrial OXPHOS gene transcription. Dexamethasone (DEX) specifically affects mitochondria in hepatocytes where it increases ATP synthesis by inducing OXPHOS enzymes increasing thermodynamic coupling and efficiency of OXPHOS (in liver, not muscle). β2-adrenergic receptors mediate cell protection via cross talk with mitochondrial apoptosis pathways (Fajardo et al. 2011).
Mitochondrial Permeability Transition (MPT)
Electrophysiologists talk of “mitochondrial membrane depolarization” that leads to an MPT, a calcium-dependent increase of mitochondrial membrane permeability and membrane depolarization (loss of mitochondrial membrane depolarization [ΔΨm]) that leads to leakage of mitochondrial proteins, matrix swelling, and rupture of the MOM. MPT is actually a cooperative event regulated by opening and closing of multiple pores/channels in both MOM and MIM. These pores are referred to collectively by electrophysiologists as the mitochondrial permeability transition pore (mPTP). In reality, ultrastructural studies reveal that the mPTP includes VDAC in the MOM, the adenine nucleotide translocator (ANT) in the MIM, and cytophilin D (CypD), a mitochondrial peptidyl prolyl-cis, trans-isomerase located in the mitochondrial matrix. Binding of CyPD to ANT converts ANT from a specific transporter to a nonspecific pore. These channels and many others cooperate to maintain osmotic balance and permeability. Uncontrolled opening of mitochondrial transition pores causes cell death. Furthermore, misfolded proteins associated with mitochondria have been shown to clog the permeability transition pore; for example, lodgment in and obstruction of transport pores by amyloid precursor protein leads to mitochondrial dysfunction.
VDAC controls calcium ion (Ca2+)-induced MPT, and when hypereactive mitochondria release O2 − from intramitochondrial spaces to the cytosol, ROS augment Ca2+-induced MPT. Closure of VDAC by drugs impedes O2 − efflux from intermembrane space (IMS); this increases oxidative stress and sensitizes mitochondria to Ca2+-induced MPT. During the MPT, translocation of proteins from cytosol to mitochondria also occurs; one of these, the E3 ubiquitin ligase PARKIN, controls degradation of membrane proteins. PARKIN activates degradation of MOM and MIM proteins differently through proteasome- and mitophagy-dependent pathways; it causes (1) mitophagy of inner membrane and matrix and (2) proteasome-dependent degradation of outer membrane proteins Tom 20, 40, and 70, and Omp25.
Mitochondrial Shape
Mitochondrial shape can shift between small fragmented units and large networks of elongated mitochondria (Figure 2). This shift is determined by fission (fragmentation) and fusion reactions that continually reshape mitochondria (Iwasawa et al. 2011). Fission and fusion are catalyzed by large dynamin-related proteins (Drp). Mitochondrial fusion requires the coordinated fusion of MOM and MIM. Three large GTPases, OPA1, Mfn1, and Mfn2, are essential for the mediation of sequential fusion. Fusion can generate highly interconnected tubular mitochondrial networks. Mfn-2 deficit leads to mitochondrial enlargement and delay of MPT—downstream of Ca2+ stimulation by local ROS (Papanicolaou et al. 2011).
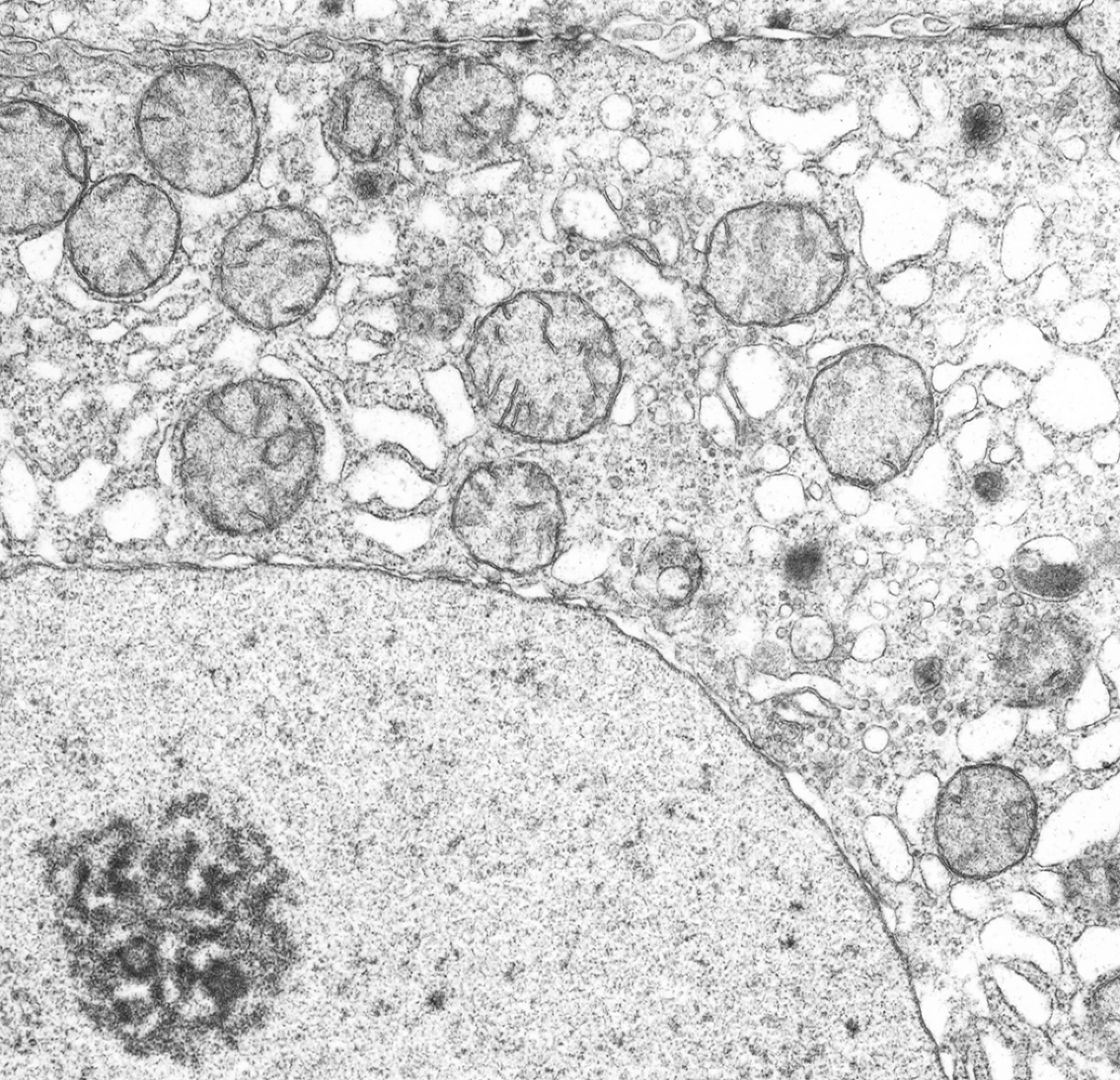
Mitochondrial fission: hepatocyte regeneration after partial hepatectomy, rat. Mitochondria are small, spherical, and uniform in size as a result of maximum fission. Nucleus has dispersed chromatin, large bipartite nucleolus, and straight nuclear envelope, all consistent with an actively transcribing cell. Cytoplasmic transport vesicles are large and prominent reflecting active metabolism and growth. Many free ribosomes and peroxisomes are evident. There is diffuse dilatation of cisternae of endoplasmic reticulum and nuclear envelope. Cell junctions (top), normally condensed and short, are long and dense. Note: wide nuclear pores.
For mitochondrial fission, the major fission protein Drp1 is recruited from the cytosol to fission sites on MOM surfaces by the Drp1 receptor, Fis1 (fission 1 or Mff). The tethering protein Mfn2 plays a role in recruitment, and ER release of Ca2+ and membrane lipid changes may be involved (Rambold et al. 2011; Rambold and Lippincott-Schwartz 2011). When activated, anchored Drp1 assembles into spirals, hydrolyzes and deforms the membrane, and then disassembles (Green, Galluzzi, and Kroemer 2011). Different conformations of Drp1 cause it to inhibit fusion and to promote fission (Huang, Galloway, and Yoon 2011). To cause mitochondrial fission, Drp1 forms contractile helices that wrap around the MOM to constrict the mitochondrion (Figure 3). Drp1 assemblies are found in mitochondrial fissions sites—sites marked by ER–mitochondrion contact sites that are characterized by ER tubules (distinct from but arising from sheet-like regions of the ER) crossing and enwrapping mitochondria. At these sites, the mitochondria first become constricted and then divide.
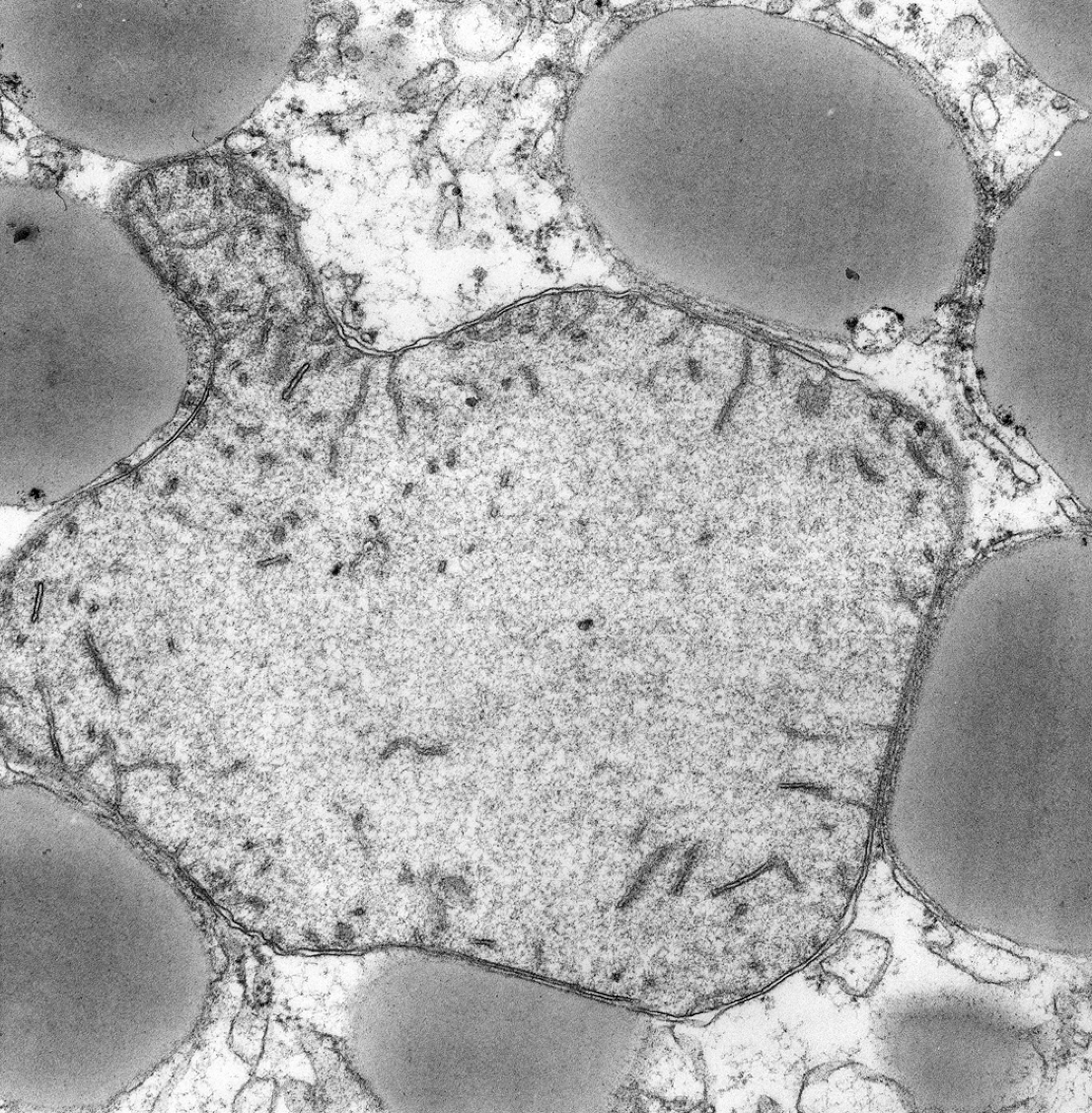
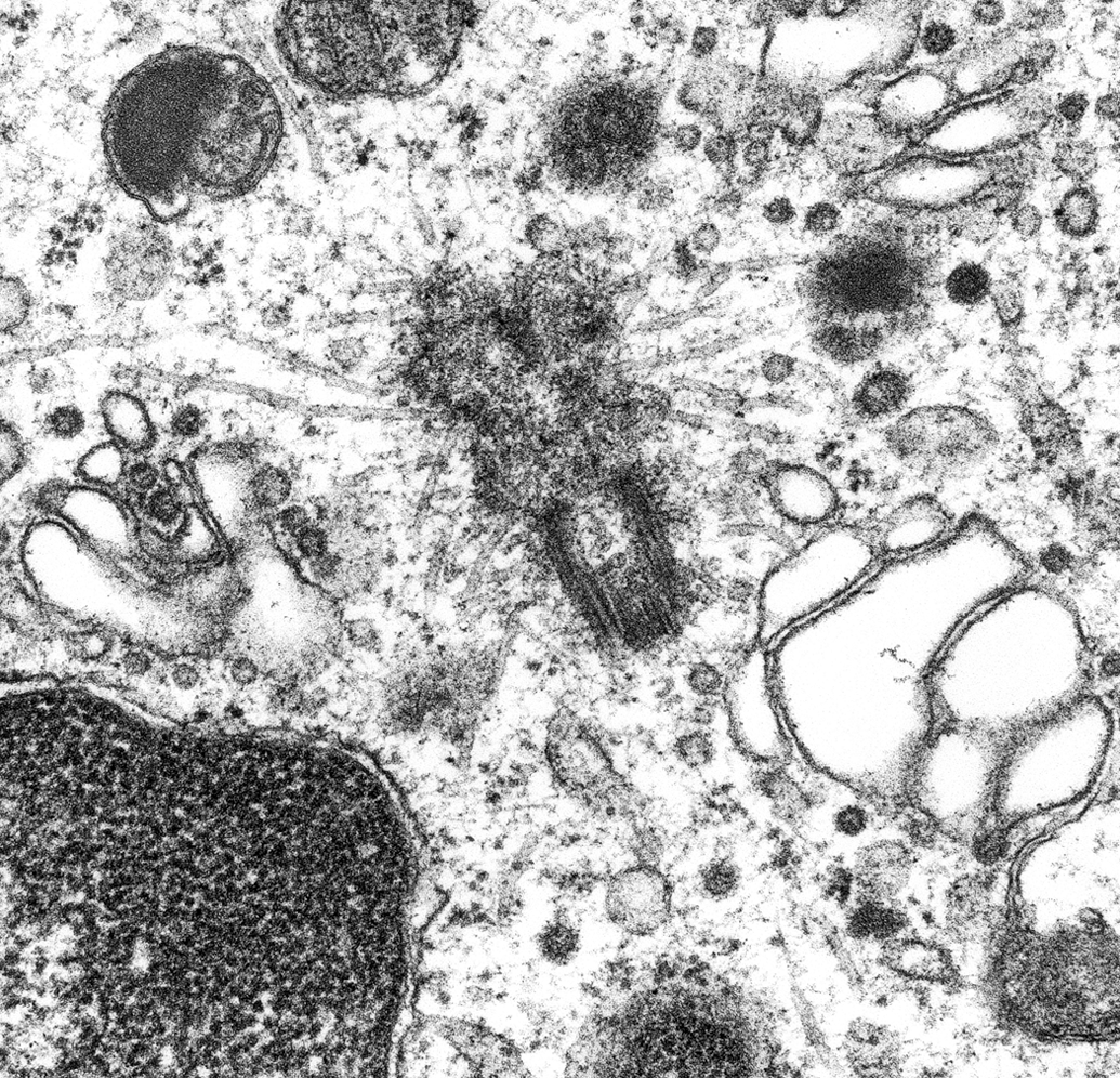
Mitochondrial fusion: mitochondria-lipid droplets (LD)-smooth endoplasmic reticulum (SER) interface, hepatocyte steatosis, diabetes mellitus, dog. Large LDs are closely apposed to mitochondrion and SER membranes. Mitochondrial matrix is massive expanded with mitochondrial ribosomes, DNA fibers, and protein filaments. Cytoplasm contains SER transport vesicles but is relatively devoid of rough endoplasmic reticulum and other organelles. Mitochondrial bleb (top left) is evidence of mitochondrial fusion activity or fission failure.
Mitochondrial Matrix and Proteostasis (Protein Homeostasis)
Peptide chains arising from cytosolic polysomes are moved to mitochondria by chaperones where they translocate across the MOM. Translocating proteins are met by their mitochondrial chaperones as they emerge from the membrane. Mitochondrial chaperones maintain the protein folding environment in the matrix and refold proteins into the appropriate conformation. When protein refolding in the mitochondrial matrix is abnormal, misfolded proteins activate special receptors in the membrane, which, in turn, activates a transcriptional program called the mitochondrial unfolded protein response (mUPR). Therapies that target mitochondrial chaperones in tumor cells can trigger autophagy and an mUPR centered on regulation of C/EBP transcription factors, repressed NF-kB-dependent gene expression, and apoptosis.
Mitochondrial Iron
The liver is the central organ of iron metabolism, and the hepatic peptide hepicidin is the systemic iron regulatory hormone. Hepicidin regulates intestinal absorption, plasma concentrations, and tissue distribution of iron by inducing degradation of its receptor, the cell iron exporter ferroportin. Transferin/transferin receptor 2 interactions mediate transport of iron into mitochondria. Intracellular free iron is toxic and is stored in nonredox-active iron-rich organelles such as mitochondria. Recycling occurs in lysosomes, which often contain large amounts of redox active iron.
In hepatocytes, mitochondria are the main consumers of iron that is used for OXPHOS components, control of iron metabolism, assembly of Fe-S protein clusters, and synthesis of heme and steroid precursors. In particular, the respiratory chain relies on iron-containing redox systems (complexes I–III) using Fe-S clusters and cytochromes with heme as prosthetic groups. Ferritins (formed when iron is added to apoferritin in the cytosol), the iron storage protein, sequester iron taken up in the cell and release stored iron for metabolic needs, using metallochaperones to deliver iron to appropriate transporters and enzymes. Structurally, ferritin is composed of hydrous iron oxide cores surrounded by subunits, proteinaceous micelles of 2.7-nm diameter arranged at apices of a regular octahedron. When taken up by lysosomes, ferritin is converted to the more complex hemosiderin. Mitochondrial ferritin (mtFerritin) is a unique ferritin type found in cells with high metabolic activities, but not in hepatocytes, macrophages, or other iron storing cells.
Ferredoxins (FDXs), the iron–sulfur proteins, facilitate monooxygenase reactions catalyzed by p450 enzymes. Mitochondria depend on two distinct ferrodoxins for specific production of either steroid hormones (FDX1) or heme and Fe/S proteins (FDX2). Mitochondrial FDX1 (adrenodoxin) is involved in steroidogenesis; it interacts with p450 enzymes to synthesize mitochondrial steroid precursors. FDX1 destruction leads to iron overload, reduced mitochondrial cytochrome p450 enxymes, and oxidative stress. Mitochondrial FDX2 functions to synthesize Fe-S clusters. Frataxin is a mitochondrial iron chaperone for iron–sulfur cluster assembly and heme biosyntheses.
Mitochondrial Oxygen Homeostasis: The Hypoxic Stress Response
Mitochondria are the organelle for oxygen sensing. ATP synthases drive the OXPHOS reactions that generate ATP (Davies et al. 2011). Within seconds of anoxia, ATP values fall to 0, mitochondrial cristae disappear, and membranes disintegrate. The effect of abolishing OXPHOS is magnified by its tight coupling with the citric acid cycle. In acute hypoxia, mitochondrial cristolysis results from uncoupling of OXPHOS and citric acid cycles, inhibition of electron transport between complex I and ubiquinone, and accelerated production of hydrogen peroxide (from nicotinamide adenine dinucleotide [NADH] and NADH oxidase activity). NADH accumulates and exerts a powerful effect to inhibit dehydrogenases of the cycle; that is, pyruvate dehydrogenase can no longer catalyze flux of pyruvate to acetyl CoenzymeA (CoA).
In chronic hypoxia, mitochondrial enlargement may be striking—the myocardium of patients with severe anemia coupled with myocardial ischemia may have increased mitochondrial mass and volume. Mitochondria in the chronically hypoxic skeletal myocytes of Sherpas and of mountaineers returned from the Himalayas have activated glycolytic pathways, reduced lipids, and accumulations of lipofuscin, a mitochondrial degradation product. When mitochondria in metabolically active cells are damaged, the glycolytic enzyme GAPDH translocates to the nucleus, where it induces gene expressions that drive autophagy to degrade damaged mitochondria. Phosphorylation of ULK1 by AMP-activated protein kinases connect energy sensing to mitophagy (Egan et al. 2011).
Hypoxia leads directly to activation of the HIF-1 gene, the pivotal regulator of hypoxic gene expression; its product, the transcription factor HIF-1 (hypoxia inducible factor-1), is a key metabolite sensor. HIF-1 activates gene transcription for glucose transporters; glycolytic enzymes (increases glucose → pyruvate flux); PDK1 (pyruvate dehydrogenase), for pyruvate → acetyl CoA formation; LDHA (lactates dehydrogenase, for pyruvate → lactate); and mitochondrial proteins BN1P3 and Bn1P3L, which induce mitophagy and autophagy. Shunting of substrates away from mitochondria reduces ATP production, but prevents excess ROS production due to inefficient electron transport in hypoxia.
ER: The Metabolic Workhorse
Membranes of the ER form a system of anastomosing canaliculi and saccules that are compartmentalized according to specific function. Protein and lipid synthesis, glycogen synthase control, and Ca2+ homeostasis, all critical interconnected functions, provide an internal trafficking system that is protected from the ions and enzymes of the cytosol. The diversity of integral and transmembrane proteins embedded in the ER membrane suggests four subcompartments: (1) rough ER (RER) for ribosomal protein synthesis, (2) smooth ER (SER) dedicated to detoxification reactions, calcium homeostasis, and other functions, (3) transitional ER (tER), and (4) nuclear envelope ER. All are interconnected and can dissociate and reassemble under the guidance of protein signal regulators in the cytosol.
RER Membrane: Protein Synthesis and Export to the Golgi Complex
As peptide chains spin out of the ribosome into the RER cisternae, they are folded into the appropriate conformation. New peptides are met by their chaperones as they emerge from the ribosome. To help peptides fold correctly, chaperones use successive rounds of ATP hydrolysis to continually bind and release new proteins until they are properly folded. Folding, glycosylation, cross-linking, and sorting of peptides into appropriate conformations are posttranslational modifications that begin in RER cisternae but are completed in the Golgi complex. For glycosylation, oligosaccharide-processing enzymes, resident in the RER membrane, remove and add terminal sugar molecules; glucose-6-phosphatase, a marker for the RER, heavily labels this organelle.
For exit and transport from the RER to the Golgi complex, tiny vesicles bud from the RER membrane carrying soluble proteins from the cisternae with it. These COPII-bearing transitional vesicles move normal proteins from RER to the Golgi complex for glycosylation and packaging. Properly folded proteins leave the RER at membrane exit sites where components of the COPII coat associate with the cytosolic surface of the RER membrane. These complexes bud off as COPII vesicles and tubular saccules to begin export to the Golgi complex. After detaching from the RER membrane, COPII vesicles form vesiculotubular clusters.
RER Trafficking: Cargo Moves to the Golgi and Endomembrane System
Vesicular transport systems move fluids, proteins, and other macromolecules among organelles and between organelles and the plasma membrane. In directed movement, cargo-bearing organelles are targeted to the appropriate pathway by specific receptors and signal transduction molecules. Migration is achieved by motor proteins and is guided through the cytoplasm by microtubules and other components of the cytoskeleton. In hepatocytes, directional movement of protein trafficking vesicles, from RER to the cis Golgi complex, is determined by proteins embedded in the vesicle membrane and coat. A specific vesicular transport pathway shuttles cargo from mitochondria to lysosomes (Soubannier et al. 2012).
Membranous saccules of the Golgi complex form a broad, continuous perinuclear network composed of three separate compartments (cis, medial, and trans), each identified by unique cytochemical reactivity according to their different functions. The ubiquination proteolytic system in the pre-Golgi ER is rapid and highly selective, and it is here that protein synthesis and autophagic degradation are regulated in opposite ways by the signal molecule mTOR. The retromere is a heteropentameric complex that associates with the cytosolic face of endosomes to mediate retrograde transport of transmembrane cargo from endosomes to the trans Golgi network (TGN). The mammalian retromere complex comprises a sorting nexin dimer, combinations of SNX 1, 2, 5, and 6, and cargo recognition trimers of Vps 26, 29, and 35 (which have arrestin, phosphoesterase, and α-solenoid folds).
tER Interfaces with Mitochondrion and Golgi Complex
The ER–mitochondrial encounter structure (ERMES) tethering complex has a major role in exchange between mitochondria and ER; the Ca2+-binding Miro GTPase GEM1 is an integral component of ERMES (Grimm 2012; Kornmann, Osman, and Walter 2011). For ER–Golgi traffic, omegasomes in the ER membrane are platforms for initiation of phagophores. Selective cargo export from ER to Golgi complex involves budding and vesicle formation, a process that requires assembly of the COPII coat complex at tER exit sites, sites where the tER scaffold for COPII is organized by membrane proteins such as Sec16. Immunogold labeling for COPII-bearing transitional vesicles and electron tomography of tER platforms reveals 50-nm diameter vesicles at the tER–Golgi interface (Zueschner et al. 2006).
The tER mitochondria-associated membrane (MAM) domain is a physical platform for interplay of ER and mitochondria. To facilitate signaling, tER signal molecules are tethered to MAM and mitochondrial membranes. MAM is the major site of a mitochondrial antiviral signaling (MAVS) protein. MAVS protein resides in mitochondria and peroxisomes and is the adaptor protein for RIG-1, a cytosol protein pathogen recognition receptor. RIG-1 engages viral RNA to trigger innate immune defenses through MAVS. RIG-1 is recruited to MAM to bind MAVS to form a signal synapase. MAVS protein forms functional prion-like aggregates in the MOM to activate and propagate antiviral innate immune responses. MAVS activates transcription factors IRF3 and NF-kB to induce interferon (IFN), a trigger of antiviral signals (Hou et al. 2011).
RER Stress: The Unfolded Protein Response (UPR)
To deal with accumulation of misfolded proteins in the ER lumen, the cell uses highly specific signaling pathways collectively called the UPR to restore normal RER function. Major sensing and signaling pathways of UPR involve Xbp1 (X-box protein 1, activating transcription factor 4 [ATF4]) and CHOP (C/EBP homologous protein). There is a close relationship of these pathways to those of mitochondria; for example, the proapoptotic mediator CHOP upregulates mitochondrial ORP 150 (oxygen-related protein 150), an inducible cytoprotective chaperone that is activated after many types of cell injury. ER stress leads to dysregulation of endogenous sterol responses and activates oxidative stress pathways.
Failure of the UPR to correct the damage of RER stress can lead to a variety of ultrastructural changes. The outcome may be adaptation with restoration of ER homeostasis or, if ER stress is unresolved, hepatocyte apoptosis from lethal calcium uptake, activation of CHOP, and premature restoration of mRNA activity (Giorgi et al. 2010; Malhi and Kaufman 2011). Prolonged UPR induces programmed cell death by activating the CHOP gene that then acts transcriptionally to inhibit protective cellular molecules such as Bcl-2 and glutathione.
In addition, there may be activation of an acute phase response with excess production of hepicin and SAA (serum amyloid protein) with activation of the inflammation via NF-κB and JNK, or induction of steatosis via dysfunctional VLDL assembly and export and activation of the sterol regulatory element binding protein SREBP1c.
Smooth Endoplasmic Reticulum (SER): Where Calcium, Lipids, and Detox Converge
Membranes of the SER form arrays and interweaving tubules and extensive lamellar arrangements that have open connections to both nuclear envelope and channels of the RER. SER tubules are sites of synthesis and metabolism of lipids, phospholipids, and steroids, and of control of calcium flow. SER increases and decreases with the metabolic demands of the cell. Lipids developed in SER cisternae are conveyed to junctions with RER where they are conjugated with peptides to form lipoproteins. SER of the hepatocyte is the dominant detoxification organelle for most drugs and toxins. Most lipophilic drugs and steroids are metabolized in the mixed function oxidase system, a chain of enzymes that includes cytochrome-P 450. Located primarily in centrolobular hepatocytes, these enzymes are associated with drug-induced SER proliferation.
Ca2+ Import: Plasma Membrane to SER to Mitochondria
Calcium homeostasis is a key component of many pathologic conditions. Any increase or decrease in cytosolic free Ca2+ activates calcium channels in the plasma membrane, SER, and mitochondria to restore resting levels of Ca2+. Cytoplasmic transport to and from these organelles occurs in calcisomes, small (50–250 nm diameter) transport vesicles in the hepatocyte cytoplasm that bear Ca2+-binding proteins; using immunogold labeling for calsequestrin-like proteins, calcisomes have been specifically identified (Hashimoto et al. 1988). Increase or decrease of cytosolic free Ca2+ also causes mitochondria to take up or release Ca2+ into the matrix where it stimulates oxidative metabolism and opening of mPTP channels. The content of Ca2+ in the cytosol and SER determines the activity to mPTP and thus the cells sensitivity to apoptotic stress. Transient mPTP opening is restorative; persistent opening is not because the deregulated release of matrix Ca2+ terminates OXPHOS and causes matrix swelling, MIM depolarization and unfolding, and MOM rupture with release of apoptogenic proteins.
Reflecting their role in Ca2 homeostasis, mitochondria possess an elaborate array of Ca2+ transport systems. Mitochondrial uptake of Ca2+ leads to an increase in mitochondrial calcium-sequestering granules in the matrix (mitochondrial Ca2+ is stored as hydroxyapatite in sequestering granules). The calcium channel mitochondrial Ca2+ uniporter (MCU) in the MIM drives rapid and massive entry of Ca2+ into mitochondria; its molecular structure is not understood, but it operates with a uniporter regulator, M1CU1, and is most efficient at high cytosolic Ca2+ levels. Calreticulin is an ER Ca2+-binding multifunctional chaperone that interacts with calnexin, an integral ER membrane chaperone, to regulate intracisternal calcium and luminal calcium buffering. The calreticulin–calnexin cycle also is an important component of ER quality control.
The IP3 receptor (IP3r), an IP3-activated Ca2+ channel in SER membranes, delivers Ca2+ to the mitochondrion. When IP3 (inositol 1,4,5-triphosphate) acts on the IP3r, it discharges Ca2+ from ER stores. ER-to-mitochondria Ca2+ transport is modulated by signaling pathways in the membranes of these organelles where Ca2+ levels are used to control diverse events. Ca2+ is a key regulator of mitochondria and acts at several levels to stimulate ATP synthesis.
Intracellular Ca2+ Rise: The Pasteurella Multocida Toxin (PMT)
Several toxins act to cause dysfunctional Ca2+ regulation. PMT, one of the several dermonecrotic bacterial toxins, alters important calcium signal transduction cascades. PMT is a glutamine-specific protein deamidase; it acts by deamidation of specific glutamine residues in the α-subunit of heterotrimeric G proteins that inhibit GTPase activity in a way that stimulates a large subset of signal transduction pathways. In the end, PMT upregulates JAK (Janus kinase) and STAT 1,3,5 (signal transducers of transcription) to activate Gαq, which leads to rise in intracellular Ca2+, accumulation of inositol phosphates, and activation of phospholipase C (Orth et al. 2007). In hepatocytes, PMT enters the cell into late endosomes, by receptor mediated endocytosis—sharing a receptor pathway with transferrin and cholera toxin (Repella et al. 2011). Once in the cytosol, the pathways of these proteins diverge: transferrin is moved to recycling endosomes, cholera toxin is directed retrograde to the ER, and PMT is trafficked to late endosomes by an endocytic pathway that involves the small regulatory GTPase Arf6. In rats with PMT-induced hepatic necrosis, the first detectable ultrastructural change is discrete foci of swelling in cisternae of the RER; this is followed by similar lesions in mitochondrial cristae (Figure 4).
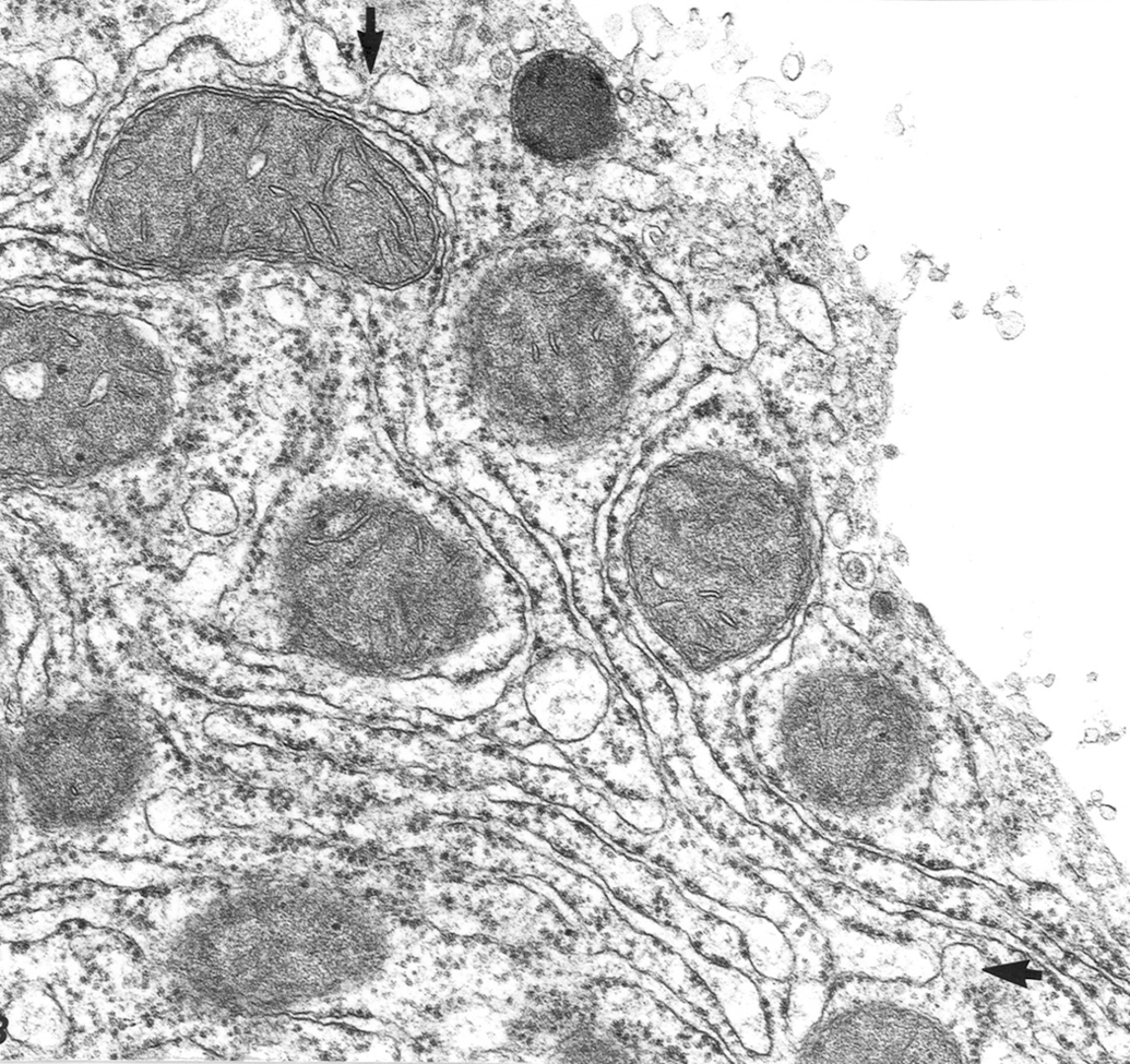
Acute cell swelling (early phase): cytoplasmic lake, hepatocyte, rat, Pasteurella type-D toxin. Dilatation of termini of rough endoplasmic reticulum (RER) sacculi (arrows) and vesiculation of membranes at the edges of the large cytoplasmic lake (upper right). Periphery of the lake has fragmented RER and is not membrane-bounded as in a true vacuole.
Lipid Droplets
The cytoplasmic LD (lipid globule) contains a core of neutral lipids surrounded by a phospholipid monolayer that harbors protein molecules and small amounts of free cholesterol. LDs develop as triglycerides and sterols (neutral lipids) are secreted into the interleaflet space (between the inner and outer leaflets) of the lipid bilayer of the SER membrane. Diacylglycerol acyltransferase (DGAT), embedded in the phospholipid blanket as an integral protein of the outer leaflet, drives lipid synthesis in the interleaflet space. As the LD appears, pinches off, and moves into the cytosol as an LD, it is surrounded by the single outer leaflet that bears DGAT and other functional proteins.
Proteins bound to or embedded in the LD surface monolayer drive interactions that not only contract and expand the LD but that change its internal composition. The major long hydrophobic helices that integrate into the phospholipid monolayer are DGAT and caveolin (from transport caveoli that fuse with the LD monolayer). Caveolins are a family of integral membrane proteins that oligomerize to coat flask-shaped caveolae thereby conferring an invagination function. Caveolin types Cav-1α, Cav-1β, Cav-2, and Cav-3 all regulate signaling proteins that initiate plasma membrane caveolae formation and direct its movement in the cell. Within the membrane, coordination of signaling and movement is achieved by the development of lipid rafts, microdomains that compartmentalize proteins and lipids (Ohsaki et al. 2009).
Perilipin and other PAT proteins and the Rab lipid anchors used in transport functions are components of the CLDs expand and grow by three ways: (1) local lipid synthesis; (2) coalescence and fusion of small LDs; and (3) vesicular transport of lipids by caveoli into the CLD. Any disturbance of the CLD coat proteins can lead to striking increases in vesicular-CLD traffic and the accumulation of dense coat protein granules on the surface of the LD.
Perilipin, the best characterized LD coat protein, is required for optimal lipid incorporation and release of lipids from the droplet. EM-immungold staining for perilipin reveals it to be evenly dispersed on the LD surface. Freeze-fracture EM analysis shows that the LD hydrophobic face contains intramembranous particles, a unique feature of hydrophobic surfaces on lipid bilayers (Robenek et al. 2011). The PAT or perilipin family, which includes perilipin, adipophilin, S3-12, and TIP47, regulates LD turnover by modulating lipolysis; they act as surfactants on LD surfaces, packing lipids in small units, and restricting access of lipases. Perilipins are found not only at the surfaces and cores of LD but also in mitochondria of cells with high OXPHOS capacity and in the cytoplasmic leaflet of the plasma membrane. At cell surfaces, upon appropriate stimulation, plasma membrane LD proteins cluster—clustering attracts and facilitates carrier-mediated lipid influx. LD coat protein perilipin 5 directs FAs from the LD to mitochondria for FA oxidation in human muscle (Bosma et al. 2012). Hepatic stellate cells, specialized for storage of retinoid (vitamin A and its metabolites), have CLDs with peripheral proteins for that storage function (Blaner et al. 2009).
Steatosis
Mitochondrial β-oxidation of FA, the major energy-yielding process in severe exercise and starvation, is critical for energy production during periods of fasting and metabolic stress. FAs taken up by the hepatocyte are shunted to mitochondria for energy production. Activated to CoA by MOM enzymes, FAs are transported across the MIM and oxidized in the matrix. Derivatives contribute electrons to the respiratory chains and carbon units to oxaloacetate of the citric acid cycle. In hepatocytes, the MOM contains glycerol 3-phosphate acyltransferase, carnitine-palmitoltransferase, and other enzymes that catalyze the early steps in phospholipid and triacylglycerol synthesis. Insufficient mitochondrial β-oxidation of FA leads to steatosis and, if severe, to lipotoxicity. Free fatty acids (FFAs) are the principle toxic triggers that mediate damage to the cell from lipids; that is, FFA-mediated cell death uses the mitochondrion to accumulate ROS. Dysfunctional FA metabolism leads to uncoupling of electron transport from OXPHOS. In contrast to insufficient β-oxidation, upregulation of mitochondrial β-oxidation of FA stimulates mitochondrial biogenesis.
FFA Overload: The High Fat Diet Model
Dietary FFAs in the bloodstream enter the hepatocyte where they are shunted to processing in lipoprotein metabolic pathways. Hepatocyte FFA overload leads to macrovesicular steatosis, the benign lesion (in the short term) that is also induced by moderate reduction of FA oxidation or by increasing hepatic de novo lipid synthesis and secretion of VLDL-associated triglycerides. This contrasts with microvesicular steatosis, the potentially severe lesion associated with liver failure and profound hypoglycemia due to major inhibition of FA oxidation.
Steatosis induced by dietary means is controlled by the signal complex mTORC1/Lipin-1 (a phosphatidic acid phosphatase that is an mTORC1 substrate); mTORC1 phosphorylates lipin-1 causing it to translocate to the nucleus. mTORC1-regulated lipin-1 nuclear translocation controls SREBP transcriptional activity. SREBP is the master regulator of lipo- and sterologenic gene transcription, and mTORC1 regulates SREBP by controlling nuclear entry of lipin-1.
The close association of peroxisomes with SER and mitochondria facilitates the shuttle of lipid molecules among these organelles. Carnitine, essential for the β-oxidation of FA, maintains homeostasis in the acyl-CoA pools of the cell, keeping acyl-CoA/CoA pool constant during high turnover and providing a constant source of CoA. Carnitine derivatives are shuttled back and forth between peroxisomes, mitochondria, and the SER. The carnitine transporter has been demonstrated in peroxisomes using immunogold techniques. Etomoxer, a carnitine palmitoyl transferase (CPT) inhibitor, decreases FA transport via the carnitine shuttle and can strikingly increase vesicular traffic to and from the CLD (Figure 5).
Lipid droplet-vesicular traffic increase: etomoxer toxicity, hepatocyte, mouse. Etomoxer is a carnitine palmitoyl transferase (CPT) inhibitor, massive increase in LD-directed vesicles from the endoplasmic reticulum and accumulation of LD coat protein granules is caused by degradation of the unfolded protein response. [Source: D. T. Rutkowski, Department of Pathology, College of Medicine, University of Iowa; previously unpublished.]
Peroxidative Injury: The Carbon Tetrachloride (CCL4) Model
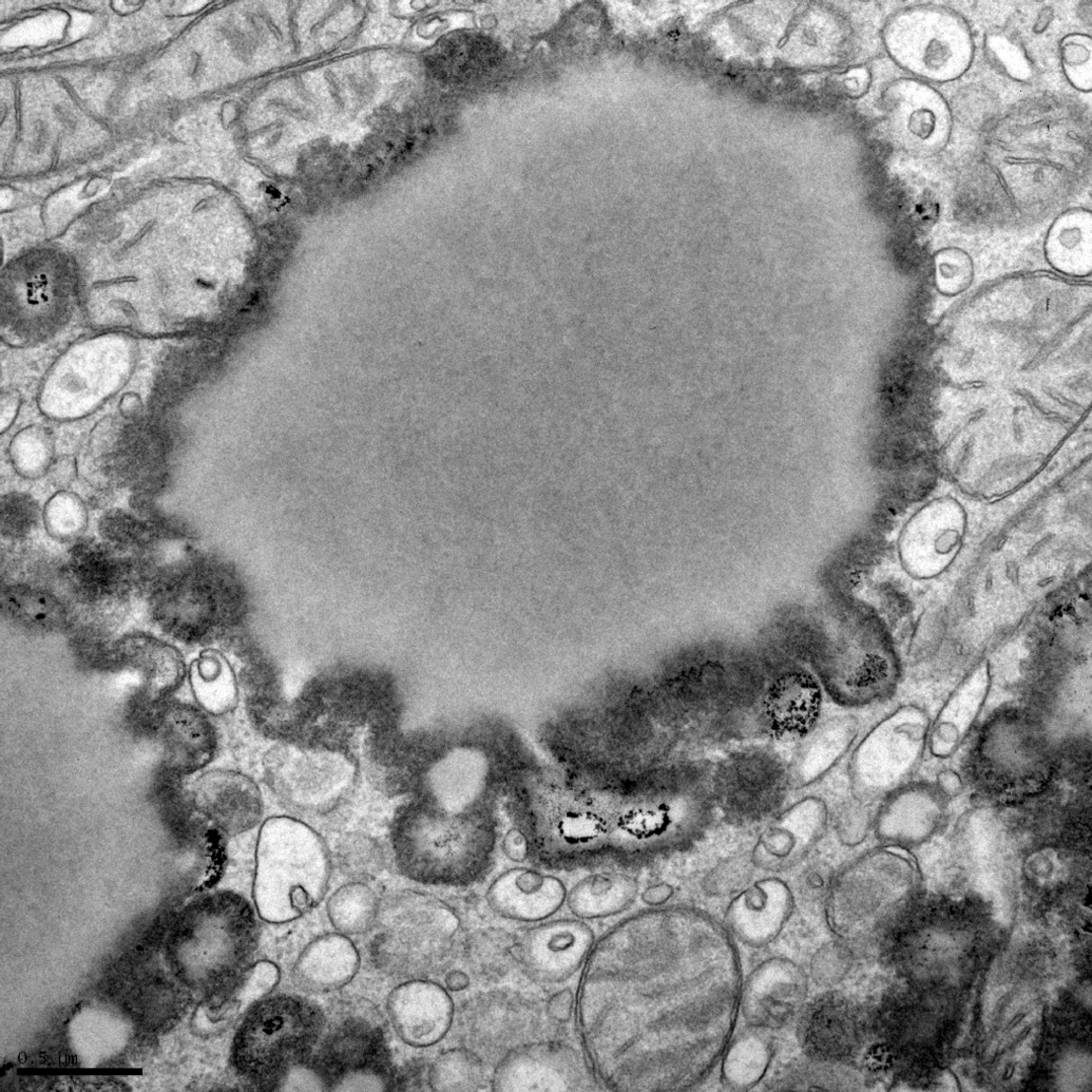
Within a few hours after experimental injection of a sublethal dose of CCl4 into a rat, hepatocytes develop dilatation of cisternae of the ER and nuclear envelope (Figure 6). The SER fragments into tiny vesicles. Disintegration of ribosomes is an early change, and ribosomal detachment from RER membranes is followed by the fragmentation of the RER—first in focal areas and then involving the entire cytoplasm. Subsequent cellular disintegration in individual hepatocytes depends on their accessibility to oxygen, nutrients, and cytokines. When rats are given a sublethal dose, centrolobular hepatocytes are most apt to undergo necrosis; portal hepatocytes survive and are most affected by steatosis. The location of carbon tetrachloride damage mirrors the oxygen gradient across the liver lobule (Weber, Boll, and Stampfl 2003). In the end, cell death occurs from the cooperative effects of many mechanisms of damage, and often the pathway to apoptosis uses activation of the cytokine TNFα.
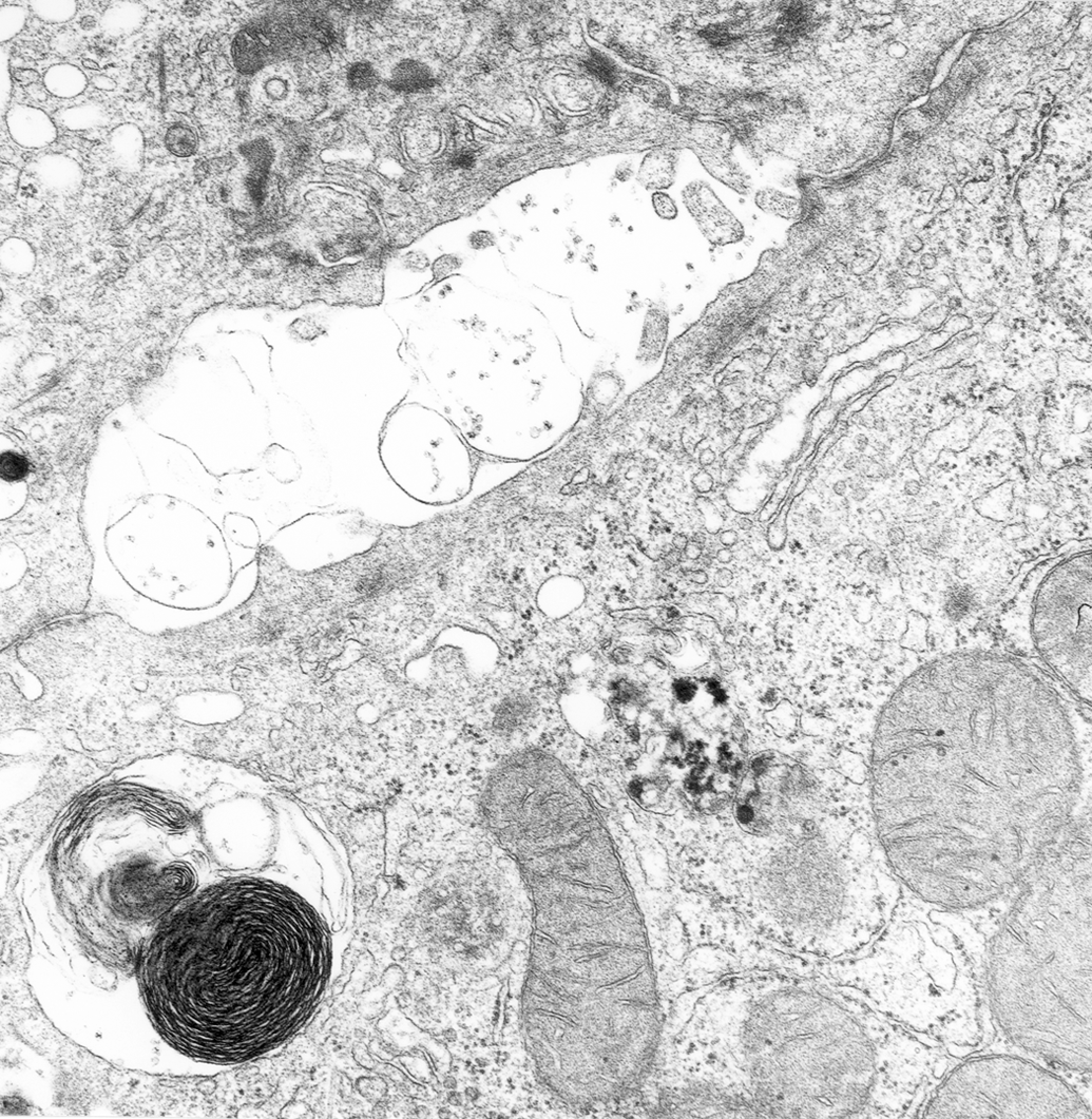
Acute cell swelling (late phase): hepatocyte, rat, CCl4 toxicity. Note fragmentation and vesiculation of rough endoplasmic reticulum (RER; left), detachment and dispersal of ribosomes (bottom center), reduplication and contraction of smooth endoplasmic reticulum (SER) membranes (right), and swollen mitochondrion (top left). Peroxisome-lipid droplet (LD) interface (bottom right) reflects repair attempt. Peroxisome–mitochondrial interfaces are associated with damaged membranes.
CCl4 is activated in the SER by cytochromes (CYP)-2E1, (CYP)-2B1, and (CYP)-2B2 to form toxic trichloromethyl radicals: CCl4 → CCl3
• + Cl•. CCl3
CCl3 • reacts with oxygen to form the highly reactive trichloromethylperoxy radical CCl3OO•. This radical initiates a chain reaction of lipid peroxidation, which attacks polyunsaturated FA, especially those associated with phospholipids as components of membranes. Membrane lipid peroxidation, in turn, leads to loss of cell Ca2+ sequestration and homeostasis. Within the membrane, the products of lipid peroxidation are also highly reactive and enhance lipoxidative damage. Degradation products of FAs are reactive aldehydes, especially 4-hydroxynonenal which binds to functional groups of protein to inhibit enzymes. There is hypomethylation of RNA and diminished protein synthesis. Peroxidized cardiolipin in mitochondrial membranes inactivates cytochrome oxidase by mechanisms similar to that of H2O2. The end result is loss of ATP, drop in pH, and rise in Ca++, all of which causes a state of irreversible injury.
Nucleolar Targeted Injury in Steatosis: The Aflatoxin Model
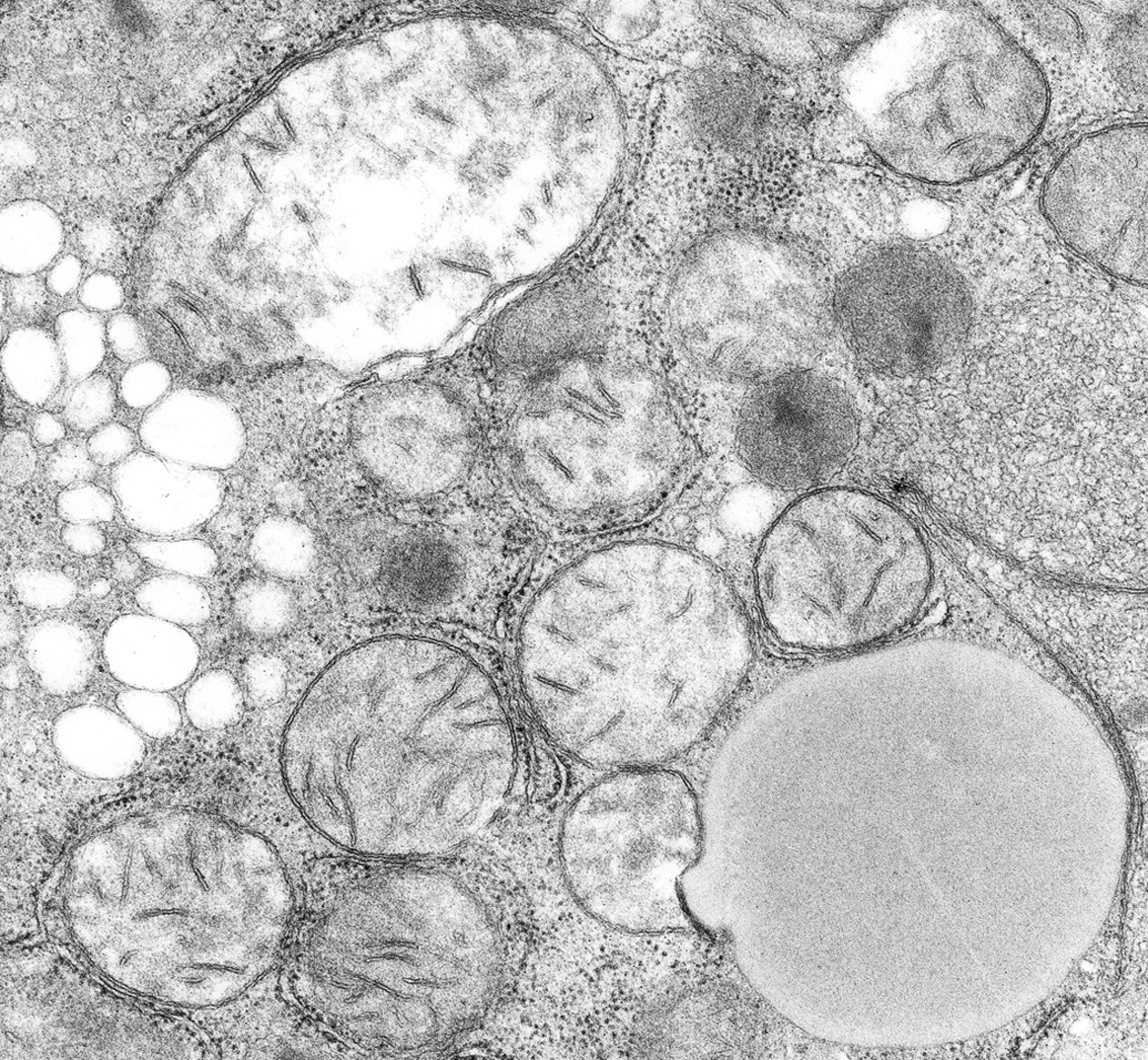
Aflatoxins, hepatotoxic metabolites produced by certain strains of Aspergillus flavus, cause direct toxic effects on DNA-dependent RNA polymerase in the nucleolus. This in turn suppresses RNA and protein synthesis. Ultrastructurally, studies in rats reveal that disaggregation of the nucleolus can be detected soon after treatment—appearance of numerous helical polysomes occurs within a few minutes of injection of aflatoxin B1. Aflatoxins also bind to hepatocyte mtDNA with preference and may have long-lasting effects because of a lack of excision repair in the mitochondrion. DNA synthesis is affected by the affinity of aflatoxin to bind to the DNA molecule. In sublethal injury, a few cells in centrolobular areas may be killed, but most hepatocytes survive with steatosis. Lesions of toxicity include reduplication of SER, increase in peroxisomes, mitochondrial swelling, and nucleolar degradation (Figure 7).
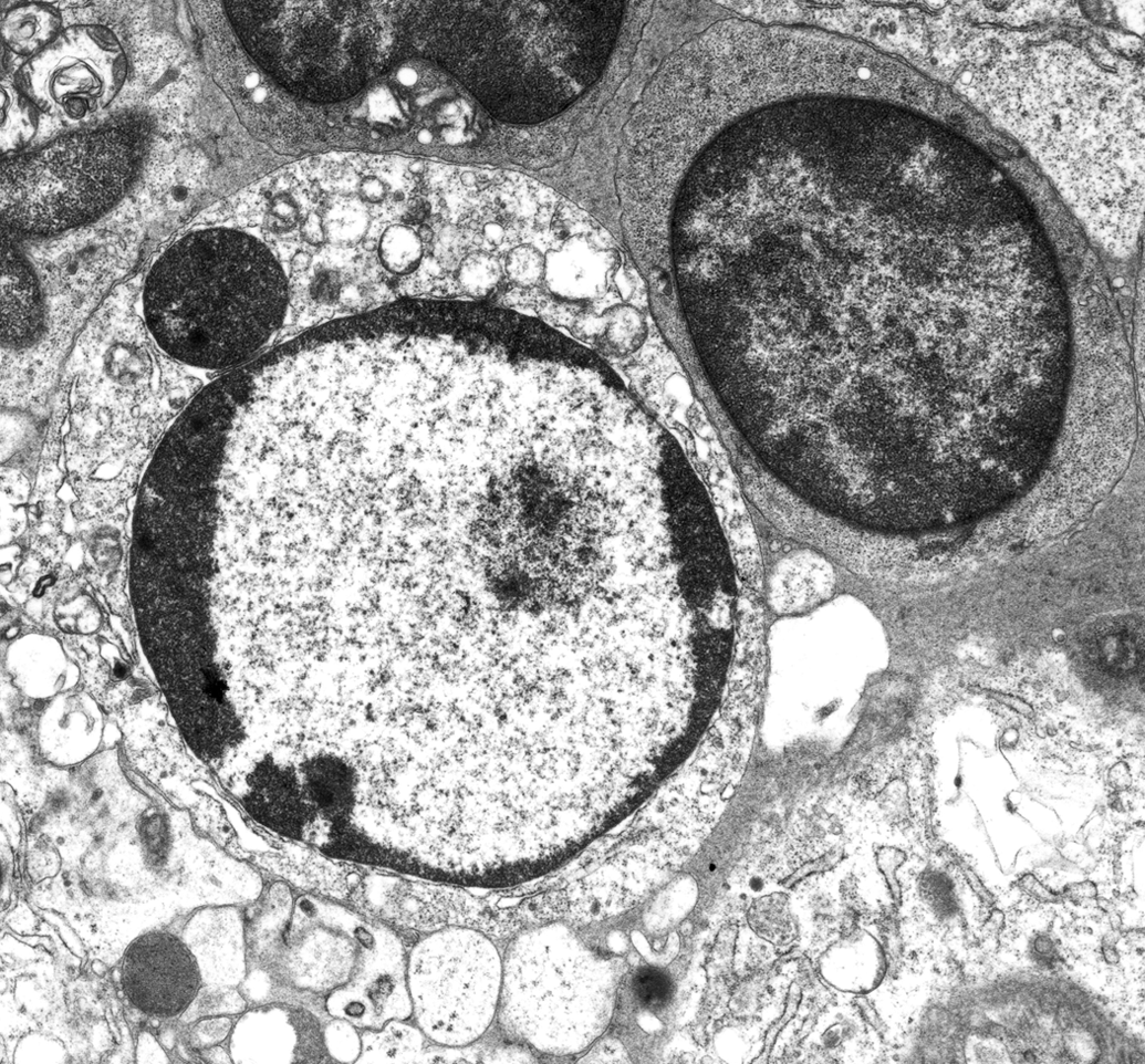
Steatosis from DNA injury: binucleate hepatocyte, horse, aflatoxin B1. The nucleolus is abnormal with separation of fibrillar and granular regions, and dispersal of perinucleolar chromatin. Mitochondria have undergone swelling, cristolysis, and precipitation of proteins in the matrix.
SER Enzymes Give Rise to Glycogen
Glycogen exists free in the hepatocyte cytosol as granules, in a size range that reflects the varying size of glycogen polymers. It develops among saccules of the SER, the sites of glycogen synthetase. Monoparticulate glycogen (10- to 30-nm diameter) is typical of most tissues, including muscle. In hepatocytes, glycogen occurs as 150 (50–200)-nm diameter glycogen rosettes (aggregates of monoparticular glycogen). Starved animals show loss of liver glycogen, and upon refeeding, rapid reduplication of SER occurs, chiefly in centrolobular hepatocytes.
In metabolically stressed hepatocytes, there is an inverse relationship between lipid and glycogen synthesis; that is, as one rises the other falls. When a cell is deprived of oxygen, ATP production in mitochondria is shifted from aerobic to anaerobic metabolism. ATP-utilizing processes such as membrane transport, filament contraction, and signaling continue for a short time, but accumulation of ADP and inorganic phosphate lead to the anaerobic shift to glycolysis, a hallmark of the hypoxic cell. Glycogen is rapidly broken down, and calcium accumulates and is redistributed. The shift from aerobic respiration in mitochondria to the more transient, inefficient anaerobic glycolysis prevents irreversible damage. Cancer cells typically use glycolysis for energy production and have short blunted mitochondrial cristae and upregulation of VDAC isomers. The hexokinase–VDAC interaction is critical for survival.
The Dexamethasone Model
Glucocorticoids have two marked anabolic effects in the liver: first, they increase mitochondrial ATP synthesis; second, they stimulate enzymes required for gluconeogenesis and for shunting of glucose to glycogen. When injected into the rat, DEX enters hepatocytes and translocates to mitochondria where it upregulates the gene for GSK-3 (glycogen synthase kinase-3) (Figure 8). DEX specifically targets mitochondria in which it activates gluconeogenesis by increasing levels of Drp1 with corresponding decreases in Mfn-1 and -2 (Hernandez-Alvarez et al. 2012). DEX increases ATP synthesis by inducing OXPHOS enzymes that are both nuclear and mitochondrially encoded, and it increases thermodynamic coupling and efficiency of OXPHOS (in liver, not muscle) (Anver et al. 2007). DEX-induced GSK-3 can trigger apoptosis in lymphocytes and DEX so used clinically.
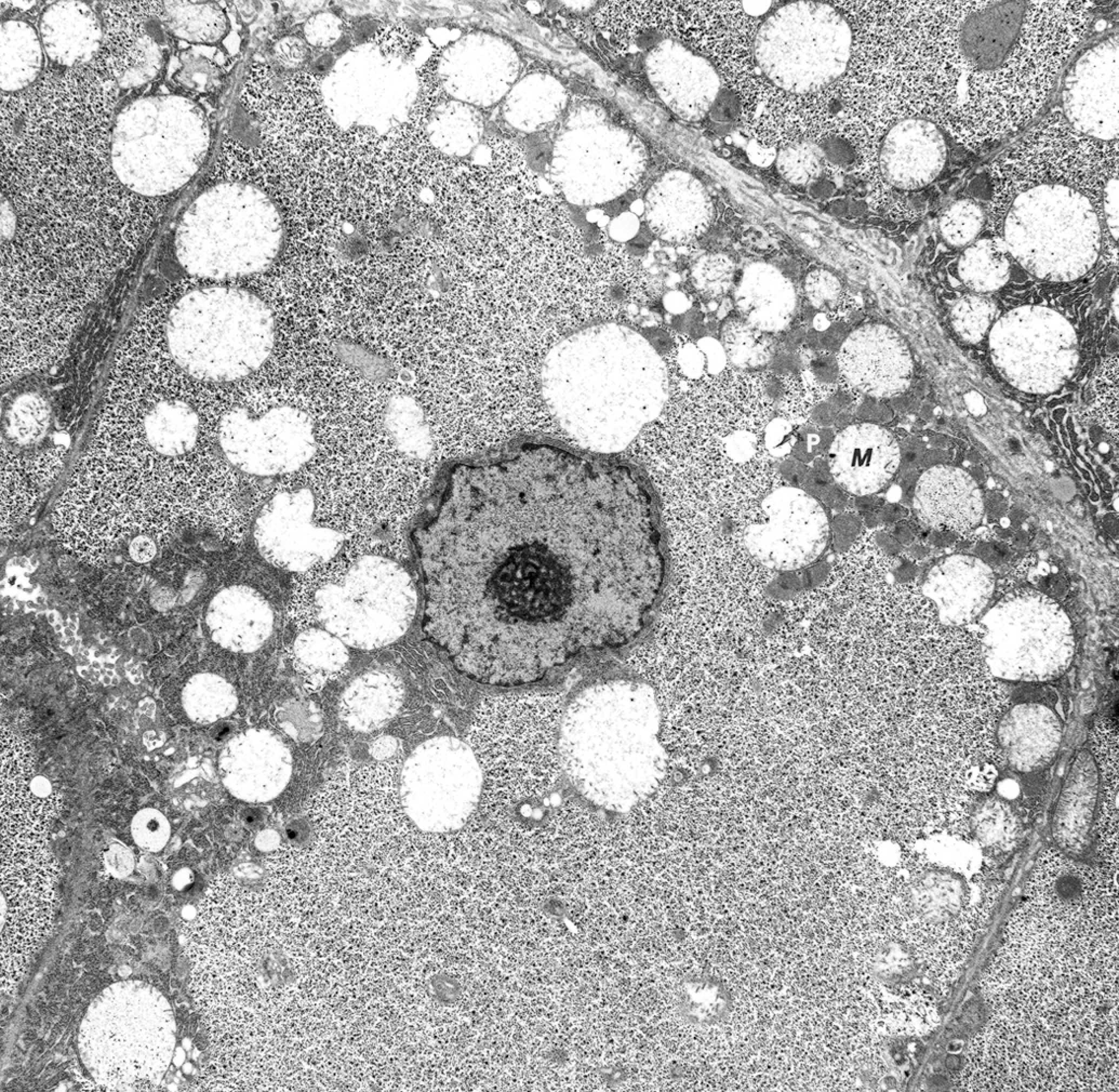
Glycogen deposition: dexamethosone toxicity, hepatocyte, rat. Cell is greatly enlarged by accumulation of glycogen. Rough endoplasmic reticulum (RER) is fragmented with remnants remaining around nuclear and plasma membranes. Mitochondria (M) are swollen and structureless, and are surrounded by peroxisomes (P).
Autophagosomes versus Proteasomes in Protein Degradation
Ubiquitination is the proteolytic process used to selectively recognize unwanted endogenous proteins and shunt them to proteasomes for degradation or to autophagic pathways for removal from the cell; that is, ubiquitination targets proteins to proteasomes or to autophagosomes. To target proteins for autophagic degradation, ubiquitin on the modified protein is recognized and bound by autophagy receptors such as p62 or Nbr, which interact with lysosomal LC3 to deliver cargo to autophagosomes. For proteosomal degradation, persistently misfolded proteins in RER cisternae are retrotranslocated to the cytosol where they are degraded by the proteasome. Unique transitional vesicles transport abnormal proteins from RER to cytosolic proteasomes via a pathway that does not use the same COPII coat protein exit mechanisms as normal transitional vesicles; this pathway uses an ER degradation–enhancing α-Mannosidase-like protein-1 (EDEM 1) that targets misfolded glycoproteins for proteosomal degradation (Perreault et al. 2009).
Mannose trimming of glycoproteins in ER cisternae is critical for sensing and sorting of terminally misfolded glycoproteins in ER-associated degradation (ERAD), a process that requires retrotranslocation and ubiquitination mediated by a luminal chaperone network.
EDEM1 is induced to target misfolded glycoproteins for proteasomal degradation by removing them from the calnexin/calreticulin cycle. Soluble EDEM1 in RER cisternae is sequestered in membrane buds that form at regions outside of the transitional RER; these buds give rise to vesicles (of ∼150 nm) that lack the canonical COPII coats (Zuber et al. 2007). The EDEM1 subgroup proteins are especially critical for ERAD in obesity, diabetes, and LD metabolism.
In posttranslational modifications, misfolded proteins are conjugated to ubiquitin through ubiquitin ligase (E3 enzymes). The extent of ubiquitinization determines intracellular traffic. Polyubiquitination, in which a chain of ubiquitins is attached, moves the ubiquinated target protein to the proteolytic degradation pathway in the proteasome. Monoubiquitination, wherein a single ubiquitin moiety is appended, is a signaling device that directs the peptide to other intracellular proteins that harbor ubiquitin-binding domains.
In addition to being assembled onto target proteins by the cell’s ubiquitin conjugation machinery and moved into 26S proteasome-mediated protein degradation pathways, ubiquitin is present in cells as free, unconjugated chains. Free ubiquitin can bind directly to activate signaling proteins. The diverse nonproteolytic functions of ubiquintin in cellular processes include regulation of kinase signaling, membrane trafficking, chromatin homeostasis, and DNA repair. The key DNA replication/repair protein PCNA (proliferating cell nuclear antigen) is ubiquinated and SUMOylated to control how cells respond to different types of DNA damage/tolerance pathways. Translesional DNA synthesis (TLS), the process whereby focal DNA damage is overridden, is regulated by PCNA ubiquitination. TLS involves PCNA monoubiquitination that is induced by DNA damage or any stall in the DNA-replicaton fork. Mammals have multiple TLS polymerases, mutations of which are associated with genetic disease and cancer.
Proteasomal Degradation
The proteasome (from protease + some, body), a hollow cylinder with multiple catalytic sites, is the structural form of the proteolytic complex that degrades ubiquinated misfolded proteins (Da Fonseca, He, and Morris 2012). (Note: the proteosome is the cluster of genes that encodes components of the cell’s proteolytic complex.) The ubiquitization process involves (1) attachment of ubiquitin to abnormal peptides to form a ubiquitin–peptide complex; (2) movement of the ubiquitin–peptide complex to a proteasome; and (3) proteasomal proteolytic complexes recognize the ubiquitin component and degrade the substrate peptide. Proteasomes in the pericentrosomal area of the cytoplasm and in PML (promyelocytic leukemia) tumor suppressor nuclear bodies function as major proteolytic centers in the cell; they are also present in cytoskeletal networks, the outer surfaces of the ER, and in nuclei throughout the nucleoplasm (but not in nucleoli). When the misfolded protein threat is resolved, proteasomal areas may be removed by autophagy (in resolving lesions, proteasomal components are found in lysosomes).
Cytoprotective pericentrosomal cytoplasmic granular aggregates that contain proteasomes, polyubiquinated misfolded proteins, chaperones, and the intermediate filament vimentin are referred to as aggresomes (Figure 9). Movement to the centrosome requires microtubule-dependent transport, and the presence of γ-tubulin is their distinctive feature. Linker proteins such as Hook2, which associate with the motor protein dynein, contribute to aggresome movement. Aggresomes are associated with a malfunction of the ubiquitin–proteasome system (UPS). In the brain, aggresomes are common in ischemia and neurodegenerative disorders. Lewy bodies of Parkinson’s disease begin as aggresomes, Mallory bodies in hepatocytes are cytokeratin aggresomes, and Lafora bodies in neurons are tubulin aggresomes. These confusing granular bodies (Table 2) must be differentiated by immunogold labeling. Nuclear aggressomes are nuclear inclusions that represent the nuclear UPS. They resemble cytoplasmic aggresomes in gathering ubiquitin and UPS components. They control the supply of ribosomal proteins for assembly of new ribosomes and are linked to defect in nuclear export.
Aggresomes: pericentriolar cytoplasmic granular inclusions of ubiquinated proteins ensheathed in a cage of intermediate filaments. Centriole (center) has radiating microtubules and is surrounded by granular debris.
Nuclear and cytoplasmic bodies.
When protein synthesis is vigorous and exceeds the carrying and folding capacity of the RER, misfolded proteins accumulate and serve as a stress signal to direct the cell to make more RER. Misfolded proteins activate a special set of receptors in the ER membrane, which in turn, activate a vast transcriptional program called the UPR, a signal transduction network from ER to nucleus (Rutkowski et al. 2008). The three branches of the UPR are ATF6 (for transcriptional response), PERK, and IRE1 (for translational control, mRNA decay, and decrease in ER protein folding load). Activation of each of these sensors produces a transcription factor that activates genes to increase the protein-folding capacity of the ER (Walter and Ron 2011). There is a close relationship of these pathways to those of mitochondria: CHOP is a proapoptotic mediator that upregulates mitochondrial ORP 150 (oxygen-related protein 150), an inducible chaperone that is activated after many types of cell injury and has cytoprotective roles (Malhi and Kaufman 2011). If the new ER cannot reestablish the proper balance of production and need, the UPR program can then direct the cell to self-destruct by undergoing apoptosis.
Autophagic Degradation
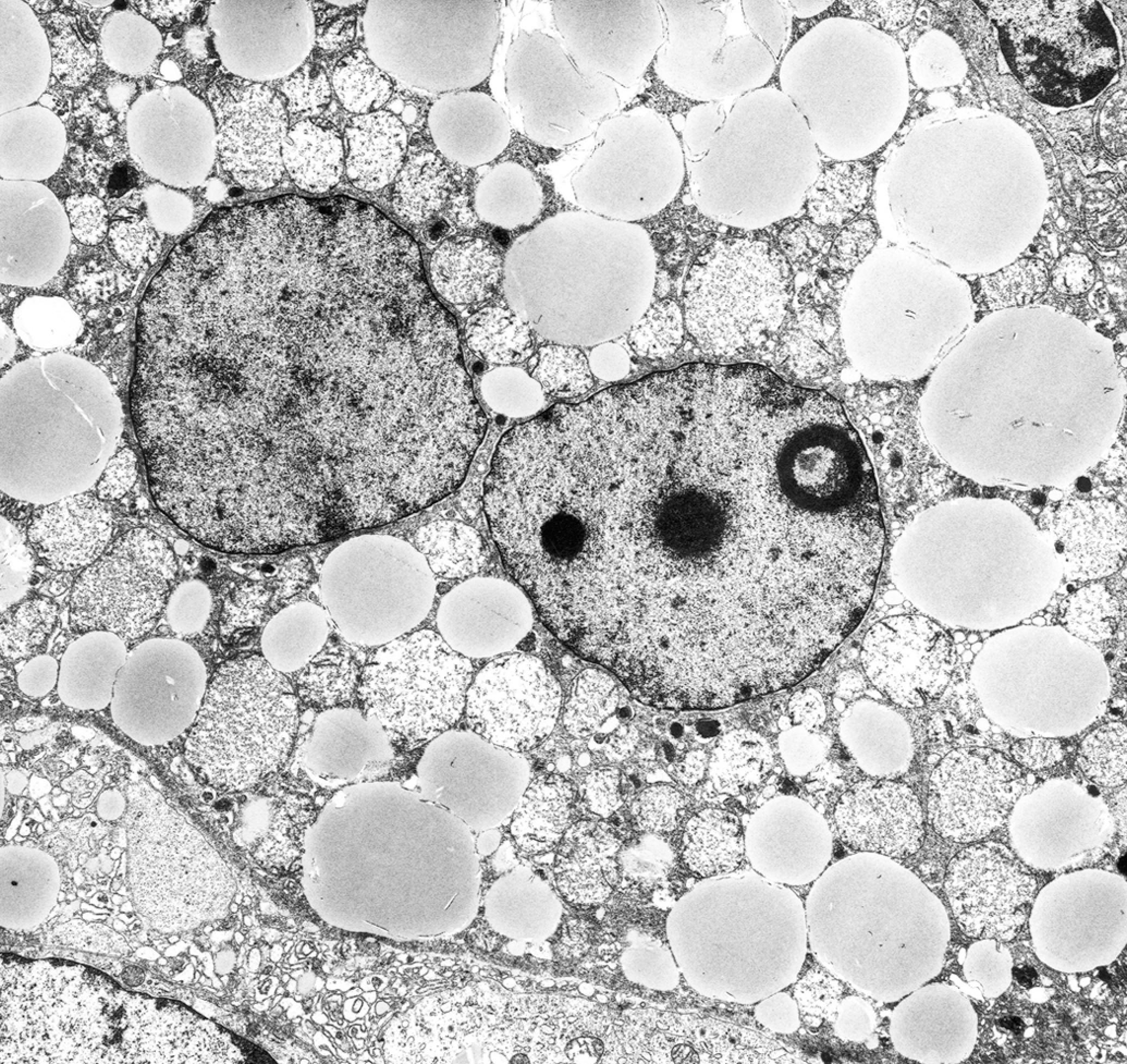
Autophagosomes are formed by unique phosphatidylinosital (PI3I)-enriched foci in membranes that bud from ER. These PI3I-enriched membrane foci, referred to as omegasomes, carry the marker protein DFCP1 and are viewed as preautophagosomal platforms that develop first into phagophores (isolation membranes) and then into autophagosomes. Omegasomes accrue the WIPI1 and WIPI2 proteins that cycle between preautophagosomes and mitochondria or Golgi complexes shuttling components into the developing autophagosome. Once free in the cytosol, the developing, progenitor autophagosome moves to, binds, and forms a membrane-cup phagophore that sequesters damaged cell proteins and organelles (Figure 10).
Autophagy: hepatocyte, horse, aflatoxin toxicity. Newly formed autophagic vacuoles are present at sites of rough endoplasmic reticulum (RER) degradation (center). Note: Residual body (bottom left) and dilatation of large canaliculus with atrophy of microvilli (upper left).
Autophagy involves about 10 lysosomal hydrolases and over 35 autophagy-related genes (ATGs), each of which generates multiprotein complexes that act sequentially. The membrane lipid phosphatidylserine, which is increased on surfaces of injured organelles, is an important “eat me” signal. LC3 (microtubule-associated protein light chain 3) is a robust marker of autophagosomes. LC3 binds p62 (p62/SQSTm1), an adaptor protein that is selectively degraded by autophagy. Aggregates of LC3 and/or p62 are visible in electron micrographs (sometimes called p62 bodies) in cells undergoing massive autophagy; for example, in chloroquine toxicity, hepatocytes will develop massive numbers of large autophagosomes that are positive for LC3 and p62 (Chien et al. 2011). Microtubules facilitate autophagosome movement and fusion with enzyme-bearing lysosomes, the merger of which forms the degradative autophagolysosome. In addition, fusion with endosomes causes the autophagosome to become more acidic.
Mitophagy, autophagy of dysfunctional mitochondria, is a cytoprotective mechanism and represents the cell’s failed attempt to adapt to stress. To initiate mitophagy, mitochondria must send two signals: one to the ER to stimulate phagosome biogenesis and another to the TGN to stimulate lysosome production. As new prelysosomal vesicles emerge from the trans face of the Golgi complex their hydrolytic enzymes, structural proteins, and glycosaminoglycans are complexed in an inactive state, awaiting the appropriate stimulus for activation; that is, the primary lysosome is a small, dense granule with its enzymes in an inactive state. Biologically active proteases in lysosomal cargos are held in check by a family of intracellular peptidase inhibitors, the serpins. Although most serpins are secreted, a unique family of serpins protects the lysosome, and the loss of serpin activity can lead to necrotic cell death.
When MOM permeability is induced and postmitochondrial caspase activation is blocked or disrupted, permeabilized mitochondria are removed by mitophagy. The kinase PINK1 is imported into healthy mitochondria (import is dependent on normal ΔΨm), where it is degraded by the protease PARL. On the surface of mitochondria with low ΔΨm, PINK1 accumulates, leading to recruitment of the ubiquitin ligase PARKIN. PARKIN suppresses mitochondrial fusion and promotes mitophagy by ubiquitinylating MOM proteins including BCL-2, VDAC1, MFN1, and MFN2.
Nutrient Deprivation Induces Autophagy: The Starvation Model
Nutrient deprivation causes livers to be small. In mammals, hepatocyte volume is reduced up to 50% and both SER and RER are reduced. Mitochondria may remain unchanged in number but are increased in size by excessive matrix material. Loss of glycogen and shutdown of cell enzyme systems and synthesis of protein and lipids are brought about in part by the lowering of the serum thyroid hormone T3. Within 24 hr of food deprivation, blood glucose and hepatic glycogen are markedly decreased; there are simultaneous increases in glycogen phosphorylase (for glycogen degradation) and decreases of glycogen synthetase (for synthesis). As fasting continues, there are profound alterations in enzyme systems relating to utilization for glucose. During fasting, plasma insulin falls (glucagon and epinephrine increase) stimulating triglyceride hydrolysis in adipocytes.
Degradation of bulk cytosol and whole organelles by autophagy provides raw materials to maintain cell activities. Mitochondrial signals drive ER functions in nutrient deprivation and are known to supply membranes for autophagy (Hailey et al. 2010); they are spared degradation by mitophagy because fusion reactions cause them to elongate (Gomes, Di Benedetto, and Scorrano 2011).
Hepatocytes have robust autophagic activity, and when mitochondria are injured, the glycolytic enzyme GADPH translocates to the nucleus where it induces gene expressions that drive autophagy to degrade damaged mitochondria. Hepatocyte autophagy is triggered by glucagon (a dominant hormone of the fasted state), adrenergic receptor activation, ammonia (a by-product of amino acid catabolism), vitamin D3, and other signals. Ablation of liver-specific autophagy leads to excess hepatic lipid accumulation. Short-chain fatty acid–induced autophagy serves as an adaptive strategy in retarding mitochondrial-induced apoptosis.
In starvation autophagy, mitochondria elongate because fission is inhibited by the phosphorylation of the profission protein Drp1; however, elongate mitochondria resist autophagy and maintain cellular energetics—they are spared from autophagy, possess increased cristae, have elevated levels of dimerization and ATP synthase activity, and maintain ATP production. Mitochondria elongate in starvation due to unopposed fusion via the PKA (protein kinase A) pathway (Karbowski et al. 2004). Following starvation-induced autophagy, lysosome homeostasis is restored by autophagosome reformation (ALR) an event that requires activation of mTOR kinase. Spinster (Spin) encodes a lysosomal efflux permease (with hallmarks of a sugar transporter); defects in Spin lead to accumulation of enlarged lysosomes. Spin is essential for mTOR and lysosome formation (Rong et al. 2011).
Mitochondrial Dysfunction Can Kill the Cell
Mitochondrial damage underlies cell death in many toxic injuries. Highly specific toxins that destroy mtDNA, components of OXPHOS, or mitochondrial membrane permeability, typically lead directly to cell death because ATP production has disappeared. There are, however, special instances where dysfunctional mitochondria directly and actively lead to cell death. Here, we examine three mechanisms by which dysfunctional mitochondria kill cells: (1) ROS overproduction, (2) production of proinflammatory signals, and (3) mitochondrial permeabilization with subsequent apoptosis. The bewildering array of special names applied to cell death (Table 3) can be avoided by simply dividing all cell death into necrosis (death of cells in the living animal) and programmed necrosis.
Names used for cell death.

Note: ATP = adenosine triphosphate; ER = endoplasmic reticulum; ROS = reactive oxygen species; VDAC = voltage-dependent anion channels.
1. Overproduction of ROS
ROS, when generated in excess in the mitochondrion, produce highly oxidizing lethal cell damage. ROS corrode molecules by snatching their electrons. The primary source of ROS occurs from transfer of e− to O2 at complex I (into the matrix) and complex III (into matrix and IMS). In normal hepatocytes, highly efficient superoxide dismutases, catalase, and other antioxidants protect mitochondria from this ever present danger. However, ROS are also produced in non-OXPHOS sites: in the MOM by monamine oxidase and Cyt c reductase; in the MIM by GPDH and some cytochrome P450 enzymes; and in the matrix by several enzymes and complexes including aconitase, pyruvate dehydrogenase, and α-ketoglutarate dehydrognease. Other sources that produce oxidants as part of normal enzymatic function include NADPH-oxidases (in phagocytes), xanthine oxidase, cyclooxygenases, and lipoxygenases.
NADPH-oxidase-induced oxidative stress, a major problem, is caused by many chemotherapeutic agents. For example, doxorubicin (DOX) causes DNA damage, nuclear translocation of p53, and mitochondrial injury; p53 regulates oxidative stress-mediated retrograde signaling and is important in mitochondrial-nuclear cross talk. Immungold labeling of a lipid peroxidation product bound to proteins, 4HNE (4-hydrox-2′-nonenol) adducted protein, reveals a marked increase of 4HNE in both nuclei and mitochondria.
Generation of toxic amounts of ROS by dysfunctional mitochondria damages DNA, both nuclear and mitochondrial. Strand breaks in mtDNA activate the gene p53, which encodes p53 protein. Ultrastructural studies detecting mitochondrial p53 by immunogold techniques show that it is protective by activating mtDNA repair. ROS are a major cause of mtDNA damage that, in cardiac failure, can lead to both necrosis and apoptosis in the cardiac myocyte. Mitochondrial p53 levels correlate with mtDNA oxidative damage and, if injury is severe, p53 triggers apoptosis using the membrane pathways BAX and PUMA (p53 upregulated modulator of apoptosis), both proapoptotic targets of p53: ↑p53 → ↑PUMA + Bax → Bax oligomerization → Cyt c dumped → caspase 3 cleaved.
Mitochondria and Peroxisomes Are Highly Coupled
Peroxisomes and mitochondria are highly coupled. Their biogenesis is linked through common transcriptional pathways; their growth and division are mediated by common fission machinery; and they have common biochemical processes of FA β-oxidation and peroxide scavenging. They both contain Mn-superoxide dismutase and catalase. Mitochondria-derived vesicles (MAV) deliver specific cargo from mitochondria into peroxisomes. MAV are enriched for the small ubiquitin-like (SUMO) E3 ligase called MAPL (for mitochondrial-anchored protein ligase) that regulates vesicular traffic (Braschi et al. 2010).
Peroxisomes, in addition to having a central role in lipid metabolism, generate ROS and RNS but can counteract oxidative stress and maintain REDOX balance. In acute toxic injury, peroxisomes protect hepatocyte mitochondria from membrane damage by moving to sites of injury on the MOM. In acute cell swelling of toxic origin, peroxisomes are not only increased in number and size but have moved to establish an interface with damaged membranes throughout the cell, particularly to mitochondria. Their potent oxidases and catalases are essential for recovery. Catalase, the classic marker of peroxisomes, has both peroxidative and catalytic activity. Peroxisomes contain H2O2-generating oxidases, and enzymes that facilitate α-, β-, and ω-oxidation of complex FA. In hepatocytes, there may be 1,000 peroxisomes per cell, all in close relation to ER. Pleomorphic peroxisomes are typically massed in groups at sites of cellular injury. In rat liver, there is a difference in the reaction of peroxisomes in different zones. Centrolobular hepatocytes are most sensitive to peroxisomal proliferation while abnormal peroxisomes with matrix tubules are apt to occur in more peripheral areas.
Peroxisomes have a close association with SER. In rapidly regenerating cells, or cells treated to increase peroxisomes, whisps of membranes or tail-like extensions are common on peroxisomal membranes—these have been shown to form in tubules that connect to other peroxisomes in a network referred to as the peroxisomal reticulum, a membrane reservoir for proliferation of peroxisomes.
2. Mitochondrial Prolinflammatory Signals: The Inflammasome
Inflammasome is the term coined by nonmorphologists to denote proinflammatory, multiprotein complexes that recognize damaged cellular components and microbes by their surface muramyldipeptides. Inflammasome formation begins with proinflammatory cytokine production and ends with cell death. All of this is a result of proteolytic processing of the proinflammatory cytokines IL-1β and IL-18 by activated caspase 1.
Inflammasomes are scaffolded by cytosolic pattern recognition receptors (PRRs) that recognize abnormal endogenous and microbe-associated proteins. Inflammasomes that promote microbial clearance are composed of oligomers of procaspase 1, a specific NLR (nucleotide binding and oligomerization, leucine-rich repeat) protein, and an adaptor protein called ASC (apoptotic-associated speck-like). Caspases 1, 4, 5 are inflammatory caspases. Over 20 NLR family members have been identified in humans. This response in pathogen infection associated with caspase 1-mediated cell death has been referred to as pyroptosis.
At the interface between mitochondria and the ER (at the mitochondria-associated ER membranes or MAMS), mitochondrial ROS can activate an inflammasome composed of NLRP3, the adapter protein ASC, and caspase 1. Many cellular receptors sense danger signals for the cell in the cargo of the phagosome, notably the Toll-like receptors (TLRs) and the NOD-like receptors (NLRs, nucleotide binding and oligomerization domain-like receptors). The NOD-like receptor Nalp3 (cryopyrin) senses components of microorganisms and large particulates.
Mitochondria play a central role in initiation of inflammasomes and other inflammatory pathways by bacteria and viruses. In viral infections, IFN, and other cytokines released into the cytoplasm translocates to mitochondria to control cell metabolism. To do this, the PRRs RIG-I and MDA-5 recognize viral RNA in the cytosol and interact with an adapter on the mitochondrial membrane, MAVS, to trigger a signal transduction cascade that drives the production of IFN. IFN-γ activates antiviral viperin (virus-inhibitory protein, ER-associated, IFN inducible) to translocate to mitochondria to reduce ATP generation (Seo et al. 2011). Viperin is an iron–sulfur (Fe-S) cluster-binding antiviral protein that can be induced not only by IFN (types I and II) but also by LPS, dsRNA, and viral DNA. In a human cytomegalovirus (CMV) model, viperin interacts with the viral protein vMIA to relocalize from the ER to mitochondria where it interacts with mTP in a way that reduces ATP generation, which results in actin cytoskeleton disruption and enhanced CMV infection.
3. Mitochondrial Permeability Transition: Mitochondrial Apoptosis
MPT involves the coordination of events in the MOM and MIM. MPT in the MOM is centered on the multimolecular signaling complex of VDAC, the ganglioside GD3, and the fission protein Fis1. The VDAC–GD3–Fis1 complex forms a microdomain in the MOM to control apoptogenic events including megapore and fission–fusion programs. Mitochondrial lesions in intrinsic apoptosis are often subtle but involve clearing of the matrix, blunting of cristae, loss of calcium sequestering granules, and swelling (Figure 11).
Apoptosis: lymphocytes, lymph nodes draining site of vaccination with modified live Brucella mellitensis, goat. Ultrastructural evidence of apoptosis: Mitochondrial degeneration with cristolysis, matrix clearing, and swelling; chromatin degradation with aggregation, peripheralization, clogging of nuclear pores, and formation of crescent-shaped masses along the nuclear membrane; nucleolar disintegration with loss of fibrillar and granular components; cytoskeletal degradation with cell rounding and surface blebbing; and endoplasmic reticulum (ER) fragmentation with formation of autophagic vacuoles.
An increase in MOM permeability facilitates mitochondrial (intrinsic) apoptosis by releasing Cyt c, inducing APAF-1 (apoptotic protease activation factor-1), and activating caspase 8. In addition, MOM permeability disrupts the electron transport chain and ΔΨm across the MIM. VDAC is the convergence point between death and survival mediated by its many ligands and proteins. VDAC oligomerization is induced by autophagic and apoptotic stimuli and is a general mechanism common to many apoptogens including TNFα, H2O2, UV-irradiation, etoposide, cisplatin, staurosporine, curcumin, and selenite (Keinan, Tyomkin, and Shoshan-Barmatz 2010)—it is a major target of anticancer drug therapy.
In the early phase of mitochondrial apoptosis, when MOM permeability is increased, the MIM compartmentalizes; that is, the MOM segregates into different compositional areas. Analysis of this change using 3-dimensional electron microscopic tomography (which gives a composite time-course overview of change) reveals swelling and vesiculation of the inner mitochondrial membrane—changes associated with release of proteins from intermembrane and intercristal spaces (see Cheville 2009).
Apoptosomes signaling platforms that initiate dismantling of the cell during apoptosis and are pivotal structures in the mitochondrial pathway of apoptosis. Cyt c released by the hyperpermeable mitochondrion binds to Apaf-1 to form the apoptosome. The binding of Cyt c to Apaf-1 (protease-activator factor 1) in the presence of ATP leads to a wheel-like heptamer of seven molecules of Apaf-1 and seven of Cyt c. The resulting complex binds and activates the initiator caspase 9 which then recruits caspase 3 which causes many of the effects of apoptosis, and signals a downstream caspase cascade. Within the apoptosome are the caspases 9, 8, and 2 (Teng and Hardwick 2010; Yuan et al. 2010). The cytotoxic effect of many chemotherapeutic drugs rests on their ability to induce apoptosomes. Apoptosome function is controlled by intracellular levels of K+, XIAP (an inhibiter of apoptosis protein), and at least two mitochondrial-released proteins, Smac/DIABLO and Omi/Hta 2, a serine protease. The type of apoptosome depends on the adaptor. There may be Apaf-1 apoptosomes, DISC (death inducing signaling complex) apoptosomes, and PIDDosomes. Cells respond to apoptotic signals according to their individual adaptor proteins and to their spectrum of caspases. In some cells, caspase 7 can be incorporated into apoptosomes, but its apoptotic effects appear to be blunted by an inhibitory molecule, X-linked inhibitor of apoptosis (XIAP).
The caspase cascade, the major death signaling pathway, plays the critical roles in both apoptosis initiation and execution. Initiator caspases (caspases 8 and 10) respond to death signals and activate execution. Executioner caspases (caspases 3, 6, 7, and 9) make specific cuts in key proteins that are required for cell survival. Procaspase 8, located primarily in mitochondria, is colocalized with Cyt c—both identified by immunogold techniques in the IMM, IMS, and matrix.
Cardiolipin is an anchor and essential platform for caspase 8, and mobilization of Cyt c is tightly regulated by the oxidative state of cardiolipin. Inactivation of caspases does not block cell death but converts it from apoptosis to necrosis. This has suggested that the caspase family of cystine proteases function not to kill but to extinguish the proinflammatory pathways of dying cells. Bcl-2 and Bclx(L), which block the MPT to initiate apoptosis, also cause necrosis by blocking the MPT.
Footnotes
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
The authors received no financial support for the research, authorship, and/or publication of this article.