
Abstract
Programmed cell death is physiological when disposing of senescent, dysfunctional, or redundant cells, but pathological if these cells cannot be replaced. Mitochondria help determine cell fate as “gatekeepers” of apoptosis and effectors of cell necrosis. Apoptosis was first described 40 years ago this year. Cell suicide (or the less emotionally charged “programmed cell death”) impacts organism development, normal organ homeostasis, and degenerative (too much cell death) or metaplastic (too little cell death) diseases. The components of apoptosis signaling through mitochondrial targeted Bcl-2 family proteins and activation of the caspase cascade and its downstream proteases and nucleases are well described. More recently, we have realized that there is a parallel cell death pathway, programmed necrosis, in which calcium cross-talk between endoplasmic reticulum and mitochondria causes mitochondrial depolarization, reversal of electron flow through the electron transport chain, and ATP depletion. Since apoptosis and programmed necrosis signaling can occur concurrently in a suicidal cell and are difficult to distinguish using conventional techniques, their relative roles in disease are still being researched and debated. Here, the different molecular mechanisms, effects, and pathophysiological implications of apoptosis and programmed necrosis are reviewed as they relate to heart failure and diabetes mediated by the Bcl-2 family protein, Nix.
Introduction
To be or not to be. That’s not really a question.
Jean-Luc Godard, French-Swiss New Wave filmmaker
Should I kill myself, or have a cup of coffee?
Albert Camus, author, philosopher, and 1957 Nobel Laureate
Elimination of cells for biological purposes, frequently referred to as “programmed cell death,” is as necessary as new cell generation. During embryonic development or homeostatic regeneration, organs are sculpted from growing tissue through directed proliferation and orchestrated elimination. The classic example is benign and rare interdigital webbing of the hand or foot, which is a consequence of impaired apoptotic removal of normal developmental interdigital tissue. More severe is failure of normal developmental apoptosis of the cardiac outflow track that can produce lethal pulmonary atresia (Fisher, Langille, and Srivastava 2000) or failure of normal restraints on cell death than induce malignant transformation (Vazquez et al. 2008). Conversely, when programmed cell death is exaggerated or inappropriately induced in a tissue that cannot replace lost cells, it can cause organ insufficiency. For this reason, pathological effects of increased programmed cell death are especially pronounced in the neurological and cardiac systems wherein terminally differentiated neurons and cardiac myocytes have limited regenerative ability (Ohsako and Elkon 1999; Shimohama 2000; Dorn 2009).
It is increasingly acknowledged that end organ function is affected not only by the quantity of cell undergoing programmed death but also by the mechanistic pathway leading to cell removal. So, how many cell death pathways are there? When discussing those mechanisms of programmed cell death whose goal is actually cell elimination (as opposed to cell autophagy wherein cells may succumb after a failed effort to survive nutrient deprivation or injury), there are three pathways of interest: extrinsic or death receptor–mediated apoptosis (Cabal-Hierro and Lazo 2012), intrinsic or mitochondrial mediated apoptosis (Munoz-Pinedo 2012), and programmed necrosis (aka necroptosis or regulated necrosis [Baines et al. 2005; Hitomi et al. 2008]). This review focuses upon the two mitochondria-centric pathways, intrinsic apoptosis and programmed cell necrosis.
Apoptotic cell death as a response to nonlethal developmental programming or environmental stimuli was conceptualized by Kerr, Wyllie, and Curie in 1972. Apoptotic cells have made the decision to disassemble their organelles, DNA, and proteins and package them for disposal with a minimum of collateral damage. The definitive biochemical pathway that mediates apoptosis consists of a series of evolutionarily conserved caspase cysteine proteases. Caspases are indirectly activated through the effects of mitochondrial localized Bcl-2 family members that are themselves activated by intrinsic transcriptional and/or posttranslational cues (reviewed in Youle and Strasser 2008). Apoptosis is conserved throughout Eukaryotes as a means of removing cells without engendering inflammation that is normally induced by the degeneration of dead cells and tissue. Because inappropriate apoptosis has massive pathological potential, its implementation is rigorously constrained and highly orchestrated. I envision apoptosis as a performance of Haydn’s 45th Symphony in F sharp minor (the “Farewell” Symphony; Figure 1, left panel). When performing this work, the entire orchestra plays in the usual manner until the last movement. During the final adagio, individual players turn off the lights on their music stands, stand up, and leave the stage one by one. By the end of the piece, only two violinists remain on an otherwise empty and darkened stage. In cells, the orchestrated bit-by-bit removal of organelles, membranes, DNA, and other contents is accomplished by apoptotic induction of specific DNases and proteases. It is important to bear in mind that all of this cellular deconstruction requires a great deal of energy, and so maintenance of mitochondrial respiration and ATP production is essential to fuel apoptosis.
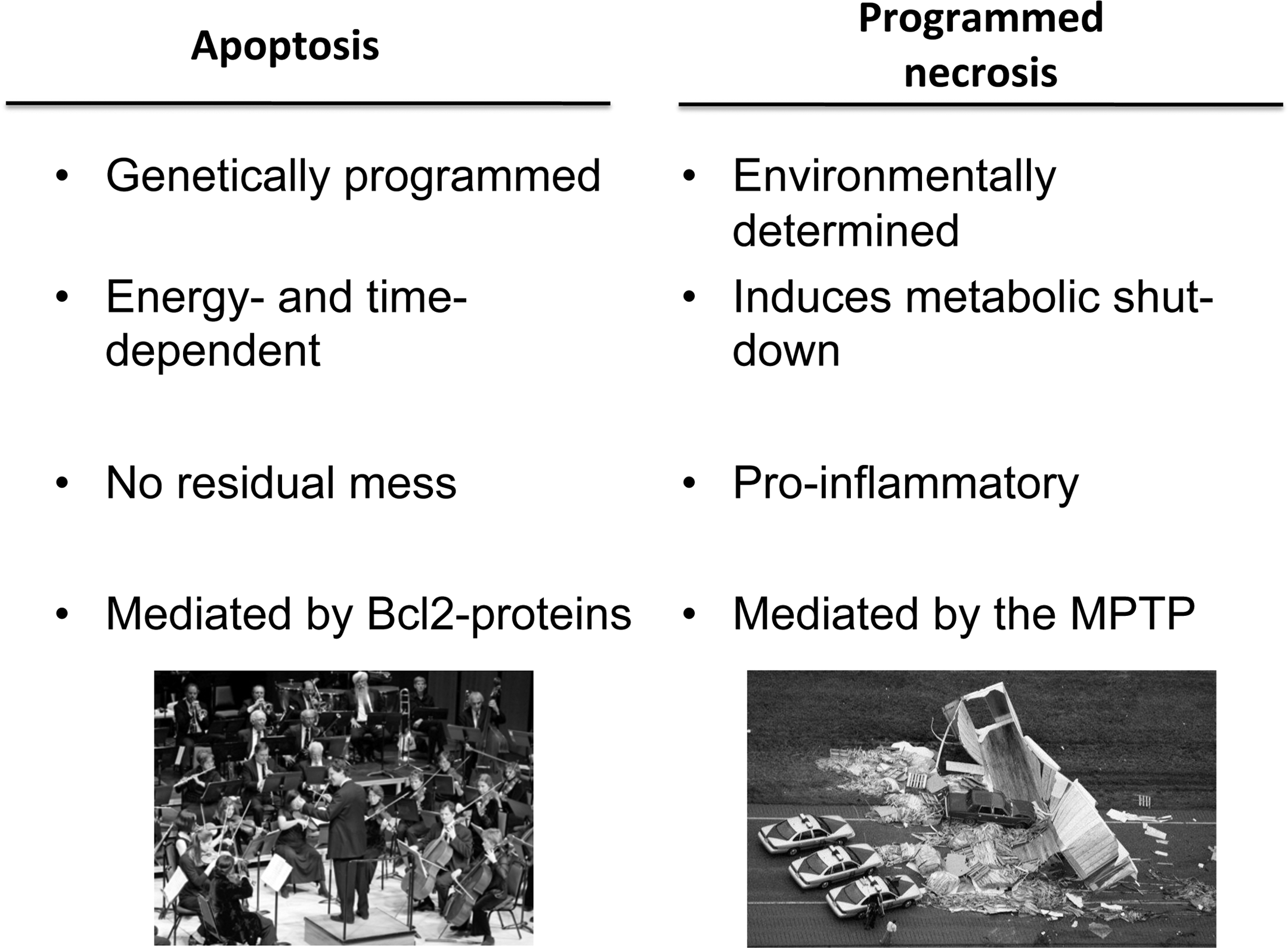
Conceptual and mechanistic differences between apoptosis (left) and programmed cell necrosis (right).
Compared to apoptosis, programmed necrosis is more like a car wreck (Figure 1, right panel). There seems to be no developmental function for programmed necrosis, which is invoked in response to external stimuli (almost always those that directly or indirectly increase calcium influx or release from intracellular stores). The essential mechanistic feature of programmed necrosis is opening of mitochondrial permeability transition pores (MPTP), which disrupts mitochondrial respiratory function. Thus, programmed cell necrosis accrues as a consequence of ATP deficiency, which is the opposite of apoptosis that requires mitochondrial–derived ATP to fuel the multiple processes leading to cell disassembly. Metabolic shutdown in programmed cell necrosis does not permit tidy removal of cell organelles, and so there is reactive inflammation. Extending the car wreck analogy, ambulances, police, and tow trucks are recruited to the scene to clean up the mess, but their presence further disrupts normal traffic flow (i.e., tissue/organ function).
Although apoptosis and programmed necrosis are mechanistically distinct, the two processes are difficult to differentiate in vivo and may occur contemporaneously within the same tissue and pathophysiological context. Absence of specific biochemical or histochemical markers for apoptosis and programmed necrosis has almost certainly contributed to confusion about the prevalence and roles of these two processes in disease. Ultrastructural studies can show chromatin condensation in apoptosis versus nuclear and mitochondrial swelling in programmed necrosis, but it is difficult to quantify the incidence or extent of apotosis and/or programmed necrosis across an entire organ or tissue based on limited sampling from transmission electron microscopy. The TUNEL assay that labels nicked DNA is commonly used to describe an apoptotic index (TUNEL positive nuclei/total nuclei) and infer rates of apoptosis. TUNEL labeling is not specific for apoptosis, however, as cytochrome c released from depolarized necrotic mitochondria can activate apoptotic caspases and downstream DNases. For this reason, cells undergoing programmed necrosis exhibit histological TUNEL positivity, even though the cell is destined to die from necrosis caused by reversal of mitochondrial ATP production. Histological identification of dystrophic calcification or complement activation has been used to identify necrotic tissue. Unfortunately, these markers are not specific for programmed cell necrosis and are also positive with conventional necrosis that is observed after ischemic cerebrovascular accident and myocardial infarction.
Here, I review recent findings that are helping to unravel the complexities of apoptosis and programmed necrosis signaling. As a relevant example, I focus on the functions of a “pro-apoptotic” Bcl-2 family protein, Nix, that can independently mediate both apoptotic and necrotic programmed cell death, therefore contributing to the pathophysiology of such conditions as heart failure and diabetes.
The Role of Programmed Cell Loss in Heart Failure and Diabetes
Shortly after birth (i.e., over the ensuing several days in mice or months in humans), cardiac myocytes lose the ability to undergo cell proliferation; further heart growth is almost entirely through hypertrophic cell enlargement. In the adult heart, normal hypertrophic growth is constrained by physical limits to oxygen diffusion that, when cell cross-sectional area exceeds the ability of oxygen to fully diffuse into the cell, produces ischemic cellular cores (Vatner 1988). Thus, myocardial homeostasis is not achieved as in most other tissues by maintaining a balanced rate of cardiomyocyte loss and renewal, but instead by minimizing the loss of cardiomyocytes that cannot be replaced. Accordingly, there seems to be no “physiological” rate for programmed cardiac myocyte death in normal hearts. Indeed, “normal” cardiomyocyte TUNEL staining is observed only in ∼1/10,000 normal human cardiomyocytes. By comparison, cardiac myocyte apoptosis is 10 or more times that normal value in failing, ischemic, and pressure overloaded hearts, and programmed loss of cardiomyocytes is believed to play a causal role in progression from functionally compensated hypertrophy to decompensated or failing cardiomyopathy (Whelan, Kaplinskiy, and Kitsis 2010). Genetic reprogramming leading to cardiomyocte death is a molecular hallmark of reactive pathological hypertrophy that is induced in response to myocardial injury or hemodynamic overload (Dorn 2005, 2009).
In contrast to terminally differentiated and nonproliferative adult cardiac myocytes, insulin producing β-cells of pancreatic islets are constantly remodeling according to the physiological requirements. β-cells are stimulated to proliferate (Kulkarni et al. 2004) or undergo programmed elimination (Bonner-Weir 2000) as needed to maintain physiological insulin homeostasis according to dietary intake and nutritional status. Diabetes occurs when endogenous insulin secretion by β-cells is insufficient to meet metabolic demands, either because of an absolute insulin deficiency in type 1 diabetes (Notkins and Lernmark 2001) or under conditions where insulin levels are normal, but are still functionally inadequate for a given body mass because of obesity and/or insulin resistance (type 2 diabetes; Pimenta et al. 1995; Rhodes 2005). Thus, both type I and type II diabetes are caused by (absolute or relative) deficiency in insulin-producing β-cells, (Mathis, Vence, and Benoist 2001). While β-cell loss in diabetes had been mechanistically linked to increased β-cell apoptosis (Johnson et al. 2003; Thomas and Biden 2009; Eizirik, Colli, and Ortis 2009), inhibiting apoptosis in β-cells has been incompletely effective as a therapeutic approach (Choi et al. 2009). This suggests that an additional or parallel mechanism leading to β-cell loss is also provoked by diabetes (Cnop et al. 2005). Recent studies have identified this mechanism as programmed necrosis.
A Brief Review of the Molecular Pathways for Apoptosis and Programmed Necrosis
Apoptosis signaling occurs via cascade activation of caspase proteases. There are two different proximal pathways leading to caspase activation: (1) the extrinsic pathway initiated via activated cytokine “death” receptors that process initiator caspase 8; and (2) the intrinsic pathway initiated after cytosolic release of mitochondrial derived cytochrome c, in which caspase 9 is activated. Both pathways terminate with activation of the executioner caspase, caspase 3. As the focus of this review is on different mitochondrial pathways leading to programmed cell death, here I review details of the intrinsic pathway only.
The sine qua non of mitochodrial pathway apoptosis is activation of Bcl-2 family of mitochondrial “death” proteins. Bcl-2 family proteins are broadly classified as pro-apoptotic or anti-apoptotic. The pro-apoptotic group is further subdivided according to structural/functional characteristics as either regulatory BH3 domain-only factors or pore-forming multidomain factors (Danial and Korsmeyer 2004). As discussed below, dynamic regulation of Bcl-2 factors can contribute to heart disease and diabetes by shifting the balance away from cell survival and toward cell apoptosis.
Cellular stress initiates an apoptotic response when transcriptional upregulation and/or posttranslational processing activates pro-apoptotic BH3-only factors. BH3-only proteins interact with the multidomain Bcl-2 proteins, Bax or Bak (Wei et al. 2001), to cause mitochondrial outer membrane permeabilization that releases mitochondrial apoptogens into the cytosol (Kluck et al. 1997). Mitochondrial outer membrane permeabilization by Bax and Bak is suppressed through the actions of antiapoptotic Bcl-2 proteins such as Bcl-2 and Bcl-xL, either indirectly through their binding and sequestration of proapoptotic BH3-only factors or directly by their binding and inhibition of pore-forming Bax and Bak (Kharbanda et al. 1997). Bax/Bak-mediated destabilization of mitochondrial outer membranes leads to cytosolic relocalization of mitochondrial cytochrome c. This is the initial step in forming the macromolecular apoptosome, consisting of cytochrome c complexed with dATP, the adaptor protein Apaf-1 (apoptotic protease activating factor-1), and caspase 9. Apoptosome formation facilitates Apaf-1 oligomerization that is required for recruitment and activation of procaspase-9, which undergoes autoproteolysis to initiate cascade activation of downstream caspases.
Because unregulated apoptosis would be catastrophic, apoptosis signaling is tightly constrained at multiple levels. As noted, mitochondrial outer membrane pore formation is limited by antiapoptotic Bcl-2 protein binding to Bax and/or Bak. Inhibitor of apoptosis (IAP) family proteins bind to and prevent substrate access to downstream caspases-3 and 7 (Riedl et al. 2001), while concomitantly ubiquitinating and targeting these caspases for proteasomal degradation (Yang et al. 2000). Thus, mitochondrial cytochrome c release may not be sufficient to cause cell apoptosis unless these and other factors that chronically restrain apoptosis are simultaneously inhibited or suppressed, shifting the overall cell homeostatic balance toward cell elimination.
Signaling for programmed necrosis is less complex than that for apoptosis, in the same way that a car wreck is mechanistically less complex than an orchestral performance of Hayden’s Farewell Symphony. The inciting event in programmed necrosis (i.e., the first automobile collision of the chain reaction) is calcium-mediated opening of the MPTP, a nonselective pore in the mitochondrial inner membrane (Haworth and Hunter 1979). Creation of an ion-permeant pore connecting mitochondrial matrix with cytosol leads to dissolution of matrix protons essential for ATP production and dissipation of the normal mitochondrial inner membrane electrochemical potential (▵ψm) that sustains oxidative phosphorylation (Krasnikov et al. 2005). The immediate metabolic effect of calcium-mediated MPTP opening is reversal of ATP synthesis, that is, the mitochondrion becomes a net consumer (rather than a producer) of ATP. Loss of cellular ATP then causes general cell dysfunction and necrotic death. An indirect consequence of MPTP opening is osmotic swelling of the mitochondrial matrix, which, because the inner mitochondrial membrane is redundantly folded into numerous cristae, expands outward and ruptures the mitochondrial outer membrane. Outer membrane rupture permits apoptogenic cytochrome c that is normally sequestered in the compartment between the mitochondrial outer and inner membranes to leak into the cytosol and activate caspases through the intrinsic pathway as described above. For this reason, programmed necrosis is frequently associated with positive biochemical (activated caspase 3) and histological (TUNEL staining) markers of apoptosis. Nevertheless, cell death after MPTP opening does not occur by apoptosis because processing and packaging of cellular DNA, organelles, and soluble proteins into apoptotic bodies require energy in the form of ATP that is critically deficient after mitochondrial depolarization. Instead, cell death occurs via metabolic shutdown.
Research is still underway identifying all of the molecular components of the MPTP (Halestrap and Pasdois 2009; Javadov, Karmazyn, and Escobales 2009). Roles in MPTP function have been established for the voltage-dependent anion channel (VDAC), the adenine nucleotide translocator (ANT), and cyclophilin D (CyP-D; Szabo, De Pinto, and Zoratti 1993; Beutner et al. 1996; Marzo et al. 1998; Baines et al. 2007; Kokoszka et al. 2004). Genetic ablation of CyP-D has been especially useful in uncovering roles for MPTP opening and resulting programmed cell necrosis in murine Alzheimer’s disease, muscular dystrophy, diabetes mellitus, and heart failure (Du et al. 2008; Millay et al. 2008; Fujimoto, Chen, et al. 2010; Chen et al. 2010).
Nix, Cardiomyocyte Apoptosis, and Heart Failure
The genetic program that is induced in adult cardiac hypertrophy and heart failure includes multiple genes normally expressed during embryonic heart development (Dorn, Robbins, and Sugden 2003). Among these are genes encoding cell growth and death factors. Over a decade ago, our laboratory took advantage of a genetic model of cardiac hypertrophy that transitions to heart failure, the Gq transgenic mouse (D'Angelo et al. 1997; Adams et al. 1998), to define specific cardiac death genes that are expressed in hypertrophy and heart failure. Cardiomyocyte-autonomous activation of Gq signaling by overexpressing the alpha subunit of the heterotrimeric Gq signaling protein, Gαq, activated pathways normally stimulated by angiotensin II, α1-adrenergic, or endothelin receptors in the heart. The phenotypic consequence was recapitulation of many molecular, histological, morphological, and functional characteristics of pressure overload hypertrophy (D'Angelo et al. 1997). We also observed that the Gq hypertrophy phenotype progressed over time to classic dilated cardiomyopathy associated with greatly increased cardiomyocyte apoptosis (Adams et al. 1998; Sakata et al. 1998). The molecular underpinnings of cardiomyocyte apoptosis were defined using transcriptional profiling by RNA expression array. Surprisingly, the mRNA signature of non-failing cardiac hypertrophy included markedly increased levels of multiple apoptosis genes (Aronow et al. 2001; Yussman et al. 2002). The most thoroughly studied and important transcriptionally induced cardiac pro-apoptosis gene encodes Nix, one of the BH3-only pro-apoptotic Bcl-2 proteins. Nix is upregulated in a protein kinase C–dependent manner (Galvez et al. 2006) and stimulates mitochondrial outer membrane permeabilization by Bax and Bak. The discovery that Nix expression is intrinsic and apparently inseparable from the genetic reprogramming that occurs in reactive hypertrophy suggested a mechanism linking altered cardiac gene expression and programmed cardiomyocyte loss to the characteristic progression of functionally compensated hypertrophy to dilated cardiomyopathy.
To interrogate the pathophysiological consequences of increased cardiomyocyte Nix, we employed cardiomyocyte-specific forced Nix gene expression using murine transgenesis. Under conditions where cardiac Nix expression was increased shortly after birth the transgenic pups died of fulminant dilated cardiomyopathy on the 7th to 10th day of life. Massive increases in TUNEL positive cardiomyocytes (15–20% of all cardiac myocytes) implicated apoptosis as the mechanism for cardiomyocyte dropout and myocardial insufficiency (Yussman et al. 2002). A follow-up study in which forced cardiomyocyte Nix expression was conditionally induced in adult mouse hearts revealed synergism between genetic reprogramming and physiological stress for inducing cardiomyocyte apoptosis in adult hearts, suggesting how cardiomyocyte apoptosis can be activated after injury or hemodynamic overload in normally apoptosis-resistant adult hearts (Syed et al. 2004).
We further examined the role of Nix as the essential mediator of programmed cardiomyocyte death by creating cardiomyocyte-specific Nix gene knockout mice. Cardiac Nix knockout mice were subjected to chronic pressure overload resembling severe human hypertension by surgical transverse aortic banding. Pressure overloaded Nix-deficient hearts hypertrophied to a normal extent, but showed a ∼50% reduction in the “normal” amount of cardiomyocyte TUNEL positivity seen in this model. Additionally, cardiac Nix knockout mice showed substantial reductions in cardiomyocyte replacement fibrosis (a late marker of apoptotic cardiomyocyte dropout) and almost none of the left ventricular chamber dilatation, wall thinning, and decrease in contractile systolic function that is typical of late decompensation after pressure overload hypertrophy (Diwan et al. 2008). We therefore concluded that Nix is one of the major stress-inducible factors responsible for apoptotic cardiomyocyte loss, ventricular remodeling, and functional decompensation of pressure overload hypertrophy. The related BH3-only protein Bnip3, which is structurally and functionally similar to Nix (but encoded by a different gene that is regulated by tissue hypoxia), plays an analogous role mediating programmed cardiomyocyte loss and adverse ventricular remodeling after myocardial infarction (Diwan, Krenz, et al. 2007).
Nix-stimulated Programmed Necrosis Identifies Parallel Cardiomyocyte Death Signaling Pathways
As reviewed above, all pro-apoptotic Bcl-2 family members (including Nix and Bnip3) promote mitochondrial outer membrane permeabilization that activates endogenous pathways leading to caspase-dependent apoptosis. Accordingly, Nix primarily localizes to mitochondria (Yussman et al. 2002). However, we unexpectedly detected a fraction of both endogenous and transfected Nix protein that localized at the endoplasmic reticulum (ER) or its analogous organelle in cardiac myocytes, the sarcoplasmic reticulum (SR; Diwan et al. 2009). We further observed that dynamic regulation of Nix affected SR calcium homeostasis: cells/hearts with greater Nix expression had a ∼20% increase in SR calcium content, whereas cells/hearts lacking Nix had a ∼20% lower SR calcium content. These results led to the hypothesis that Nix-mediated regulation of SR calcium was increasing cardiomyocyte sensitivity to programmed cell necrosis through calcium-dependent MPTP opening. Consistent with this paradigm, genetic manipulations that normalized the characteristically depressed SR calcium levels of Nix knockout mice largely abolished the cardioprotection typically conferred by Nix gene ablation (Diwan et al. 2009). We further observed that Nix mutants engineered to localize specifically and exclusively to either cell/cardiomyocyte mitochondria or to ER/SR activated only one of the two different programmed cell death pathways, apoptosis and programmed necrosis, respectively (Chen et al. 2010). Transfecting cultured fibroblasts with mitochondrial–targeted Nix induced pure apoptosis defined as caspase activation without dissipation of the mitochondrial inner membrane electrochemical gradient, ▵ψm. By comparison, forced expression of ER-targeted Nix caused MPTPs to open, revealed by dissipation of mitochondrial ▵ψm. Pharmacological (cyclosporin A) or genetic (CyP-D/Ppif ablation) inhibition of MPTP opening largely prevented programmed cell death induced by ER-targeted Nix without affecting apoptotic death induced by mitochondrial targeted Nix (Chen et al. 2010).
Concomitant activation of parallel but independent apoptotic and necrotic cell death pathways by translationally upregulated Nix in hypertrophied hearts raised the question as to which pathway of cardiomyocyte elimination is more important for the pathogenesis of heart failure. To answer, we engineered transgenic mice in which we could force in vivo cardiomyocyte-specific expression of either mitochondrial–targeted or SR-targeted Nix, and examined the consequences on cardiac remodeling, function, and development of overt heart failure (Chen et al. 2010). Because apoptosis and programmed necrosis can both increase TUNEL positivity and biochemical caspase activity (see above), we assayed cardiac myocyte necrosis using histological staining for complement 9 protein, a marker of local inflammation. Anti-complement 9 labelling or mitochondrial swelling, matrix degeneration, and outer membrane disruption (visualized by transmission electron microscopy) were observed only in cardiomyocytes isolated from SR-targeted Nix expressing mice. This result established an in vivo link between SR localization of Nix and programmed necrosis. We further determined that MPTP opening is an essential part of the mechanism of cardiomyocyte death induced by SR-targeted Nix by genetically ablating ppif (encoding CyP-D) in mice expressing the mitochondrial or ER/SR-targeted Nix mutant. Abrogation of the MPTP by ppif ablation prevented complement 9 staining, normalized mitochondrial ultrastructure, and prevented the dilated cardiomyopathy typically induced by cardiac expression of SR-targeted Nix. In contrast, ppif ablation only slightly improved apoptotic cardiomyopathy induced by mitochondrial targeted Nix. To our knowledge, these are the first findings to prove that programmed necrosis mediated by a SR/ER localized Bcl-2 factor can significantly impact end-organ function in vivo.
β-Cell Apoptosis and Programmed Necrosis in Diabetes
As introduced earlier, programmed elimination of β-cells from pancreatic islets and resulting insulin insufficiency are implicated in the pathogenesis of diabetes. We have studied apoptosis and programmed necrosis in a genetic model of diabetes, the Pdx1 haploinsufficient (+/−) mouse, that mimics the human conditions of heritable maturity onset diabetes of the young type 4 and type 2 diabetes (Stoffers et al. 1997; Stoffers, Stanojevic, and Habener 1998; Macfarlane et al. 1999). Whereas homozygous germ-line Pdx1 gene deletion produces lethal pancreatic agenesis (Jonsson et al. 1994), heterozygous germ-line or β-cell-specific Pdx1 null mutations induce a diabetic phenotype (Brissova et al. 2002; Gannon et al. 2008) that has been mechanistically attributed to both decreased β-cell proliferation (Sharma et al. 1999) and increased β-cell apoptosis and/or autophagy (Johnson et al. 2003, 2006; Fujimoto et al. 2009). (The consensus opinion seems to be that increased apoptotic β-cell death, and not decreased proliferation, is the most important mechanism; Thomas and Biden 2009.) Because the commonly used research markers do not faithfully distinguish between apoptotic and necrotic cell death, we addressed the question of “By what mechanism do β-cells die?” using pharmacological or genetic inhibition of the essential MPTP regulator, CyP-D. Our first studies were performed in vitro using cultured mouse insulinoma-derived β-cells (MIN6 cells). In these cells, Pdx1 expression can be suppressed using specific shRNA, thereby mimicking Pdx1 insufficiency caused by loss-of-function mutations in the human disease (Fujimoto, Chen, et al. 2010). Pdx1 shRNA increased MIN6 cell death and was associated with loss of MIN6 mitochondrial inner membrane potential, that is, with opening of MPTP. Treatment of MIN6 cells with cyclosporin A, a pharmacological inhibitor of CyP-D, prevented MPTP opening and protected Pdx1-deficient cells from programmed death.
The above results suggested a role for programmed necrosis in β-cell demise. However, one cannot evaluate diabetes in a petri dish. To translate the cell biology findings to an experimental disease model, we designed parallel studies that used analogous approaches in genetically manipulated mice. Pdx1+/− mice develop diabetes (measured as abnormal glucose tolerance) when fed a high fat diet: Pancreatic islets of Pdx1+/− mice are abnormally small with decreased insulin staining, indicating loss of pancreatic β-cells. The rate of Pdx1+/− β-cell TUNEL positivity is abnormally high, supporting conclusions that apoptosis is increased by Pdx1 insufficiency. We also observed increased complement 9 staining of Pdx1+/− islets, revealing local inflammation that would be generated by programmed necrosis, but not from apoptosis.
We developed Pdx1+/− diabetic mice with either functional MPTP or nonfunctional MPTP by cross-breeding to ppif null (i.e., CyP-D knockout) mice. Genetic ablation of CyP-D (and the resulting loss of functional MPTPs) restored Pdx1+/− pancreatic islet size and insulin staining while decreasing β-cell TUNEL labeling and islet complement 9 staining, demonstrating at least a partial role for programmed necrosis in β-cell loss. Finally, we found that CyP-D ablation almost fully normalized the characteristic decrease in circulating insulin and increase in blood glucose (at baseline and in response to an acute glucose challenge) seen in the Pdx1 haploinsufficient mouse model of diabetes. These results reveal an important, perhaps even dominant, role for MPTP-mediated cell necrosis in β-cell death caused by Pdx1 deficiency.
Unexpectedly, the pathways leading to programmed β-cell death in diabetes caused by Pdx1 deficiency were found to be directed by Nix. Unbiased transcriptional profiling of Pdx1-shRNA treated cultured MIN6 cells identified Nix as one of the upregulated mRNAs, and forced Nix expression was sufficient to induce MIN6 cell death. We also detected increased Nix mRNA in pancreatic islets isolated from Pdx1+/− mice, suggesting a role for Nix in the in vivo diabetic phenotype (Fujimoto, Ford, et al. 2010). We examined this by deleting Nix in Pdx1+/− mice, that is, we crossed the two knockout models to achieve concomitant hemizygous (+/−) Pdx1 deletion and homozygous (−/−) Nix ablation (Diwan, Koesters, et al. 2007) in the same animal. In this manner, we prevented upregulation of Nix in Pdx1+/− mice by interrupting its gene. Nix ablation eliminated the high pancreatic islet β-cell TUNEL labeling characteristic of Pdx1+/− mice and normalized islet size and insulin staining. Insulin secretion was normalized (i.e., increased) and reactive hyperglycemia limited by Nix ablation, thus “rescuing” or “curing” the diabetes phenotype.
Summary
The findings reviewed herein place Nix at the apex of multiple signaling pathways leading to programmed death of cardiac myocytes and pancreatic β-cells. Although these data may provide compelling reasons to therapeutically target Nix in heart failure and diabetes, the take-away message is not that Nix is important in this or that disease, but that “pro-apoptotic” Bcl-2 factors seem to have been misnamed. Certainly Nix, Bnip3, Bax, Bak, and the other pro-apoptotic Bcl-2 proteins can and do promote mitochondrial outer membrane destabilization leading to caspase activation that produces apoptosis. But they can also localize to SR and ER where they promote calcium release that opens MPTPs, create mitochondrial dysfunction, and lead to programmed necrosis. In general, researchers have not paid sufficient attention to programmed cell death from MPTP-dependent necrosis because we have not recognized it as such; TUNEL staining does not reliably distinguish between the two processes and caspases may be activated during both. Nevertheless, the mechanisms and consequences of cell death differ, as will effective preventative measures. When considering appropriate therapeutics, one wonders whether interfering with one pathway or the other will be effective in most conditions, or whether cells that have decided to die using mitochondrial mechanisms will, for example, simply shift from apoptosis to necrosis if caspase activation is blocked. This paradigm may explain the limited success of pharmacological caspase inhibition for most experimental conditions characterized by “increased apoptosis.”
Footnotes
The author declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
The author received no financial support for the research, authorship, and/or publication of this article.