
Abstract
p53 is well known as a regulator of apoptosis and autophagy. In addition, a recent study showed that p53 is a modulator of the opening of the mitochondrial permeability transition pore (mPTP), a trigger event of necrosis, but the role of p53 in necrosis induced by myocardial ischemia–reperfusion (I/R) remains unclear. The aim of this study was to determine the role of p53 in acute myocardial I/R injury in perfused mouse hearts. In male C57BL6 mice between 12 and 15 weeks of age, 2 types of p53 inhibitors were used to suppress p53 function during I/R: pifithrin-α, an inhibitor of transcriptional functions of p53, and pifithrin-μ, an inhibitor of p53 translocation from the cytosol to mitochondria. Neither infusion of these inhibitors before ischemia nor infusion for the first 30-minute period of reperfusion reduced infarct size after 20-minute ischemia/120-minute reperfusion. Infarct sizes were similar in p53 heterozygous knockout mice (p53+/−) and wild-type mice (WT), but recovery of rate pressure product (RRP) 120 minutes after reperfusion was higher in p53+/− than in WT. The protein expression of p53 in WT was negligible under baseline conditions, during ischemia, and at 10 minutes after the start of reperfusion, but it became detectable at 120 minutes after reperfusion. In conclusion, upregulation of p53 during the late phase of reperfusion plays a significant role in contractile dysfunction after reperfusion, although p53 is not involved in cardiomyocyte necrosis during ischemia or in the early phase of reperfusion.
Introduction
Opening of the mitochondrial permeability transition pore (mPTP) is a critical event in necrosis induced by myocardial ischemia/reperfusion (I/R). 1 –3 Myocardial ischemia primes the mPTP for opening via adenosine triphosphate (ATP) depletion, accumulation of inorganic phosphate, and Na+ overload. Subsequent reperfusion induces Ca2+ overload, overproduction of reactive oxygen species (ROS), and recovery of pH, which trigger mPTP opening. Cyclophilin D (CypD) plays a regulatory role in mPTP opening. 3 In addition, there is a tight association between GSK-3β activity and the threshold for mPTP opening. 1,2,4
Although p53 is known to regulate apoptotic and autophagic pathways, through mechanisms that are both transcription dependent and transcription independent, a few lines of evidence suggest that p53 potentially modulates I/R-induced necrosis. First, cerebral infarct size after 1-hour cerebral artery occlusion (CeAO)/24-hour reperfusion was reduced in p53 heterozygous knockout mice (p53+/−), and a similar effect was seen in wild-type mice (WT) pretreated with cyclosporine A (CsA), an inhibitor of CypD. 5 p53 was translocated to mitochondria and associated with CypD at 24 hours after reperfusion. Furthermore, reduction in p53–CypD complex formation by p53 deletion suppressed mPTP opening in some types of cells, 5 suggesting that p53 directly regulates CypD-mediated mPTP opening. Second, active GSK3β, which promotes mPTP opening, interacts with p53 in the mitochondria and enhances p53 function. 6 Finally, p53 localizes on the outer membrane of mitochondria and associates with voltage-dependent anion channel (VDAC), 7 and adenine nucleotide transport through VDAC was implicated in ATP depletion during ischemia. 2,8 These findings led us to examine the role of p53 in myocardial I/R injury.
To inhibit p53 function selectively during ischemia or upon reperfusion, we used a buffer-perfused heart preparation, which allows us to analyze the relationship between timing of p53 inhibition and its impact on I/R injury. The effect of genetic ablation of p53 on I/R injury was also examined.
Methods
Animals
This study was conducted in accordance with the Guide for the Care and Use of Laboratory Animals published by National Research Council of the National Academies, United States (2011) and approved by the Institutional Laboratory Animal Care and Use Committee. Male C57BL/6 mice (12-15 weeks of age) and p53 heterozygous knockout mice (p53+/−) were obtained from The Jackson Laboratory (Bar Harbor, Maine, USA).
Perfusion Protocols
Anesthetization and preparation of isolated hearts were performed as described previously with slight modification. 9 In brief, mice were anesthetized with a mixture of ketamine (90 mg/kg, intraperitoneally [ip]) and xylazine (10 mg/kg, ip), and hearts were quickly excised and perfused at a pressure of 100 cm with noncirculating Krebs-Henseleit buffer (NaCl 120, KCl 4.7, MgSO4 1.2, KH2PO4 1.2, NaHCO3 25, CaCl2 1.75 and glucose 10 mmol/L). The buffer was gassed with 95% O2/5% CO2, and the temperature of the perfusate was maintained at 37°C. A fluid-filled latex balloon was inserted into the left ventricle to monitor hemodynamics.
After equilibrium perfusion for 30 minutes, the hearts were subjected to 20 minutes of global ischemia followed by 120 minutes of reperfusion for infarct size determination. The hearts were assigned to 1 of 8 treatment groups: dimethyl sulfoxide (DMSO) control (no inhibitor), 15-minute infusion of 2.5 μmol/L pifithrin-α prior to ischemia, 30-minute infusion of 2.5 μmol/L pifithrin-α upon reperfusion, 15-minute infusion of 25 μmol/L pifithrin-μ prior to ischemia, 30-minute infusion of 25 μmol/L pifithrin-μ upon reperfusion, and 5-minute infusion of 25 μmol/L or 10 μmol/L or 1 μmol/L of pifithrin-μ prior to ischemia as shown in Figure 1A. Left ventricular developed pressure (LVDP) and heart rate (HR) were recorded and digitized using a power lab system (ADInstruments, Colorado Springs, Colorado). After 2 hours of reperfusion, hearts were perfused with 1% 2,3,5-triphenyltetrazolium chloride (TTC) and incubated in TTC at 37°C for 15 minutes, followed by fixation in 10% formaldehyde. Infarct size was determined as the percentage of total ventricular area. 9

Effect of p53 inhibition on ischemia/reperfusion (I/R) injury. A, Experimental protocol (study using inhibitors). Myocardial infarction was induced by 20-minute global ischemia/2-hour reperfusion. Two types of p53 inhibitors (pifithrin-α and pifithrin-μ) were infused at the indicated doses and for the indicated time periods. B, Effects of p53 inhibitors on infarct size and functional recovery after I/R (study using inhibitors). Infarct size was expressed as a percentage of the left ventricular area. Rate pressure product (RPP) was calculated by multiplying left ventricular developed pressure (LVDP) by heart rate (HR). NS indicates not significant.
Immunoblotting
To obtain a total homogenate, frozen heart samples were homogenized in ice-cold buffer containing 20 mmol/L Tris-HCl (pH 7.5), 150 mmol/L NaCl, 1 mmol/L Na2EDTA, 1 mmol/L EGTA, 1% Triton X-100, 2.5 mmol/L sodium pyrophosphate, 1 mmol/L β-glycerophosphate, 1 mmol/L Na3VO4, 1 μg/mL leupeptin, 50 μg/mL phenylmethylsulfonyl fluoride (PMSF), a protease inhibitor cocktail (Complete mini, Roche Molecular Biochemicals, Mannheim, Germany), and a phosphatase inhibitor cocktail (PhosSTOP, Roche Molecular Biochemicals, Mannheim, Germany). The homogenate was centrifuged at 13000g for 15 minutes to obtain the supernatant. Protein concentration was determined using the Bradford assay.
Equal amounts of proteins (80 μg) were electrophoresed on 4% to 12% polyacrylamide gels and then blotted onto PVDF membranes (Millipore, Bedford, Massachusetts). After blocking had been performed with a tris-buffered saline (TBS-T) buffer containing 5% bovine serum albumin (BSA), the blots were incubated with antibodies that recognize the following: p53 (1C12, Cell Signaling Technology, Beverly, Massachusetts), p53 (PAb122, BD Biosciences, San Jose, California), and α-tubulin (Abcam, Cambridge, Massachusetts). Immunoblotted proteins were visualized using an Immobilon Western Detection Kit (Millipore, Billerica, Massachusetts).
Statistics
All data are presented as means ± standard error of mean (SEM). Differences in the parameters between study groups were analyzed by 1-way analysis of variance (ANOVA). When overall ANOVA indicated a significant difference, multiple comparisons were conducted by the Tukey post hoc test. A P-value <.05 was considered to be statistically significant.
Results
Effects of p53 Inhibitors on I/R Injury
Two types of p53 inhibitors, pifithrin-α and pifithrin-μ, were used in this study. Pifithrin-α mainly blocks the transcriptional activity of p53, whereas pifithrin-μ inhibits the translocation of p53 to the mitochondria, resulting in suppression of transcription-independent apoptosis. In a preliminary experiment, infusion of 10 μmol/L pifithrin-α for 15 minutes clearly decreased HR, LVDP, and rate pressure product (RPP) as shown in Table 1, but 2.5 μmol/L pifithrin-α did not change hemodynamic parameters. In addition, the protein level of p53 upregulated modulator of apoptosis (PUMA) was increased after 1-hour perfusion with 2 μmol/L of adriamycin, which was completely blocked by perfusion with a combination of 2 μmol/L of adriamycin and 2.5 μmol/L of pifithrin-α (data not shown). Therefore, 2.5 μmol/L of pifithrin-α was selected for infarct size experiments. Doses of pifithrin-μ were selected based on a previous study. 10 To inhibit the function of p53 before ischemia or upon reperfusion, inhibitors were infused for 15 minutes or 5 minutes before ischemia or for 30 minutes commencing upon reperfusion as shown in Figure 1A.
Hemodynamic Parameters.a

Abbreviations: HR, heart rate; LVDP, left ventricular developed pressure; RPP, rate pressure product; CF, coronary flow; PFT, pifithrin.
aData are mean ± SEM. End of perfusion = 15 minutes after perfusion with each drug.
b P < 0.05 vs Control (DMSO).
There were no significant intergroup differences in HR, LVDP, RPP, and coronary flow under baseline conditions (Table 2). Infarct size as a percentage of left ventricular area was 54.3% ± 3.0% in the control group. As shown in Figure 1B and Table 3, infusion of 2.5 μmol/L pifithrin-α before ischemia or during the first 30 minutes of reperfusion did not reduce infarct size. Infarct size was also not modified by 25 μmol/L of pifithrin-μ before ischemia or during reperfusion. Similar results were obtained when different doses of pifithrin-μ were infused (Figure 1B and Table 3). The LVDP and RPP at 120 minutes after reperfusion were not significantly changed by pifithrin-α or pifithrin-μ (Figure 1B and Table 2).
Hemodynamic Parameters (Inhibitors Study).a
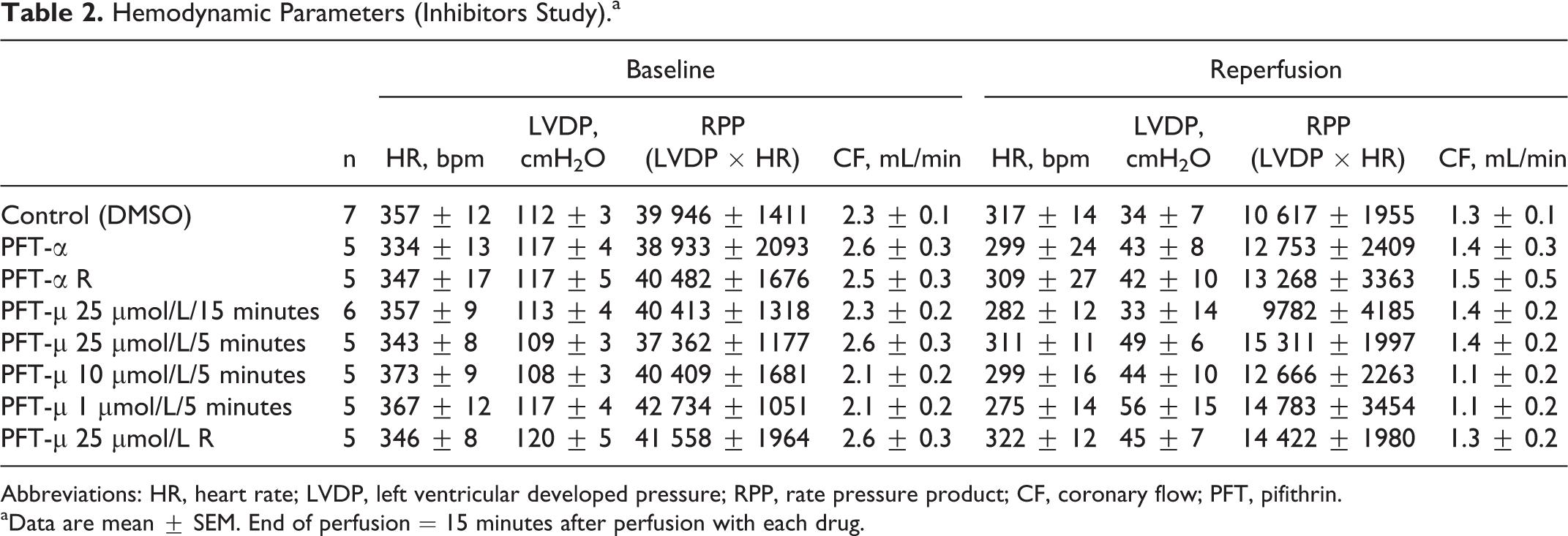
Abbreviations: HR, heart rate; LVDP, left ventricular developed pressure; RPP, rate pressure product; CF, coronary flow; PFT, pifithrin.
aData are mean ± SEM. End of perfusion = 15 minutes after perfusion with each drug.
Infarct Size Data (Inhibitors Study).a
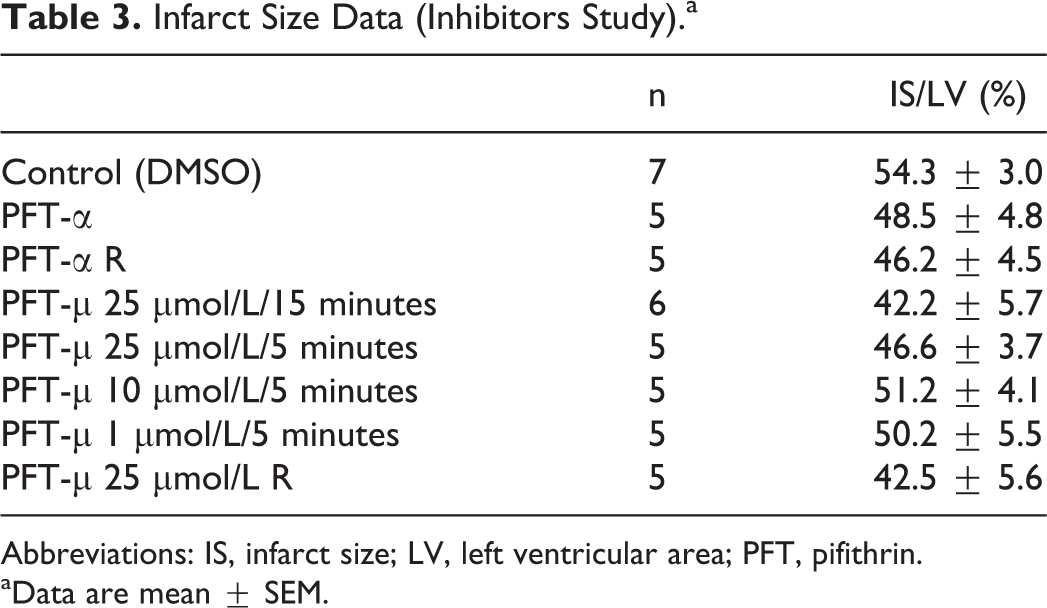
Abbreviations: IS, infarct size; LV, left ventricular area; PFT, pifithrin.
aData are mean ± SEM.
Immunoblotting Experiments
The change in p53 expression level during I/R was examined in perfused mouse hearts. A previous study by Shimizu et al showed that p53 antibodies detected a nonspecific band of approximately 55 kDa, which was derived from blood. 11 This finding was confirmed in our experiments because a 55-kDa band that was detected using p53 antibodies was lost when mouse hearts were perfused with Krebs-Henseleit buffer for 5 minutes (Figure 2B). Because a nonspecific band of 55 kDa seems to be the heavy chains of immunoglobulin as shown by Shimizu et al, 11 a secondary antibody that specifically recognizes the light chains of immunoglobulin was used for Western blot analysis.

Change in p53 expression during ischemia/reperfusion. A, Experimental protocol. Ventricular tissues were sampled at the indicated time points. Myocardial infarction was induced by 20-minute global ischemia/2-hour reperfusion. per = perfusion, I = ischemia, R = reperfusion. B-D, Representative immunoblots for p53 using antibodies that recognize 2 different amino acid sequences of p53. Ventricular tissues sampled from mice that received intraperitoneal injection of adriamycin (ADR) served as positive controls.
In ventricular tissues sampled at 30 minutes after the onset of perfusion, p53 was barely detected (Figure 2C). Expression of p53 was still negligible after 20 minutes of global ischemia or at 10 minutes after the start of reperfusion, but it became modestly detectable after 120 minutes of reperfusion but less than is observed in ventricular tissues sampled from mice that received an intraperitoneal injection of adriamycin (Figure 2C). These results were confirmed using 2 antibodies that recognize different amino acid sequences of p53 (Figure 2D).
Effect of Targeted Deletion of p53 on I/R Injury
Since immunoblotting experiments suggested the possible contribution of p53 upregulation at the late phase of reperfusion to I/R injury, the effect of targeted deletion of p53 on myocardial I/R injury was examined. Since p53 null mice display early tumor development, the effect of p53 suppression on I/R injury was examined using p53+/− mice. Baseline hemodynamic parameters were comparable in WT and p53+/− (Table 4). As shown in Table 4 and Figure 3, LVDP and RPP after 120 minutes of reperfusion were significantly higher in p53+/− than in WT, indicating that targeted deletion of p53 improved functional recovery after I/R. On the other hand, there was no significant difference in infarct size between WT and p53+/− (49.9% ± 5.4% vs. 41.6% ± 4.3%, P = .25).
Hemodynamic Parameters (Knockout Study).a

Abbreviations: HR, heart rate; LVDP, left ventricular developed pressure; RPP, rate pressure product; CF, coronary flow; WT, wild type, p53+/−, p53 heterozygous knockout mouse; baseline, 25 minutes after the start of perfusion, reperfusion, 120 minutes after reperfusion. aData are mean ± SEM.
b P < .05 vs WT.

Effect of genetic deletion of p53 on ischemia/reperfusion (I/R) injury (knockout study). A, B Effects of genetic deletion of p53 on infarct size (A, B) and functional recovery (C) after I/R. Infarct size was expressed as a percentage of the left ventricular area. Rate pressure product (RPP) was calculated by multiplying left ventricular developed pressure (LVDP) by heart rate (HR). Hatched bar = Wild type (WT), open bar = p53 heterozygous knockout mice (p53+/−). *P < .05 vs. WT.
Discussion
Favorable effects of p53 inhibition on remodeling after myocardial infarction and pressure overload-induced heart failure have been established, 12,13 but the role of p53 in acute myocardial I/R injury remains unclear. In this study, we investigated the effect of p53 inhibition on cell death induced by myocardial I/R. Neither infusion of p53 inhibitors before ischemia nor infusion during reperfusion reduced infarct size. A study by Halestrap’s group demonstrated that the mPTP remained closed during global ischemia but opened within 5 minutes after reperfusion in an isolated rat heart. 3 Therefore, the critical timing for mPTP opening after myocardial I/R is the early phase of reperfusion. However, p53 expression was undetectable at 10 minutes after the start of reperfusion but was clearly detected after 120 minutes of reperfusion. Upregulated p53 at the late phase of reperfusion may increase cardiomyocyte death through an mPTP opening-independent necrotic program such as necroptosis, a programed necrosis induced by cytokines, or damage-associated molecular pattern molecules. However, the possible contribution of p53 upregulation at the late phase of reperfusion to infarct size must be small because infarct sizes were similar in p53+/− and WT. These results indicate that p53 is not involved in myocyte necrosis induced by acute I/R in perfused mouse hearts.
A recent study showed that p53 deletion reduced cerebral infarct size induced by CeAO through reduction in p53-CypD interaction at 24 hours after reperfusion. 5 In that study, upregulation of p53-CypD binding was detected at the same time point as infarct size was determined. However, it was not examined whether the p53-CypD binding precedes the development of cerebral infarction. Thus, the causal relationship between p53-CypD binding and cell death induced by I/R remains unclear.
In this study, infarct sizes were comparable in WT and p53+/−, but functional recovery after reperfusion was better in p53+/− than in WT. Since functional recovery after I/R is an index of combined irreversible (cell death) and reversible (myocardial stunning) injuries of cardiomyocytes, a plausible explanation for the discrepancy in the effects of p53 inhibition on infarct size and functional recovery is significant contribution of p53 to the pathogenesis of stunning but not cell death of cardiomyocytes. How p53 contributes to myocardial stunning is unclear. However, a few speculations are possible. Because the present protocol of p53 inhibition appears to be too short to modify the transcriptional function of p53, inhibition of cytosolic/mitochondrial p53, but not nuclear p53, might have been involved. Overproduction of ROS after reperfusion plays a crucial role in the development of myocardial stunning, 14 and p53 is known to bind with MnSOD, reducing its ROS-scavenging effect. 15 p53 is also known to directly downregulate phosphoglycerate mutase, resulting in suppression of glycolysis. 16 The effect of p53 on glycolysis would have been accentuated in this study because glucose is the major ATP source in the Krebs-Henseleit buffer-perfused heart. Thus, enhanced production of ROS and suppression of glycolysis during the recovery process from I/R injury are plausible mechanisms by which p53 contributes to myocardial stunning. Another possibility is suppression of I/R-induced apoptosis in p53+/− because p53 is a well-known executor of apoptosis in transcription-dependent and -independent manners. Previous studies demonstrated that inhibition of the apoptotic pathway mitigated myocardial I/R injury, but conflicting data were also published. 17 –19 In addition, evidence of apoptosis in the reperfused myocardium relied on detection of DNA fragmentation by a DNA ladder assay and by in situ terminal deoxynucleotidyl transferase-mediated dUTP nick end-labeling (TUNEL) in previous studies. 17 Interestingly, Ohno et al showed that TUNEL-positive cardiomyocytes in the reperfused myocardium have ultrastructual features of necrosis but not apoptosis. 20 Thus, the contribution of apoptosis to myocardial I/R injury remains controversial.
In contrast to the results of the present study, pifithrin-α was cardioprotective against I/R injury in earlier studies as shown in Table 5. 21 –23 Several studies showed that intraperitoneal injection of pifithrin-α before ischemia reduced myocardial infarct size in rabbits and rats in situ. 21,22 Since p53 is highly expressed in inflammatory cells, the protective effect of pifithrin-α in situ might have been achieved by modifying the inflammatory response in the reperfused myocardium. In a study by Mocanu et al, infusion of 10 μmol/L pifithrin-α reduced infarct size in perfused rat hearts. 23 However, as stated in the Results section and shown in Table 1, infusion of this same dose of pifithrin-α significantly reduced LVDP and HR in the present mouse preparation. Although the possibility of a type 2 error cannot be excluded, differences in pifithrin-α dose and an involvement of the inflammatory response in the reperfused myocardium might be responsible for the differences in the results between the present study and previous studies as shown in Table 5. 21 –23
Summary of the Literature Regarding Effects of p53 Inhibition on Acute Myocardial Ischemia/Reperfusion Injury.
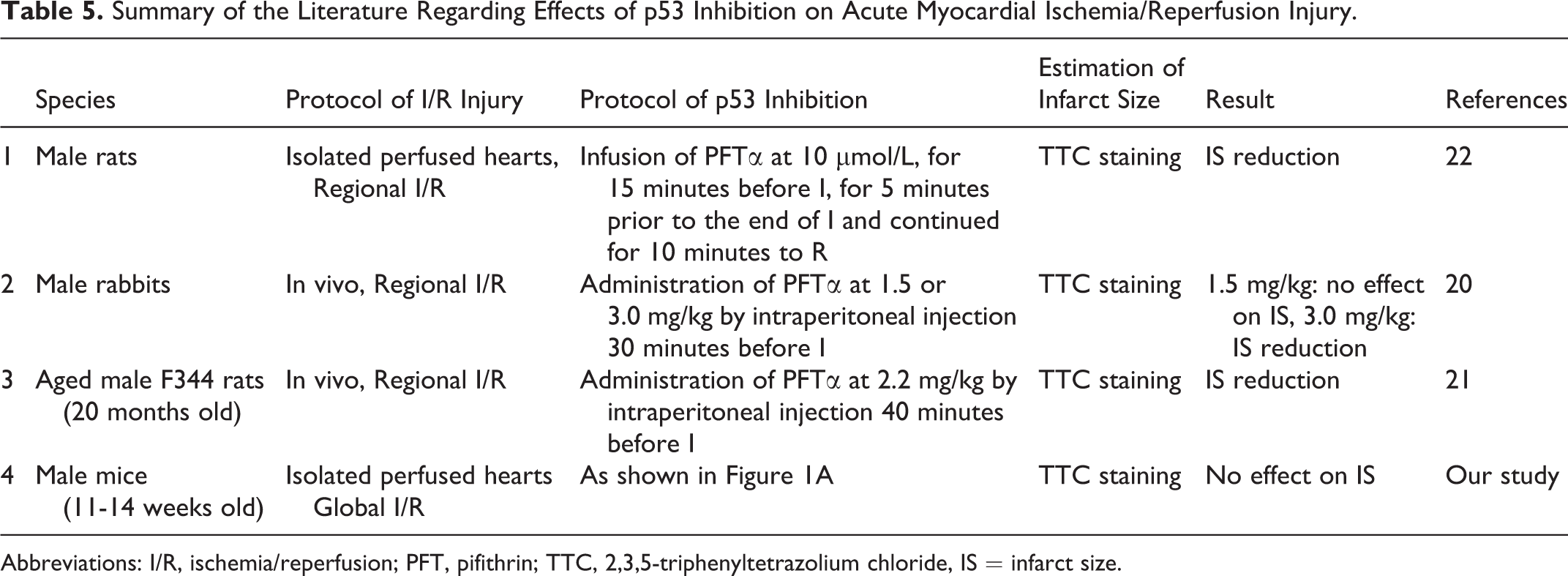
Abbreviations: I/R, ischemia/reperfusion; PFT, pifithrin; TTC, 2,3,5-triphenyltetrazolium chloride, IS = infarct size.
The level of p53 expression was negligible in the myocardium of mice at this age as shown in a previous study. 24 However, p53 is upregulated by diabetes and pressure overload in the myocardium, 13,25 and our recent studies showed that the myocardium of some models of diabetes and hypertension have increased susceptibility to I/R injury. 26 –30 Although a contribution of p53 to I/R-induced infarction was not detected in healthy mice, the possibility of a contribution of p53 to I/R injury in the myocardium with diabetes and other comorbidities cannot be ruled out.
Conclusion
In conclusion, p53 is not involved in cardiomyocyte death induced by I/R injury in perfused mouse hearts. However, upregulation of p53 in the late phase of reperfusion may suppress recovery of the contractile function from I/R injury in the noninfarcted myocardium.
Footnotes
Declaration of Conflicting Interests
The author(s) declared no potential conflict of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research and/or authorship of this article: This study was supported by National Institutes of Health (NIH) grants HL039752 (to C.S.).