
Abstract
Preclinical toxicity studies have demonstrated that exposure of laboratory animals to liver enzyme inducers during preclinical safety assessment results in a signature of toxicological changes characterized by an increase in liver weight, hepatocellular hypertrophy, cell proliferation, and, frequently in long-term (life-time) studies, hepatocarcinogenesis. Recent advances over the last decade have revealed that for many xenobiotics, these changes may be induced through a common mechanism of action involving activation of the nuclear hormone receptors CAR, PXR, or PPARα. The generation of genetically engineered mice that express altered versions of these nuclear hormone receptors, together with other avenues of investigation, have now demonstrated that sensitivity to many of these effects is rodent-specific. These data are consistent with the available epidemiological and empirical human evidence and lend support to the scientific opinion that these changes have little relevance to man. The ESTP therefore convened an international panel of experts to debate the evidence in order to more clearly define for toxicologic pathologists what is considered adverse in the context of hepatocellular hypertrophy. The results of this workshop concluded that hepatomegaly as a consequence of hepatocellular hypertrophy without histologic or clinical pathology alterations indicative of liver toxicity was considered an adaptive and a non-adverse reaction. This conclusion should normally be reached by an integrative weight of evidence approach.
Introduction
Drug and chemically induced liver enlargement in subchronic and chronic toxicology studies in rodents has, for many years, taxed the toxicology profession in terms of its perceived relevance to hepatotoxicity, to carcinogenicity in lifetime bioassays at similar dose levels, and in terms of its relevance to man (Cohen and Grasso 1981; Elcombe, Rose, and Pratt 1985; Grasso and Hinton 1991). The fact that after more than 50 years of debate (Gilbert and Golberg 1965; Rowe et al. 1959; Weil and McCollister 1963) the issue remains as contentious as ever prompted the European Society of Toxicologic Pathology (ESTP) to convene an expert opinion group to discuss the current state of the science. The purpose of the workshop was to discuss the significance of hepatocellular hypertrophy in rodents, define more clearly when adaptive responses become adverse, and understand the long-term consequences of hepatocellular hypertrophy in order to guide scientific opinion for risk assessment in man and dose setting for longer term animal studies.
One critical aspect of hepatic hypertrophy which continues to pose serious questions for toxicologists is the definition of what constitutes an adverse hepatic effect, versus a non-adverse or adaptive effect. This was a major focus of discussion by the expert opinion group resulting in a consensus opinion detailed in this article.
Hepatic Hypertrophy—What Is It?
While to a histopathologist the term hepatic hypertrophy is well recognized and readily defined histologically, to a toxicologist, the term can have various connotations including an increase in the weight of the organ (liver hypertrophy), an increase in the average size of the hepatocytes (hepatocellular hypertrophy), and even hepatic enzyme induction (functionally sometimes referred to as “work” hypertrophy).
In practice, drug- or chemical-induced hepatic hypertrophy tends to be a combination of all of these parameters in addition to others including profound changes in the intracellular enzymes involved not only in phase 1 and 2 drug metabolism (Crampton et al. 1977b; Lake, Longland, et al. 1976; Maronpot et al. 2010) but also in other more fundamental cell processes such as altered oxidative status, fatty acid metabolism, energy production and utilization, cell turnover and altered hepatocellular cytoplasmic, and nuclear morphology (Cattley and Popp 1989; Crampton et al. 1977b; Grasso et al. 1974; Grasso and Hinton 1991).
Archetypical changes that often accompany this phenomenon are increases in hepatic-derived enzymes (transaminases, alkaline phosphatase, and γ-glutamyltransferase) that may appear in the plasma following liver enlargement (Ennulat, Magid-Slav, et al. 2010). Of prime importance for interpretation of these changes with regard to risk assessment are the significant species differences shown in response to chemicals that induce a classic hypertrophic response in the rodent liver (Lake 1995; Lake, Brantom, et al. 1976; Rhodes et al. 1986; Williams and Perrone 1996).
Liver Weight Increases
Increases in liver weight in rodents due to exposure to chemicals can be achieved through a number of mechanisms and can be accompanied by a range of differing histological appearances, some of which clearly show cytotoxicity and cell death, such as carbon tetrachloride and chloroform (Edwards and Dalton 1942; Eschenbrenner and Miller 1945; Grasso and Hinton 1991; Reuber and Glover 1968), while other chemicals such as sodium phenobarbitone, the PPARα (peroxisome activated receptor alpha) agonists, Nafenopin and Clofibrate, and trichloroethylene, induce an increase in liver weight without overt cell necrosis (Lake et al. 1989; Mitchell et al. 1985; Price et al. 1986). While there is an undoubted relationship between hepatic necrosis and its consequences on the eventual increased incidence of neoplasia of the liver (Eschenbrenner and Miller 1945; Grasso and Hinton 1991), this is less clear for chemicals, or rather for doses of chemicals, that induce liver weight increases in the absence of overt hepatocellular damage. The focus of the workshop was on those chemicals that are not traditionally associated with acute hepatic necrosis but rather exert their effect by increasing the weight of the liver through other means.
Organ weight data can provide sensitive indices of toxicologic change where it can correlate and confirm changes seen down the microscope. To this end, the STP have recently published a position paper (Sellers et al. 2007) that gives careful guidance to the collection and weighing of organs in routine toxicology studies. The authors recommend that organ weight data should be routinely expressed as an organ-to-body weight ratio to avoid large variations in body weight skewing organ weight interpretation. In this way, increases in liver organ weight can be accurately correlated with hepatocellular hypertrophy. In rodent studies, where routinely larger numbers of animals are used, these data can be compared with concurrent controls and statistically interrogated to derive p values that may increase confidence through weight of evidence to allow a judgment of hepatocellular hypertrophy when the histological changes appear to be relatively small.
This is particularly important since the magnitude of the increased liver weight can vary considerably between different chemicals. Liver weight changes may also demonstrate a clear dose relationship (Table 1; typically between 110% and 150% of control liver weight). However, it should be noted that although liver weight increases are correlated with microsomal enzyme induction, the degree of microsomal enzyme induction may not be closely correlated with either the magnitude of the liver weight increase or the degree of hepatocyte hypertrophy in rats, dogs, or monkeys (Amacher, Schomaker, and Burkhardt 1998, 2001; Amacher et al. 2006; Ennulat, Walker, et al. 2010).
Dose response relationship for liver weight increase for the PPARα agonist, Fenofibrate (adapted from Price et al. 1986).

Increases in liver weights have been shown to be associated with the induction of increased incidences of hepatocellular neoplasia in 2-year carcinogenicity studies in rodents (Allen et al. 2004; Carmichael et al. 1997) and while considered to be of little relevance to man for some chemicals (Butler and Newberne 1975; Ito and Sugano 1991; McClain et al. 1995; Stevenson et al. 1990), dose levels of a chemical inducing this degree of liver weight increase would be considered to carry an increased risk of inducing hepatic neoplasms in these types of study. In a survey of 138 chemicals used in the agrochemical industry, a relative increase in liver weight of ≥150% of control values after 1 year of treatment was positively correlated with the induction of liver tumors in mice (Carmichael et al. 1997). Similarly, in another survey of mouse NTP studies where correlations between liver weight increases and histological parameters and carcinogenesis were assessed, the authors concluded that “the best single predictor of liver cancer in mice was hepatocellular hypertrophy” (Allen et al. 2004). This study demonstrates a highly significant relationship between increases in liver weight and the future outcome of hepatic neoplasia (p < .001; Table 2). In a similar review of the rat, a less statistically significant relationship (p = .018) between liver weight and hepatocarcinogenesis was also noted, whereby liver weight increases alone correctly predicted eight of the eleven liver carcinogens (but overpredicted twenty-six false positives and failed to predict three true positives; Allen et al. 2004).
Correlation between liver toxicity and carcinogenesis in mouse National Toxicology Program studies (adapted from Allen et al. 2004).
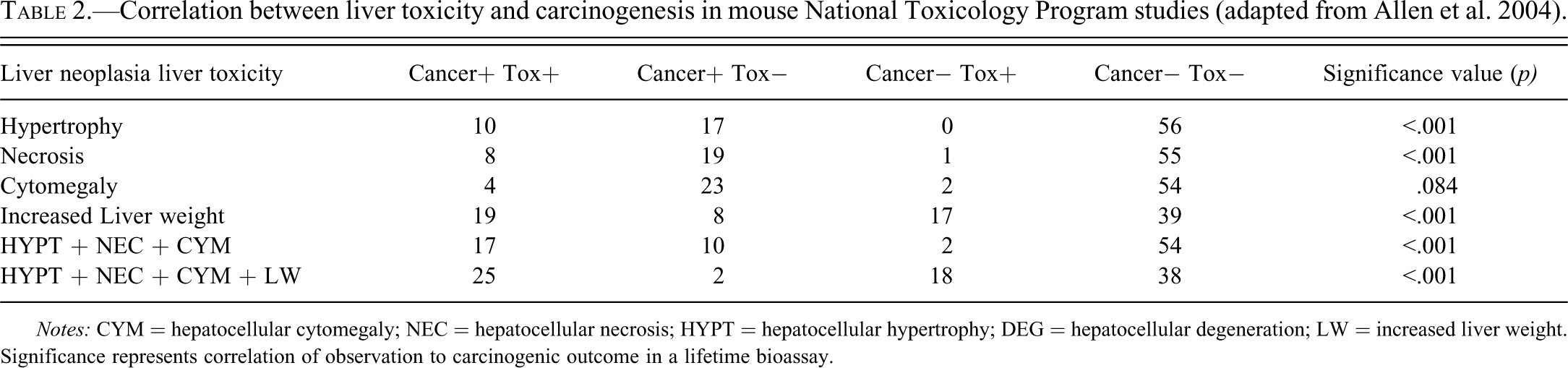
Notes: CYM = hepatocellular cytomegaly; NEC = hepatocellular necrosis; HYPT = hepatocellular hypertrophy; DEG = hepatocellular degeneration; LW = increased liver weight. Significance represents correlation of observation to carcinogenic outcome in a lifetime bioassay.
Effect of Fasting on Liver Weights
A potentially important confounder in terms of the evaluation of liver weight changes following chemical treatment is the practice of fasting animals overnight prior to sacrifice. It is likely that fasting alters the resulting organ weights in rodents over those not fasted (Chatamra, Daniel, and Lam 1984; Rothacker et al. 1988). Studies have shown that the rodent liver weight increases in fed animals are maintained for up to 8 hr after feeding. Water and glycogen account for the major portion of this increase with the former comprising over 66% of the increase (Leveille and Chakrabarty 1967). Overnight fasting will rapidly deplete both components (in preference to other tissues of the body) and accounts for the loss of liver weight seen under fasting conditions (Rothacker et al. 1988). It is currently unclear as to what percentage of laboratories utilizes fasting, but what was apparent from the workshop was that there exists considerable inter-laboratory variation in the practice of overnight fasting of animals prior to necropsy.
Welfare reasons prohibit the overnight fasting of pregnant animals but a systematic study looking into the differences between fasted and non-fasted animals would seem a worthwhile exercise if the former leads to better discrimination of effects provided animal welfare guidelines permit this (Animal and Plant Health Inspection Services [APHIS] 1997; Animal Welfare Information Center 2005). However, it is clear that overnight fasting does have some distinct advantages. First, overnight fasting decreases the variation seen in some clinical pathology parameters and therefore increases the probability of identifying statistically significant changes between control and treated animals (Matsuzawa and Sakazume 1994). Second, animals exposed to high levels of a xenobiotic very frequently experience decreased food intake due to inanition/toxicity which is often noted in toxicological studies as a treatment, and dose related, decrease in hepatocellular glycogen storage (Agren, Wilander, and Jorpes 1931; Lockard et al. 1983). It is possible therefore that loss of glycogen in animals exposed to a xenobiotic might mask statistically significant increases in liver weight. Since liver weight increases are generally considered a sensitive measure of hepatocellular hypertrophy, small changes in these parameters may not be visible using H&E stained sections. This is especially so if animals are not fasted, as a relative loss of glycogen due to toxicity in the high-dose groups is likely to reduce hepatocellular volume and obscure the change in dimensions which helps inform the diagnosis of hypertrophy when comparing treated with concurrent control animals (Li et al. 2003). However, there are clearly well-described changes in gene expression in fasted animals (Bauer et al. 2004; Lkhagvadorj et al. 2009) and further work to assess the relative merits of overnight fasting for subsequent histopathology evaluation would help assess the importance or otherwise of this effect.
The practice of fasting rodents before necropsy was one that was debated at the workshop but because of the differing experiences of the group a recommendation was made for a comparison dose–response study with a known hepatotrophic agent, such as sodium phenobarbitone, in the rat where groups would be fasted and the results obtained compared to non-fasted groups. The NOEL for the histopathology of hepatocellular hypertrophy would then be compared between the fasted and non-fasted groups.
Clinical Pathology
The following clinical pathology parameters can help in the assessment of adverse effects on the liver evolving during enzyme induction:
Alanine Aminotransferase and Aspartate Aminotransferase (ALT/AST)
Several studies in rats dosed with liver enzyme–inducing compounds have been published. In these studies ALT and AST activity increases were below twofold of the controls: sodium phenobarbitone (an archetypical mixed CYP 2B/3A inducer) given to rats at dose levels where centrilobular hypertrophy was present without accompanying degenerative changes induced small increases in both hepatic ALT (Boll et al. 1998) and serum ALT and AST (Lake and Evans 1993). In contrast, administration of the CYP 1A inducer, 3-methylcholanthrene to rats, at dose levels that induced significant levels of drug-metabolizing enzymes, failed to show any increase in serum ALT or AST (Lake and Evans 1993). Peroxisome proliferators (PPARα agonists and CYP4A inducers), similarly show minimally increased levels of serum ALT and AST even when given at dose levels that induce significant increases in liver weight, provided that the only histological effect seen in the liver is hepatocellular hypertrophy (Eacho et al. 1985; Huang and Shaw 2002; Kramer et al. 2003; Peterson et al. 2004).
Increases in serum ALT activity without increases in hepatic ALT activity are thought to be due to damage to, and leakage from, hepatocyte cell membranes, resulting in a release of enzymes from the cytosol into the blood (Amacher 1998). Alternatively, increased enzyme synthesis, as a consequence of liver enzyme induction, may also lead to higher ALT levels in both the liver and the serum. In this scenario, moderately higher serum ALT levels, especially in mice treated with liver enzyme inducers, might occur without cell membrane damage (Strauss, pers. comm. 2011). Boone et al. (1985) concluded that increases in serum ALT activity in the range of 2 to 4× or higher in individual or group mean data when compared with concurrent controls should raise concern as an indicator of potential hepatic injury unless a clear alternative explanation is found. Based on the recommendations of regulatory authorities, (EMEA 2010; FDA 2009; HED 2002) increases in ALT activity of two- to threefold should be considered as indicative of hepatocellular damage. Distinguishing irreversible and reversible liver injury is pragmatically dealt with by assessing the magnitude of the transaminase elevation where minimal and reversible hepatic injury is commonly accompanied by small increases in transaminase levels of less than twofold (Kramer et al. 2003; Lassen 2004; Peterson et al. 2004; Satoh et al. 1982; Solter 2005). However, considerably larger ALT increases in humans (30- to 100-fold of the upper limit of normal) can also be accompanied by full recovery of hepatic function (Koch et al. 1997).
Alternatively, small decreases in serum ALT might also be associated with liver enzyme induction. In a rat study, where sodium phenobarbitone was given at dose levels of 80 mg/kg, and β-naphthoflavone (a CYP1A inducer) was given at 100 mg/kg, there were no recorded elevations in serum transaminases, but instead (statistically insignificant) decreases in ALT or AST activities were recorded (Arvela, Reinila, and Pelkonen 1981). In these experiments, the only histopathology recorded was centrilobular hepatocellular hypertrophy. Small decreases in ALT/AST activity may usually be regarded as a “non-adverse” effect because they cannot be correlated with a toxicologically relevant finding. Large changes (>50%), however, may result not only from induction of hepatic drug metabolism but from deficiencies in pyridoxal 5′-phosphate. This may be confirmed or excluded by measuring enzyme activities with and without pyridoxal-phosphate addition as a cofactor to the reaction mixture (PSD Guidance Document 2007).
Alkaline Phosphatase (ALP)
Increases in ALP activity associated with hepatic microsomal enzyme induction, in the absence of accompanying degenerative histopathological findings, have been reported in dog by a number of workers (Conning and Litchfield. 1971; Leeling et al. 1975; Robertson et al. 1993). These studies correlated an increase in ALP activity with increased microsomal enzyme activity and demonstrated that the source of the ALP increase was of hepatic origin in the absence of histologically detectable hepatobiliary injury. Increases in total serum ALP activity up to approximately 2.5-fold (Leeling et al. 1975), 3-fold (Conning and Litchfield 1971), and 5-fold (Robertson et al. 1993) were observed during the course of these studies, which were noted to recover after an 8-day treatment-free period in concordance with a reduction in hepatic microsomal enzyme activity (Litchfield and Conning 1972). Additionally, the ALP change reported by Robertson et al. (1993) was not associated with any other changes in clinical pathology parameters. Leeling et al. (1975) concluded that marked changes in ALP levels during drug treatment should not automatically be assumed to have toxicological implications.
During the current ESTP-sponsored workshop, case study material was presented where a dose-related increase in plasma ALP activity (up to 10-fold) was shown to be of hepatic origin in the dog and followed for up to 52 weeks. Increases in ALP activity were evident at 4 weeks (with maximal comparable levels observed at 13, 26, and 52 weeks). ALP activity correlated with hepatocyte hypertrophy and increases in liver weight up to 1.5-fold of control values. One individual animal showed evidence of hepatocellular degeneration and atrophy at week 52, but this change was also associated with moderate increases in AST (5-fold), ALT (2-fold), and GLDH (glutamate dehydrogenase; 15-fold). Therefore, it is considered that increases in circulating ALP activity in the dog, with associated increased liver weight and histological hepatocellular hypertrophy but without hepatocellular degeneration could be interpreted as an adaptive, rather than an adverse response to chemical exposure.
γ-Glutamyltransferase (γGT)
Induction of hepatocyte γGT has also been reported with several chemicals, including sodium phenobarbitone which is known to cause liver weight increases and induce hepatic metabolism as well as various CYPs
However, cholestasis due to partial obstruction of intrahepatic bile ducts may also result in similar changes in γGT enzyme activities. The decision therefore to ascribe increases in γGT to enzyme induction should be made on a weight-of-evidence approach with full consideration of increases in liver weight, the absence of biologically significant increases in transaminase activity, as well as the absence of adverse histopathological changes.
Bilirubin/Bile Acids
Total bilirubin, as well as bile acid levels, can be reduced as a consequence of higher conjugation rates and excretion via bile when hepatic enzyme–inducing chemicals are administered (Boiteux-antoine et al. 1989; Echchgadda et al. 2004; Gógl et al. 1979). An increase in plasma bilirubin levels is generally not seen following liver enzyme induction, but is usually indicative of impaired hepatic bile flow, accelerated red blood cell destruction, or decreased bilirubin metabolism (Boone et al. 2005; Jonker, Liddleb, and Downes 2011). Additionally, competitive inhibition of bilirubin conjugation by drugs (such as Atazanavir that inhibits UDP-glucuronyltransferase isoenzyme, 1A1) may lead to a dose-related, asymptomatic, unconjugated hyperbilirubinemia in humans and in rats (Neely et al. 2010; Zucker et al. 2001), which is clearly non-adverse and normally not accompanied by histopathological evidence of hepatic degeneration (Neely et al. 2010). When accompanied by increased serum bile acids, increased bilirubin is a reliable indicator of hepatic toxicity and loss of hepatic function (Levin and Schwartz 1965).
Other Clinical Pathology Parameters
Global coagulation tests, such as prothrombin time and activated partial thromboplastin time, can be shortened by drugs/chemicals that induce hepatic drug metabolism due to increased synthesis of coagulation factors by liver cells (Niemegeers et al. 1981; Poulsen, Lerche, and Pedersen 1985). Triglyceride levels can be decreased or increased during enzyme induction depending on the administered compound (Amacher, Schomaker, and Burkhardt 1998). In the experience of the authors, administration of enzyme inducers may also alter protein levels—the change often being correlated with changes in lipid levels (Strauss, pers. comm. 2011).
In addition to the parameters discussed above, there are several other clinical pathology changes that reflect alterations in the functional/anatomical status of the liver. In addition to cholesterol, glucose, urea, and ammonia, many enzymes can be measured that are localized in different intracellular sites within the hepatocyte. Enzymes such as lactate dehydrogenase (LDH), sorbitol dehydrogenase (SDH), and α-glutathione S-transferase (αGST) are located in the cytosol; malate dehydrogenase (MDH) is in the cytosol and mitochondria; ornithine carbamoyl transferase (OCT) and GLDH are in the mitochondria and pseudocholinesterase (CHE) is in vesicles. These enzymes are not measured routinely, but they may be useful on occasion to substitute for other more commonly used enzymes or to provide additional information—for example, SDH may be used in the guinea pig because of its greater liver specificity (Clampitt and Hart 1978). Additionally, GLDH and OCT may be measured to estimate the grade of liver cell damage, while CHE can be used as an additional marker of the synthetic capacity of the liver (Giffen et al. 2002; Kutty and Payne 1994; Litchfield and Gartland 1974; O’Brien et al. 2002).
The measurement of more specific biomarkers of hepatic damage in liver tissue, serum, or urine may help distinguish adaptive enzyme induction from adverse liver cell toxicity. Studies measuring transcriptome profiles in liver tissue (Ellinger-Ziegelbauer et al. 2011; Kramer et al. 2003; Peterson et al. 2004) as well as metabolome patterns in plasma (van Ravenzwaay et al. 2010) and urine (Robertson et al. 2000) are aimed at fulfilling this aspiration.
If there are no accompanying histopathological changes, then the default assumption is that any consistent, and significant, adverse change in clinical pathology parameters can be regarded as an adverse effect with relevance to human health. However, small clinical chemistry changes beyond historical control ranges can be regarded as adaptive, if sufficient evidence exists that these effects are due to altered metabolism, if the change by its own is doubtful or of minimal toxicological importance, and the finding alone is not predictive for an adverse liver effect (Regulation EC No. 1772/2008, Annex 1, chapter 3.9)—for example—decreases in total bilirubin levels without any sign of hypoproliferative anemia, accompanied by decreases in total bile acid concentrations, may be considered adaptive provided that the synthetic pathway for these molecules remains unaffected (Kuipers et al. 1989) since these changes may be due to an increase in the conjugation rate and excretion via bile. Other confounding examples include reductions in global coagulation tests, which result from increased coagulation factor synthesis as a consequence of enzyme induction (Poulsen, Lerche, and Pedersen 1985) but, alternatively, could result from an acute phase reaction or thrombophilia.
A general rule of thumb is that changes affecting a single clinical chemistry parameter beyond historical control values can normally be regarded as “non-adverse” when no histopathological correlate can be found (ECETOC Technical Report No. 85, 2002).
Pitfalls When Interpreting Clinical Pathology Parameters
Different results in studies with liver enzyme–inducing compounds administered to various animal species as well as literature studies of different facilities have to be cautiously interpreted.
Although serum alanine aminotransferase (ALT) activity in the rat, mouse, dog, and humans mainly originates from liver tissue, species-specific differences exist regarding plasma half-life of the enzyme. In humans, the half-life of ALT in the plasma is 47 hr, in dogs it is reported to be 17 hr, while in rats it is approximately 3 to 4 hr (Boyd 1983). Depending on the enzyme kinetic ALT activity increases may be detectable in one species but not in the other in repeated dose studies.
γ-glutamyltranspeptidase (γGT) and alkaline phosphatase (ALP) have different tissue distributions in the rat, mouse, and dog (Clampitt and Hart 1978; Keller 1981). For example, the hepatic isoform is the main contribution to total circulating ALP activity in the dog, but has comparatively lower activity in the rat and hence has lower diagnostic sensitivity in this species. Moreover, in young animals that are generally used in regulatory toxicology studies, serum ALP activity derives mainly from the bone isoenzyme and not from the liver (Hoffmann et al. 1994). Therefore, in addition to liver enzyme induction, changes in other organs may need to be assessed.
Induction of the aforementioned enzymes by chemicals also appears to be species-specific: in dogs, corticoid-induced ALP isoenzyme activities are often increased (Gaskill et al. 2005), whereas in mice, hepatic ALP is increased (Kawasaki, Mataki, and.Takano 1994). In rats, hepatic γGT is induced in response to liver enzyme inducers (Gallagher et al. 1998; Satoh et al. 1982). In pregnant rats from gestation day 6 (GD6) until GD20, ALP, instead of γGT activity is increased when enzyme inducers are administered (Strauss, pers. comm. 2011). In mice, in contrast to rats, ALT is more often increased when hepatic enzyme inducers are administered without any histopathological indication of liver cell toxicity (Strauss, pers. comm. 2011).
When comparing the above with older published studies, it is apparent that different methods can lead to widely different results. There is general agreement that ALT and AST activities should be measured according to The International Federation of Clinical Chemistry and Laboratory Medicine (IFCC) methods without addition of the cofactor, pyridoxal 5′-phosphate. If decreased transaminase activities are observed, a repetition with addition of the cofactor should be performed in order to exclude/confirm alternative reasons for the decreased activity other than enzyme induction (Evans and Whitehorn 1995).
In addition, one should be aware that commercial γGT kits for human medicine have a limit of detection of about 3 U/L (at 37°C, i.e., 50 nkat/L, e.g., Roche instruction manual γGT Szasz mod., 2010-06, V7). Normal rat serum γGT activities are generally below this limit of detection, so that moderate γGT increases cannot be measured with these tests. Also, total bilirubin kits (e.g., Roche instruction manual total bilirubin Diazo method 2011-5, V8) for human medicine may also not be adequate for measuring rodent bilirubin levels, particularly where decreases in bilirubin occur, unless reagents and procedures are appropriately modified.
Serum Enzyme Activity Elevation in Human Studies
Hepatic enzyme inducers, such as sodium phenobarbitone, have been reported to increase the serum enzyme levels in human patient populations, and elevations in ALT, AST, and γGT were seen in patients following treatment with several anticonvulsant drugs (Aiges et al. 1980; Wall et al. 1992). In a study of epileptic patients receiving drug therapy, elevations in γGT were seen in almost all individuals leading the authors of the study to conclude that this did not indicate hepatic damage, since none of the patients exhibited clinical symptoms. Instead, these elevations in enzymes merely confirmed that the patients were receiving the drugs (Hirayanagi, Fujii, and Teshirogi 1991). Elevated γGT levels, in the absence of other markers of hepatic damage, were reported to be commonly elevated in patients taking rifampicin (Davis 1989), carbamazepine, zarontin, phenobarbital, and phenytoin (Knight 2005; Ohta and Toda 2001; Rosalki, Tarlow, and Rau 1971; Vandenberghe 1996; Whitfield et al. 1973). Much of this elevated enzyme activity was considered due to hepatic induction of the enzyme, and its subsequent loss into the blood, rather than an indicator of frank toxicity to the liver (Ohta and Toda 2001).
Finally, anti-epileptic drugs in man have also been reported to increase serum ALT up to 3-fold the upper limit of normality (ULN), and AST up to 2-fold the ULN in approximately 25% of the patient cohort studied (Haidukewych and John 1986). Once again these were not considered to be indicative of hepatotoxicity but to be a consequence of hepatic enzyme induction and hepatocellular hypertrophy. Indeed, liver biopsies taken from patients on long-term therapy failed to show any histological evidence of hepatotoxicity (Jacobsen et al. 1976). However despite this, it should be noted that there is a correlation between drug-induced hepatic CYP induction and documented idiosyncratic hepatotoxicity in man (Li 2002).
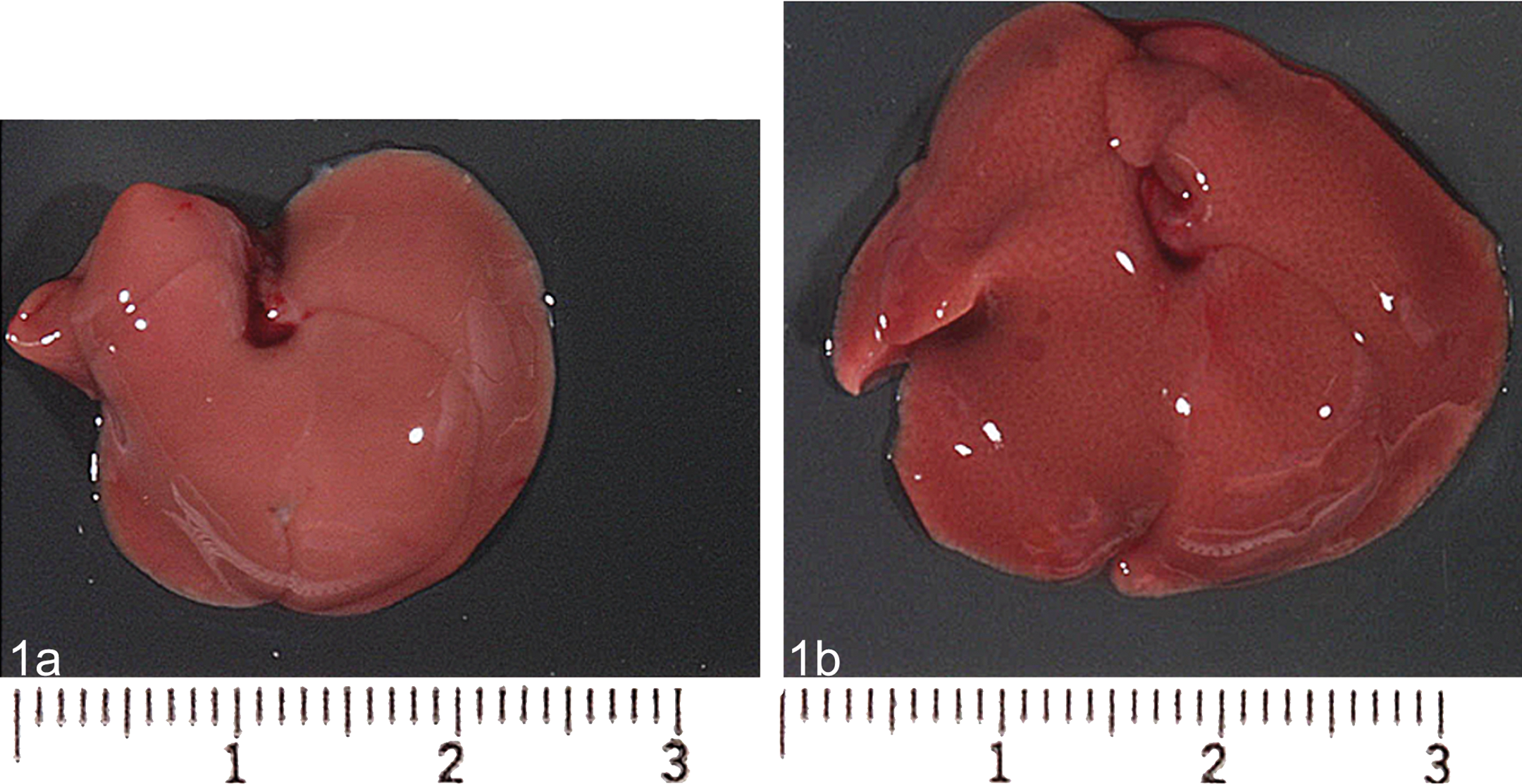
Gross photograph of a control mouse (left panel, 1a) and treated (right panel, 1b) liver demonstrating an increase in liver volume characterized by a fine reticular pattern after treatment with the enzyme inducer phenobarbital.
Histological Assessment
The first indication of hepatomegaly is usually noted at necropsy by the measurement of an increase in organ weight, and in extreme cases, by the concomitant visual observation of an increase in liver size (volume; Figure 1). In small animals where experiments can be powered to gather statistically significant data, the assessment of an increase in liver weight is relatively straightforward. However, in dogs and other larger animals, interindividual variation combined with smaller group sizes can make the assessment of meaningful increases in liver organ weight more uncertain.
Clinical pathology can provide valuable ancillary data to indicate alterations in liver function; however, the diagnosis of morphological or structural changes in the liver is ultimately made by a trained pathologist assessing formalin fixed, paraffin embedded, haematoxylin and eosin (H&E) stained sections. To this end, the National Toxicology Program (NTP) for 2-year rat carcinogenicity studies advocates two liver samples taken at the widest part of the left lobe and right median lobes (Maronpot et al. 1989; NTP 2006). The RITA trimming guide recommends to sample in addition the caudate lobe in rats and mice (Ruehl-Fehlert et al. 2003).
Treatment-related increases in liver weight may result from a wide variety of causes such as hyperplasia (of any of the resident cell types), hypertrophy, inflammation, fibrosis, abnormal storage of metabolism products, particles, or cleavage products, neoplasia, and congestion (Carthew et al. 1996; Greaves 2007). Typically, these changes do not occur in isolation, so in the absence of overt adverse changes such as inflammation, necrosis, or degeneration, it is important to recognize that increases in liver weight may be induced by hypertrophy, hyperplasia, or very frequently a combination of the two (Maronpot et al. 2010).
An increase in liver cell size can result from accumulation of glycogen, lipid, and water (hydropic degeneration) or the proliferation of subcellular organelles (typically smooth endoplasmic reticulum [sER] and/or peroxisomes). Experiments in which hepatocellular hypertrophy was induced in mice with phenobarbital suggest that polyploidization is an additional and inevitable component of liver cell hypertrophy caused by these types of drugs (possibly as an adaptive response to increased metabolic demands —RNA and protein synthesis; Böhm and Noltemeyer 1981; Bursch et al. 2004). Morphologically in both rats and mice, hepatocellular hypertrophy may present as a diffuse change affecting all zones of the liver lobule, or a regional change usually affecting the centrilobular region, but frequently extending to the mid-zonal region (and further) with increasing exposure (Figure 2). Occasionally, periportal hypertrophy without evidence of centrilobular hypertrophy is also seen—for example, both fenofibrate and ciprofibrate induced periportal hypertrophy in the cynomologus monkey as a result of both peroxisomal and mitochondrial proliferation (Hoivik et al. 2004).
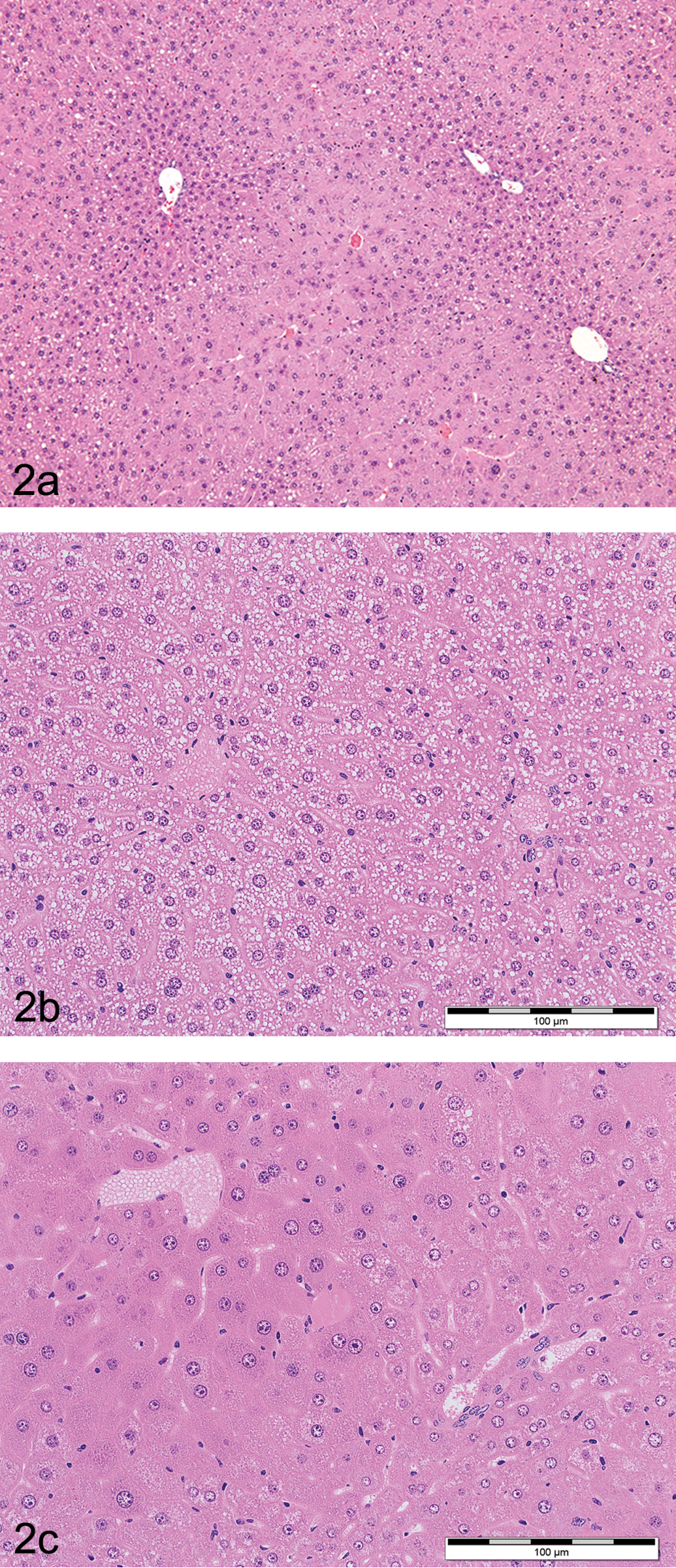
(a) H&E stained low power photomicrograph of a male B6C3F1 mouse liver treated with 100 mg/kg/day of pentabromodiphenyl oxide for 90 days showing centrilobular hepatocyte hypertrophy with characteristic enlargement of hepatocytes noted by the decreased number of nuclei per unit area and pale staining centrilobular eosinophilic cytoplasm. (b and c). H&E stained low power photomicrograph of a control male C57Bl mouse liver (left panel, 2b) and a treated C57Bl mouse liver (right panel, 2c) on a 4-week feeding study showing diffuse macrovesicular fatty vacuolation (control) and diffuse hepatocyte hypertrophy (treated) characterized by a diffuse enlargement of hepatocytes. Note that diffuse changes tend to be more subtle in appearance compared to regional changes and require careful comparison to concurrent control samples and correlation with liver organ weight data to accurately identify.
At the ESTP liver hypertrophy expert group, it was generally agreed that an increase in liver weight of at least 20% was required to histologically detect a change in hepatocyte cell size (corresponding with other publications; Ennulat, Walker, et al. 2010). However, whereas the assessment of zonal changes in liver cell size is relatively straightforward, diffuse changes are considerably more difficult to detect by examination of standard H&E stained sections without reliance upon morphometry (Greaves 2007).
In H&E stained sections at low power, hepatocellular hypertrophy is usually recognized by a zonal increase in liver cell size and eosinophilia with a reciprocal decrease in nuclear density (i.e., a decrease in the number of hepatocyte nuclei per unit area of tissue; Figure 2).
The staining characteristics of the hepatocyte cytoplasm vary depending on the mechanism of cellular hypertrophy and consequently the organelle responsible for the increase in cytoplasmic volume.
Phenobarbitone and other microsomal drug-metabolizing enzyme inducers induce hepatocyte hypertrophy through sER proliferation. This results in the characteristic eosinophilic “ground glass” appearance of hepatocyte cytoplasm (Figure 2). Chronic administration of certain enzyme inducers, such as chlordane, can result in extreme hypertrophy characterized by bizarrely enlarged hepatocytes with significant increases in hepatocyte ploidy (Figure 3). If electron microscopy is performed, induction by typical microsomal enzyme inducers such as phenobarbital reveals characteristic stacks of smooth ER that crowd out other organelles (Figure 4).
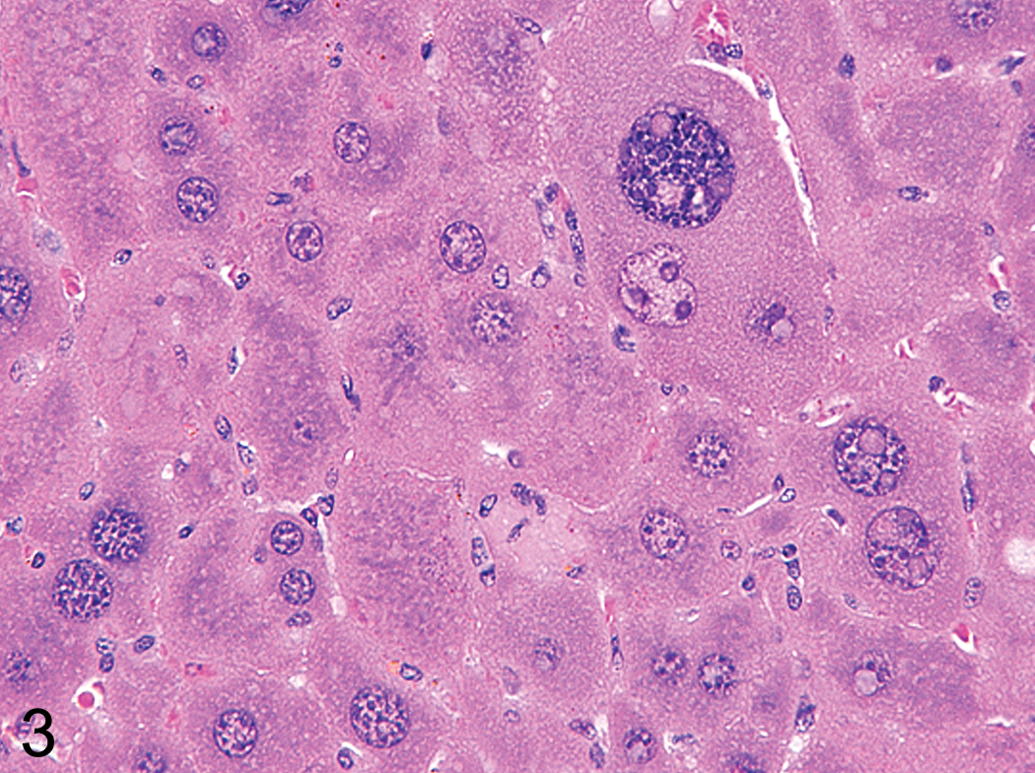
H&E stained high-power photomicrograph of a male B6C3F1 mouse liver chronically (2-year carcinogenicity study) treated with chlordane showing the cellular features of extreme hypertrophy characterized by a notable increase in cellular size accompanied by binucleate hepatocytes with enlarged hepatocyte nuclei (polyploidy).
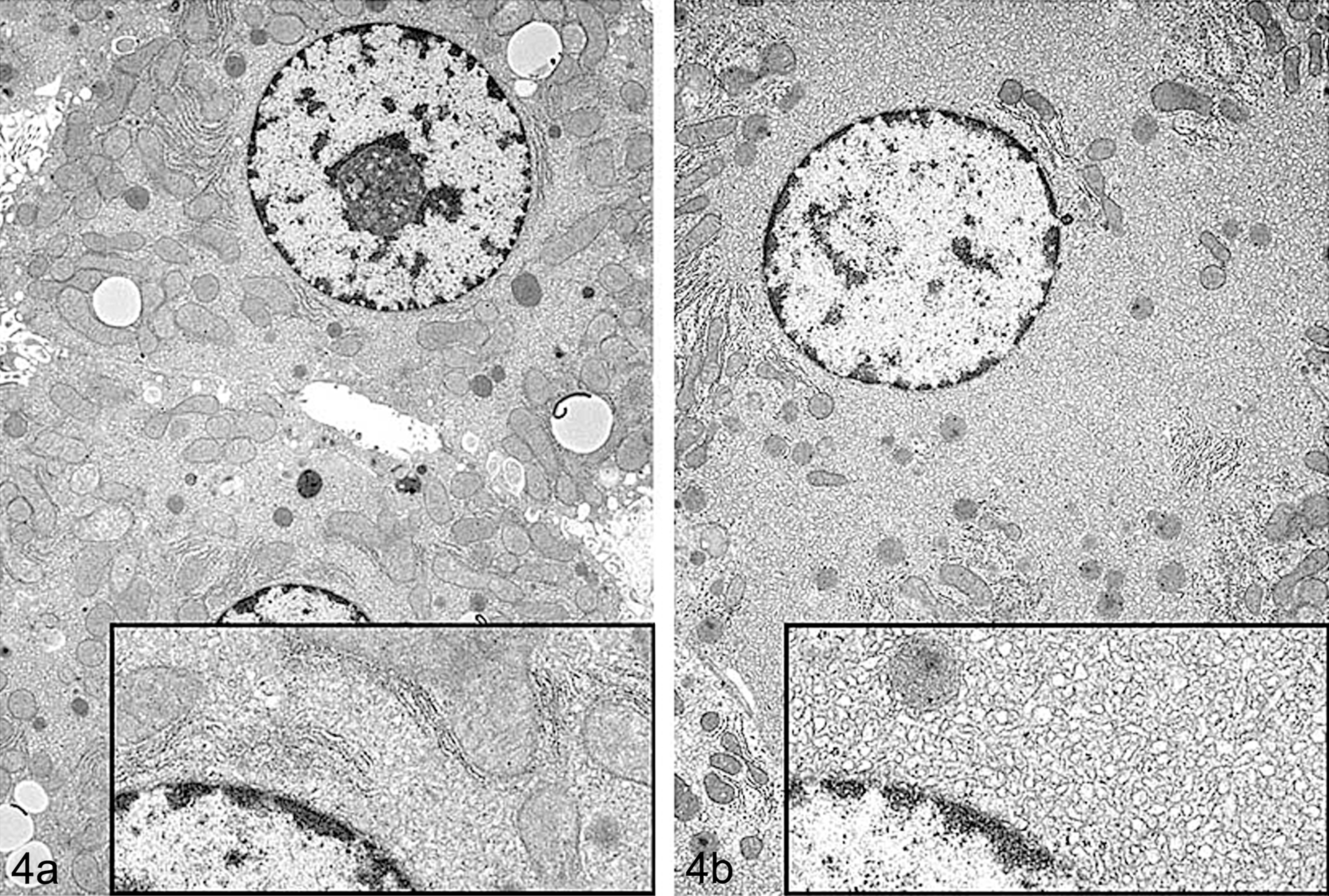
(a and b) Transmission electron microscope photograph of control (left panel, 4a) and treated (right panel, 4b) rat hepatocytes showing hypertrophy after treatment with a COX inhibitor. Treated hepatocytes are characterized by increased amount of sER (sER proliferation) that crowds out and peripherally compresses other organelles in the cell. Inset shows a higher magnification of the cytosol immediately adjacent to the nucleus. (Note: Figures 2a, 3, and 4 were previously published in Toxicologic Pathology, 38: 776–95, 2010, and used by kind permission of Sage Publishing and courtesy of NTP. Figure 4 courtesy of Dr Katsuhiko Yoshizawa [with permission].)
Other classes of xenobiotics such as Clofibrate (and similar hypolipidaemic fibrates) as well as ether/phenoxy herbicides induce hepatocyte hypertrophy through a different mechanism. These chemicals activate the PPARα (peroxisome proliferator-activated receptor alpha) resulting in peroxisome proliferation (together with sER proliferation). Histologically, these samples stain with an intensely eosinophilic granular cytoplasm in H&E sections (Figure 5). Electron microscopy has demonstrated that the granules are in fact single catalase positive electron dense vesicles bounded by a single plasma membrane (Figure 6).
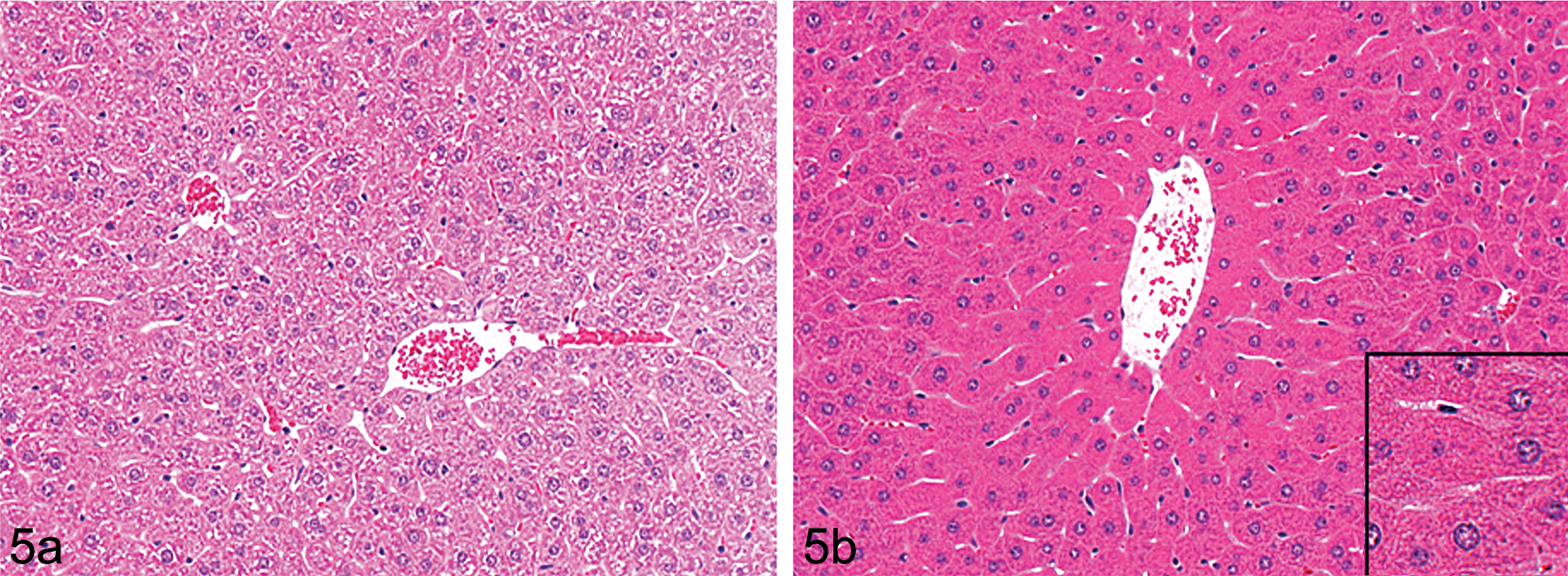
(a and b) H&E stained low-power photomicrograph of Wistar rat liver showing control (left panel 5a) and treated (right panel 5b) livers from animals treated with a peroxisome proliferating agent (phenoxy herbicide) characterized by centrilobular hepatocyte hypertrophy and intensely eosinophilic cytoplasm. Inset: oversharpened area of image to demonstrate fine cytoplasmic granularity.
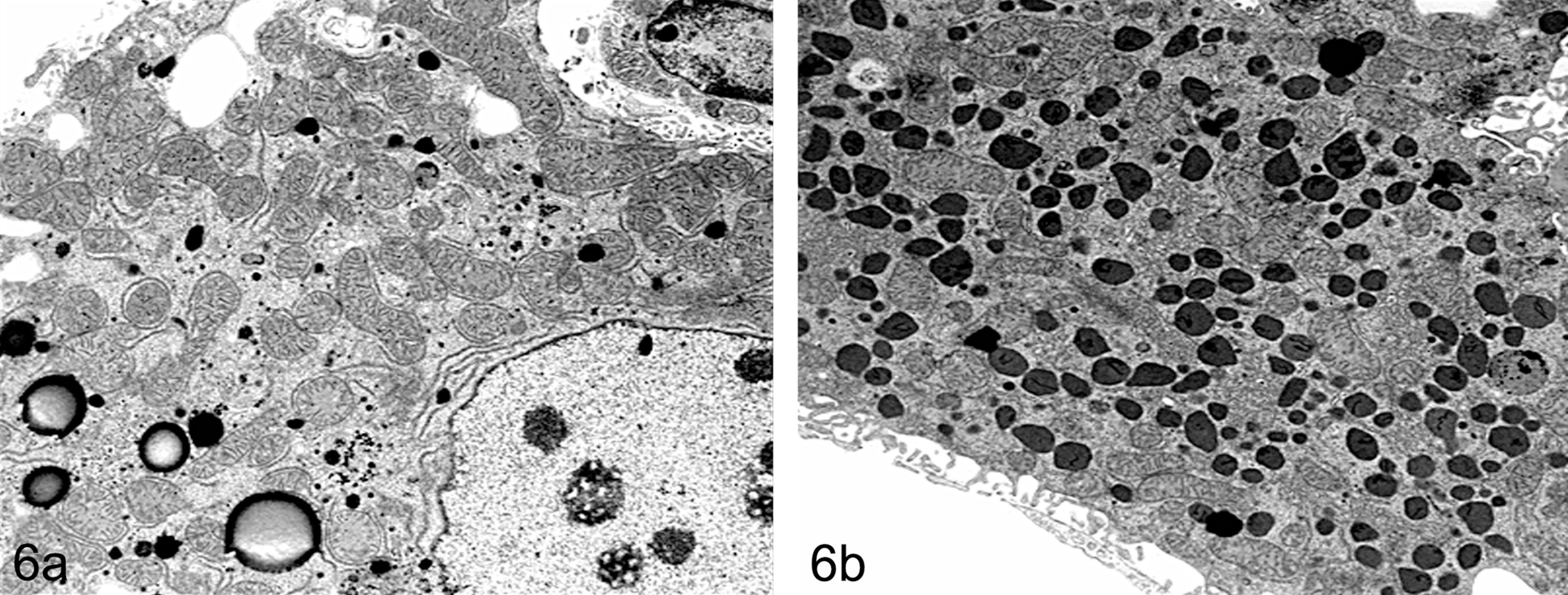
(a and b) Transmission electron microscope photograph of B6C3F1 mouse liver showing control (left panel, 6a) and treated (right panel, 6b) hepatocytes characterized by centrilobular hypertrophy and electron dense membrane bound granules in hepatocytes after treatment with a peroxisome proliferator agent (di-isononyl phthalate, DINP).
Molecular Methods to Assess Liver Enzyme Induction
A variety of methods are available to assay drug-induced hepatocyte enzyme induction (reviewed in Fretland and Monshouwer 2010; Figure 7). Many of these methods such as in silico modeling, receptor binding assays, and immortalized cell lines have proven valuable in identifying potential induction liabilities. They have also proven pivotal in increasing our knowledge of ligand–receptor binding interactions. For example, in silico modeling of PXR–ligand crystal structure complexes have elucidated the molecular basis for PXR receptor promiscuity allowing it to interact with such diverse ligand binding partners as phenobarbital and rifampicin (Watkins et al. 2001). However, these methods, although still widely used and of general applicability, possess significant limitations—for example—immortalized cell lines tend to express the major CYPs at lower levels than fresh hepatocyte preparations (Fretland and Monshouwer 2010).
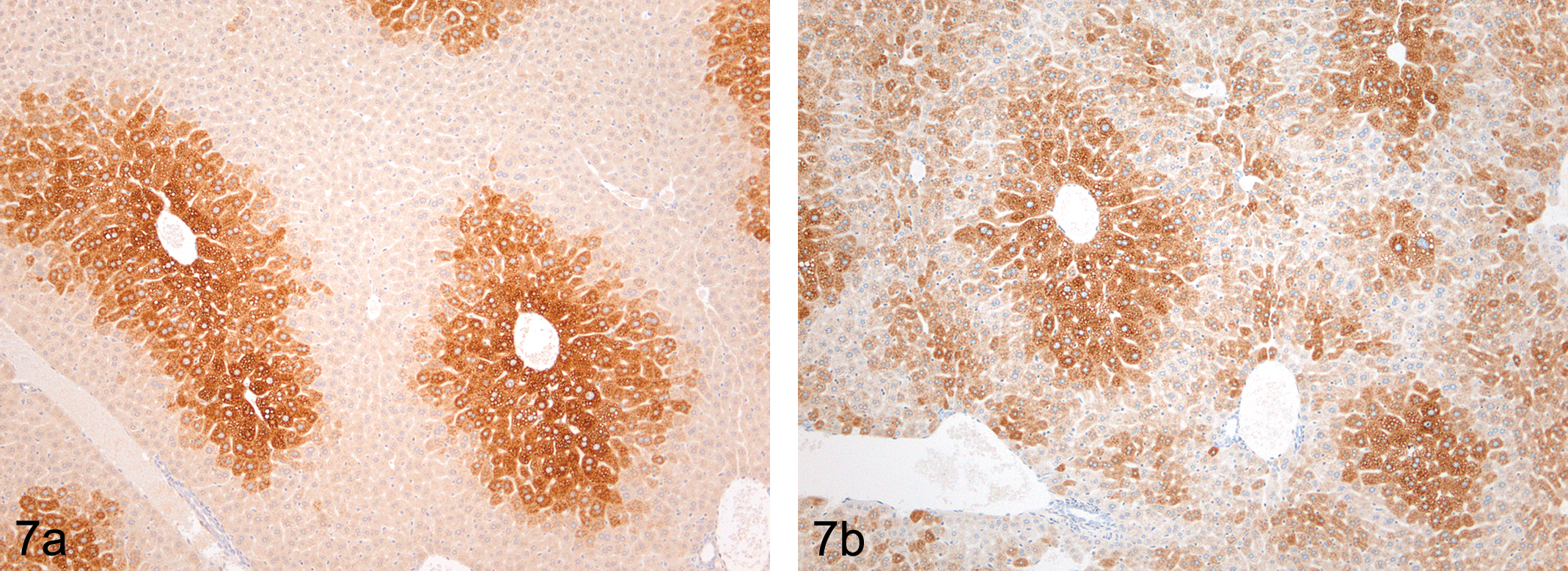
(a) Immunolocalization of CYP 1A1 in untreated CD1 mouse liver demonstrating centrilobular expression of the enzyme using immunocytochemistry. (b) Immunolocalization of CYP 3A2 in untreated CD1 mouse liver showing a predominantly centrilobular localization of the enzyme but with random cells throughout the lobule showing expression of the enzyme.
The use of newer techniques and, in particular, the use of cryopreserved human hepatocytes can now provide more reliable data to build confidence for human risk assessment and to predict the effects of novel chemicals on drug-metabolizing enzyme induction in man (Fretland and Monshouwer 2010). This type of information can also be supplemented by experiments generated in laboratory species including nonhuman primates that show considerable sequence homology in their nuclear hormone receptors—for example, the ligand binding domain of PXR in the rhesus monkey possesses 96% sequence identity with the human receptor at the amino acid level (Moore et al. 2002). More recently, with the advent of humanized mice which express the human form of one or more of the nuclear receptors PXR (Gonzalez 2007), CAR (Ross et al. 2010), or PPARα receptor (Morimura et al. 2006), investigators can compare the role of the human receptor with that of the rodent receptor to determine the species-specific responses to prototypic and novel xenobiotics.
Enzyme Induction in Safety Assessment
The determination of enzyme induction is now carried out routinely during the safety assessment of new chemical entities in the pharmaceutical sector. In one study where rats were dosed with RO9210, a non-nucleoside reverse transcriptase inhibitor for the treatment of HIV infection, Zabka et al. (2011) demonstrated by routine examination of H&E stained slides, centrilobular hypertrophy with secondary thyroid follicular hypertrophy and pituitary gland pars distalis hypertrophy. IHC analysis of formalin fixed paraffin embedded sections of the pituitary and thyroid glands demonstrated thyrotroph hypertrophy with granule depletion and thyroid follicular epithelial cell hyperplasia. Image analysis was able to confirm and quantify the extent of the centrilobular hypertrophy identified by histological examination of the H&E stained slides.
IHC analysis of the liver demonstrated increased CYP 2B1/2 and CYP 3A2 staining intensity with extension of staining from the centrilobular to the periportal region. Frozen rat liver samples as well as cultured rat hepatocytes were used to quantify the extent of mRNA induction of drug-metabolizing enzymes (UGT1A1, UGT 1A2, CYP1A1, CYP 1A2, CYP2B1 3A1, and CYP2B1 3A2) by Taqman quantitative RT-PCR.
These data illustrate the utility of routine histological assessment, IHC and qRT-PCR to assess the nature and extent of drug-metabolizing enzyme induction to help demonstrate the adaptive nature of the toxicologic changes induced by RO2910. Induction of hepatocyte drug-metabolizing enzymes in this case resulted in increased turnover of T4 (thyroxine) and secondary thyroid hypertrophy/hyperplasia due to stimulation of the pituitary–thyroid–endocrine axis. An ELISA method for total thyroxine (T4), total tri-iodothryonine (T3), and thyroid stimulating hormone (TSH) confirmed the increase in TSH and decrease in T4 (with no change in T3).
Mechanisms of Hypertrophy
Nuclear Hormone Receptors
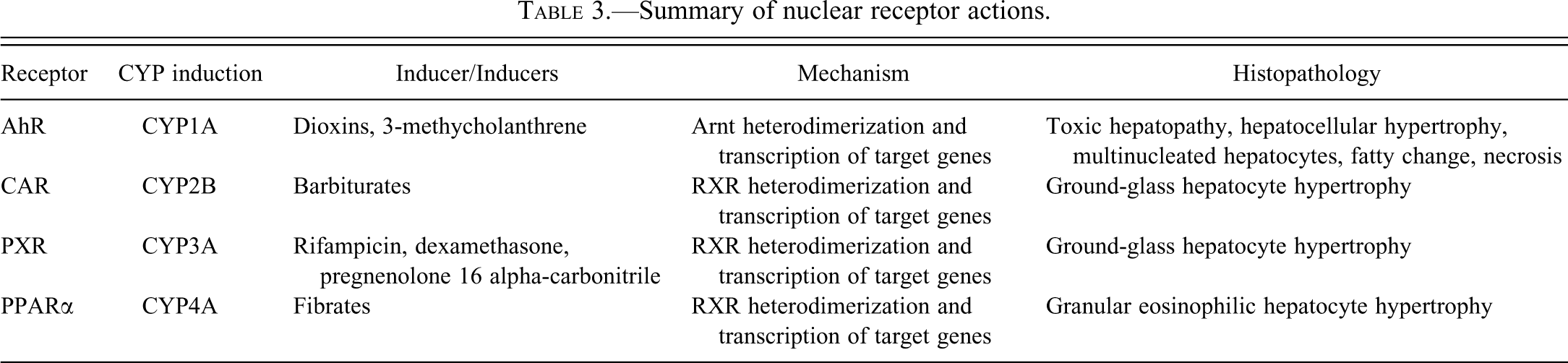
The nuclear hormone receptors constitute a superfamily of xenosensing receptors that specifically interact with endogenous and exogenous chemicals. Receptor–ligand binding results in the activation of a battery of genes mediating oxidative drug metabolism, conjugation, and transport, which serves to eliminate the xenobiotic from the organism to maintain homeostasis. The majority of these nuclear receptors are expressed on the liver (Bookout et al. 2006) and can be functionally divided into those that are generally associated with drug metabolism—the pregnane X receptor (PXR) and the constitutive androstane receptor (CAR)—and those that are involved in the metabolic regulation of endogenous compounds—principally glucocorticoids (glucocorticoid receptor), lipid oxyseroids (liver X receptor (LXR)), bile acids (farnesoid X receptor (FXR)), and lipid metabolism (peroxisome proliferator-activated receptor (PPARα)) (reviewed in Plant and Aouabdi 2009; Table 3).
Summary of nuclear receptor actions.
Peroxisome Proliferator-activated Receptor Alpha
Currently, there are three peroxisome proliferators activated receptors, termed peroxisome proliferator-activated receptor alpha (PPARα), PPARγ and PPARb/d which heterodimerize with RXR to transactivate distinct but overlapping gene targets (Plant and Aouabdi 2009). PPARα is the major PPAR found in the liver, but is also distributed and highly expressed in the kidney and intestine (Bookout et al. 2006) as well as heart and brown adipose tissue (Xu, Li, and Kong 2005). PPARγ is expressed predominantly in the adipose tissues but also in pancreatic beta-cells, intestines and spleen whereas PPARb/d is expressed nearly ubiquitously (Bookout et al. 2006; Greaves 2007).
PPARs regulate lipid and cholesterol metabolism through induction of (peroxisome proliferator response element (PPRE)) containing target genes resulting in increased beta-oxidation of fatty acids (Xu, Li, and Kong 2005). Natural ligands for PPARα include saturated and unsaturated fatty acids, eicosinoids, and linoleic acid metabolites. However, a diverse range of xenobiotics from many classes and structures are also able to activate PPARα such as the fibrate hypolipidaemic agents (clofibrate, fenofibrate, gemfibrozil amongst others), methaphenilene, thromboxane synthetase inhibitors, dehydroepiandosterone, non-steroidal anti-oestrogens, ibuprofen, Wy-14,643, diphenyl ether herbicides, and phenoxy herbicides (Greaves 2007).
Peroxisome Proliferator-activated Receptor Alpha Phenotype
Activation of PPARα by hypolipidaemic agents results in liver enlargement through peroxisome proliferation (which appears morphologically as an increase in peroxisomal volume density) as well as proliferation of sER and increases in hepatic cytochrome P450 enzyme activity (Cattley 2003; Walker et al. 1996). In rodents, these changes are highly correlated with liver tumors which has led to the general view that dose rates that induce >3 fold increases in peroxisomes and >1.5 fold increases in liver weight in short term studies are sufficient to induce hepatocellular neoplasia in long term studies (Cattley 2004; Walker et al. 1996).
Increases in drug-metabolizing enzymes induced by PPARα activators are typically characterized by increased activity of CYP4A and acyl CoA oxidase genes (Bell et al. 1991). However, more recent studies have demonstrated increased activity of other targets such as human SULT2A1 (Fang et al. 2005) and human/murine UGT2B4 (Barbier et al. 2003) as well as decreased bile acid secretion due to decreased activity of cholesterol 7a-hydroxylase (CYP7A1) and sterol 27-hydroxylase (CYP27 ) (Li and Chiang 2009; Post et al. 2001).
Hepatocarcinogenicity
Humans are regularly exposed to peroxisome proliferating chemicals either in the form of environmental exposure, or intentionally through drug treatment. Given the clear association between these agents and the induction of hepatocarcinogenesis in long-term rodent studies, there is a clear concern over the potential risk to human health.
Hepatocarcinogenesis is thought to primarily result from an increase in cell division and a reciprocal decrease in apoptosis induced upon treatment with peroxisomal proliferators resulting in hepatocyte hyperplasia and fixation of spontaneous mutations (Plant et al. 1998). Oxidative damage due to acyl CoA oxidase induction and escape of H2O2 from peroxisomes has also been proposed as an indirect mechanism of genotoxicity and carcinogencitiy (Conway et al. 1989). However it is uncertain whether this leads to DNA damage in hepatocytes and if it is causally related to the development of tumors (Cattley 2004).
The Role of the PPARα Receptor
The importance of the role of the PPARα receptor in mediating hepatocarcinogenesis in response to peroxisomal proliferators was initially raised by the observation that the relative expression level of the receptor in responder species such as the rat and mouse where liver expression is high compared to non-responsive species such as the guinea pig, non-human primate, and human where expression (in the human) is an order of magnitude lower (Choudhury et al. 2000; Palmer et al. 1998).
The relatively recent development of PPARα gene knockout mice has now confirmed that the PPARα receptor is responsible for mediating the effects induced by the peroxisome proliferator–inducing agents clofibrate, WY-14,643 and DEHP (i.e., hepatomegaly, increases in peroxisomes and enzyme induction) (Lee et al. 1995; Ward et al. 1998). It is also responsible for the transient increase in cell proliferation commonly observed immediately after treatment as well as the more variable low level sustained increases in proliferation occasionally observed following chronic treatment (Cattley 2004).
Not only do PPARα knockout mice not develop hepatocellular tumors in response to long-term treatment with peroxisome proliferators, (Lake 1995; Peters, Cattley, and Gonzalez 1997), but PPARα humanized (PPARα knockout/knockin) mice also fail to develop tumors in response to these agents suggesting that the human receptor is structurally different from the murine receptor (Morimura et al. 2006). Since induction of cell proliferation (with a concomitant decrease in apoptosis) is thought to be the mechanistic basis for the induction of liver tumors by these non-genotoxic agents, it therefore probable that humans would be resistant to this form of promotion.
Epidemiology and Risk to Man
Studies using human hepatocytes (Bentley et al. 1993; Lake 2009) as well as human biopsy material exposed to gemfibrozil for up to 27 months (Greaves 2007) have generally concluded that humans are insensitive and unresponsive to peroxisomal proliferators at therapeutic dose levels and that humans are not at an increased risk of developing liver tumors (Cattley et al. 1998; International Agency for Research on Cancer [IARC] 1995). In one study, humans were given oral fibrate therapy for 8 weeks and no differences in peroxisomal acyl CoA oxidase mRNA transcripts were detected in groups receiving either fenofibrate, bezafibrate, or gemfibrozil compared to placebo controls (Roglans et al. 2002).
These epidemiological data, however, have been collected from relatively small data sets and may therefore not be robust enough to detect small increases in human liver carcinogenesis. Further work over the past decade in nonhuman primates therefore has been conducted and confirms that they are relatively insensitive to the effects of PPARα agonists compared to rodent species. Exposure to DEHP, diisononyl phthalate, or clofibrate for 14 days failed to induce increases in peroxisomal fatty acid beta-oxidation or liver weight in the cynomolgus monkey (Pugh et al. 2000). In another study conducted in the nonhuman primate, exposure to K-111, a potent PPARα agonist elicited only modest increases in lipid beta-oxidation (up to 3-fold) and peroxisome volume density (1.5- to 2-fold increase) compared to 50- to 100-fold increase in lipid beta-oxidation and 10-fold increases in peroxisome volume density that can occur in rats and mice (Schäfer et al. 2004). Finally, another recent report demonstrated that exposure to the potent PPARα/g agonist, ciprofibrate, is capable of eliciting peroxisome proliferation (Hoivik et al. 2004) in higher primates but that this modest induction was associated with transcript profiles indicative of an antiproliferative and a pro-apoptotic effect with no evidence of oxidative stress (Cariello et al. 2005).
Constitutive Androstane Receptor
CAR is an orphan nuclear receptor that forms heterodimers with RXRa (retinoic X receptor) leading to nuclear translocation and transactivation of target genes. This can occur even in the absence of ligand (Honkakoski and Negishi 1998). CAR, however, is activated by a diverse range of ligands including 1,4-bis(2-(3,5-dichloropyridoxyloxy)) benzene (TCPOBOP) (murine CAR specific; Wei et al. 2000), CICO (human CAR specific, Maglich et al. 2003), phenobarbital, and clotrimazole (the latter two activators of both CAR and PXR, Moore et al. 2000). Activated CAR heterodimers bind to a number of regulatory regions of target genes eliciting transactivation of CYP2B genes.
CAR Phenotype
Activation of CAR by potent drug-metabolizing enzyme inducers such as Phenobarbitone causes hepatomegaly due to a profound induction of sER proliferation that occurs in combination with an early and transient hepatocyte hyperplasia/suppression of apoptosis (Huang et al. 2005) and an enlargement of the hepatic blood space (Massey and Butler 1979). Morphological changes are characterized by a centrilobular hypertrophy that is apparent after 1 week of treatment and can persist for 80 weeks without overt signs of liver cell damage (Crampton et al. 1977a).
Hepatocarcinogenesis
Long-term treatment with CAR activators such as TCPOBOP and Phenobarbitone results in a strain- and species-specific induction of liver tumors in rats and mice (Diwan et al. 1992) that is dependent upon the fixation of somatic mutations due to the stimulation of S-phase DNA synthesis leading to hepatocyte proliferation and a decrease in apoptosis (Hasmall and Roberts 1999). This process is well characterized and appears morphologically as a temporal progression from centrilobular hepatocyte hypertrophy (with a concomitant induction of Cyp2b enzymes) to the development of altered hepatic foci, and ultimately the appearance of adenomas and carcinomas (Ross et al. 2010). Hand-in-hand with acute increases in cell proliferation is a CAR-dependent increase in endoreduplication in which Phenobarbitone and TCPOBOP stimulate an increase in the ratio of 8N:4N hepatocytes (Huang et al. 2005), which appears morphologically as an increase in ploidy/nuclear size. Increases in functional drug-metabolizing enzyme capacity through cell hypertrophy and hyperplasia act to promote clearance of xenobiotics and return the organism to homeostasis. In this respect, increases in hepatocyte ploidy have been correlated with increased cytochrome P450 activity (Rajvanshi et al. 1988).
Of note, recovery from Phenobarbitone or TCPOBOP treatment results in complete reversal of changes marked by a return to normal liver cell size and an associated increase in apoptosis (Huang et al. 2005). Reversal may even extend to regression of a proportion (30%) of liver tumors (adenomas and adenocarcinomas) observed after the withdrawal of treatment with chlordane in B6C3F1 mice (Malarkey et al. 1995).
The Role of the CAR Receptor
It is now well established that the CAR, in a similar manner to other nuclear receptors, mediates the acute and chronic effects induced by Phenobarbitone and TCPOBOP. CAR knockout mice are resistant to the enlargement of liver cell size and induction of drug-metabolizing enzymes/transporters as well as the acute and chronic increases in DNA synthesis (and reciprocal p53-mediated decreases in apoptosis) that leads to hepatocyte proliferation and ultimately hepatocarcinogenesis (Huang et al. 2005).
Epidemiology and Risk to Man
Liver enlargement after 1 year of treatment with nongenotoxic xenobiotics in rodents correlates with an increase in liver tumors in chronic toxicology studies (Carmichael et al. 1997; Ross et al. 2010). In a review of 138 pesticide carcinogenicity studies in mice, induction of liver tumors was very closely correlated at 1 year with a mean relative liver weight of 150% and the presence of significant liver pathology (Carmichael et al. 1997).
In humans, liver enlargement associated with an increase in cytochrome P-450 levels is seen following exposure to drug-metabolizing enzyme inducers such as Phenobarbital and other anti-epileptic drugs (Pirttiaho et al. 1978) as well as alcohol (Pirttiaho et al. 1982). Therefore, clear concerns are raised for the safety of human health and the risk of hepatocarcinogenesis in people exposed to these types of agents.
Whereas treatment of CAR knockout mice clearly demonstrated the dependence of hepatocarcinogenesis on the presence of the CAR receptor, it has only recently been demonstrated that the human CAR receptor is relatively resistant to the mitogenic effects of CAR pathway activation and, therefore, less likely to induce liver tumors through this mechanism. This observation is based on the resistance of cultured human hepatocytes to induction of replicative DNA synthesis (Hirose et al. 2009; Lake 2009; Parzefall et al. 1991) as well as similar observations in huPXR/huCAR receptor humanized mice.
In these later studies, treatment of humanized mice (huPXR/huCAR) with either chlordane or Phenobarbitone resulted in marked liver weight increases, hepatocellular hypertrophy, and induction of cytochrome P450 in agreement with clinical data derived from human liver samples. However, no increases in replicative DNA synthesis were observed either by direct measurement of BrdU labeling index or by microarray analysis of cell proliferation associated gene expression changes (Ross et al. 2010). These experiments also demonstrated species differences in response to Phenobarbitone and chlordane in that the human receptor had greater sensitivity for Phenobarbitone-mediated increases in Cyp2b10 and Cyp3a11 compared to the mouse receptors, but the converse was true for chlordane.
These data are in agreement with a number of epidemiological studies that demonstrate no linkage between hepatocarcinogenesis and chronic exposure to Phenobarbitone, phenytoin, and other anticonvulsants at exposures similar to those that produce liver tumors in rodents (IARC 2001; Olsen et al. 1989; Whysner, Ross, and Williams 1996).
The mechanistic basis underlying the differences in sensitivity between the rodent and human receptor to induction of proliferation in response to CAR activation is still unclear. Microarray analysis of huPXR/huCAR mice has shown that these mice are resistant to the upregulation of the polo-like kinase (Plk1)-mediated cell proliferation pathway induced by Phenobarbitone treatment (Elcombe et al. 2010; Ross et al. 2010). Upregulation of this pathway in wild-type mice implies that structural differences in the human receptor may account for critical differences in the types of responder genes transactivated by CAR activation and induction of proliferation.
Pregnane X Receptor
PXR Promiscuity and Cross-talk
PXR, like CAR, plays a central role in xenobiotic clearance. CAR and PXR are structurally well conserved with a high degree of similarity in their DNA-binding domains but significant divergence in their ligand binding domains (Kliewer et al. 2002). This enables considerable “cross-talk” between PXR and CAR due to the sharing of ligands, coregulators, or DNA-binding elements resulting in a battery of genes coordinately regulated by both nuclear receptors. To this end, Ueda and Maglich identified sixty-nine genes regulated by CAR (Ueda et al. 2002) and forty genes regulated by PXR (Maglich et al. 2003) with many of them being coregulated by both PXR and CAR (Plant and Aouabdi 2009).
Not only can PXR interact with other nuclear hormone receptors to provide functional redundancy in xenobiotic metabolism, but it is also regulated by a number of other receptors including PPARα, glucocorticoid receptor, and CAR (reviewed in Plant and Aouabdi 2009) to help integrate an organism’s response to xenobiotic exposure.
This functional cross-talk is also reflected in the wide range of structurally diverse ligands that PXR can bind due to the markedly flexible pocket domain (Kliewer, Goodwin, and Willson 2002). This results in activation by numerous xenobiotics from different chemical series such as steroids, antibiotics (including the prototypic inducer rifampicin), antimycotics, bile acids, aflatoxins (Ratajewski et al. 2011), and the herbal antidepressant St. John’s wort (Kliewer, Goodwin, and Willson 2002). As with other nuclear hormone receptors, species differences exist in sensitivity to different PXR activators. However, since human and mouse PXR only share 80% amino acid homology in their ligand-binding domain compared to 96% amino acid identity in the DNA-binding domain, mouse and human PXR differ markedly in their responses to PXR ligands (Lehmann et al. 1998; Ma, Idle, and Gonzalez 2008). For example, human PXR is not activated by pregnenolone 16 alpha-carbonitrile (PCN, a potent inducer of murine CYP3A and PXR) whereas rifampicin is a selective activator of human PXR but not murine PXR (Bertilsson et al. 1998).
PXR Phenotype
PXR is a master regulator of CYP3A transcription (Bertilsson et al. 1998; Gibson, El-sankary, and Plant 2002) as well as other genes involved in phase I oxidation, phase II conjugation, and phase III transport. The growing list of PXR target genes include, in addition to CYP3A family members, CYP2B6 (Wang et al. 2003), Cyp7a1 (cholesterol 7 a-hydroxylase; Staudinger et al. 2001), UDP-glucuronosyltransferase 1A1 (Hartley et al. 2004), intestinal P-glycoprotein (Geick, Eichelbaum, and Burk 2001), OATP2 (Hartley et al. 2004), and MRP2 (Fromm et al. 2000).
Activation of PXR by PCN and cyproterone acetate (CPA), a synthetic steroid with anti-androgenic and contraceptive activity, results in liver hyperplasia and hypertrophy (Japundzić et al. 1974) in line with the response seen with other nuclear hormone receptors. This effect is visible within 3 days of treatment in rats (Schulte-Hermann et al. 1980) and is accompanied by a decrease in the frequency of binucleated hepatocytes and a concomitant increase in nuclear ploidy (Schulte-Hermann, Hoffmann, and Landgraf 1980). As with other nuclear hormone receptor agonists, long-term administration of CPA results in hepatocarcinogenesis in the mouse (Schuppler and Günzel 1979) and rat (Tucker, Kalinowski, and Orton 1996).
Role of the PXR Receptor
The generation of PXR knockout mice demonstrated that the PXR receptor is necessary for the induction of cellular hypertrophy and hyperplasia in response to PCN (as well as other PXR dependent functions such as OATP2 upregulation and bile acid excretion; Staudinger et al. 2001). This is consistent with similar observations made in CAR and PPARα knockout mice and underlines the central coordination of hepatocellular proliferation through the nuclear hormone receptor superfamily in response to a diverse range of xenobiotics.
Epidemiology and Risk to Man
The risk that PXR activators pose to the development of liver tumors in man has not been extensively researched. Isolated reports have noted that CPA is hepatotoxic in man (Savidou et al. 2006) but despite its possible mutagenic potential, long-term follow-up of a group of fifty-three patients revealed no increase in liver cell carcinomas (Regidor et al. 2000).
Instead, attention has focused on the role that PXR plays in drug–drug interactions through enhanced PXR-mediated metabolism resulting in either decreased exposure/efficacy or increased toxicity through metabolic activation (reviewed in Ma, Idle, and Gonzalez 2008). More recent research has also highlighted the role of PXR in bile acid homeostasis, PXR-mediated hepatic steatosis, steroid hormone homeostasis, and inflammatory bowel disease (Ma, Idle, and Gonzalez 2008).
Aryl Hydrocarbon Receptor
Aryl hydrocarbon receptor (AhR) is a member of the bHLH-PAS (basic helix-loop-helix/Per-Arnt-Sim) gene family of transcription factors (reviewed in Schwarz and Appel 2005). It is activated by polychlorinated dibenzo(p)dioxins (PCDD) and coplanar polychlorinated biphenyls (PCBs) resulting in nuclear translocation, heterodimerization with its binding partner Arnt (AhR nuclear translocator), and transcription of target genes (Landers and Bruce 1991).
2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) is a potent activator of AhR, and like other AhR receptor agonists, results in induction of multiple drug-metabolizing enzymes such as CYP1A1, CYP1A2, CYP2B1, and UGT1A6 (Schwarz and Appel 2005). In parallel with hepatocyte drug-metabolizing enzyme induction, there is liver enlargement due to hepatocyte hypertrophy, multinucleated hepatocytes, fatty change, necrosis, and increased cellular replication with suppression of apoptosis resulting in the eventual outgrowth of enzyme altered hepatic foci and hepatocarcinogenesis (Bock and Köhle 2005; Kasai et al. 2009; Maronpot et al. 2010; Yoshizawa et al. 2007).
Role of AhR
TCDD and other polyhalogenated dibenzodioxins as well as dibenzofuranes and dioxin-like PCBs are non-genotoxic. Studies in AhR-receptor-deficient mice have demonstrated that the acute toxic effects of TCDD are mediated via the AhR receptor (Fernandez-Salguero et al.1996). Similar work in mice mutated for the AhR receptor has also demonstrated that promotion of hepatocarcinogenesis by TCDD is dependent upon sustained activation of the AhR receptor (Beebe et al. 1995). In line with this, mice that have constitutively active AhR receptor show increased promotion and yield of liver tumors following N-nitrosodiethylamine initiation (Moennikes et al. 2004).
Epidemiology and Risk to Man
TCDD was first classified as a human carcinogen in 1997 (IARC 1997) and since then epidemiological studies have linked dioxin exposure to cancers in man (Ma et al. 2006; Schwarz and Appel 2005). However, the carcinogenic effect of TCDD and dioxin-like molecules is weak and there is some uncertainty as to their true carcinogenic effect (Schwarz and Appel 2005). This uncertainty is partly due to the binding characteristics of the human AhR receptor which shows approximately a 10-fold lower affinity to TCDD than laboratory animal strains (Connor, and Aylward 2006). At this stage, therefore, it is not possible to definitively define what the adverse effects or risk is to human health, especially with regard to hepatocarcinogenesis from exposure to xenobiotics that induce liver enlargement through this mechanism. This view is in line with that from a recent AhR Receptor expert panel workshop that concluded that the mode of action for AhR mediated carcinogenesis could not be excluded for humans (Elcombe, pers. comm. 2011).
The Application of Newer Toxicogenomic Technologies
Advanced technologies such as microarray analysis and quantitative real-time PCR are now being used in conjunction with more traditional approaches such as Western blotting, immunocytochemistry, and electron microscopy to better evaluate the toxicological response to xenobiotics (Miyawaki et al. 2011). This has led to the evolution of the “omics” (metabonomics, transcriptomics, and proteomics) technologies from an observational science that essentially reported lists of differentially expressed genes to one in which Principal Component Analysis can now be used to better understand the effects of drug treatment upon biologically relevant signaling networks.
The rodent liver transcriptome has been studied by numerous researchers to gain mechanistic insights into the complex biological processes involved in drug-induced liver injury, repair, and hepatocarcinogenesis (Au, Navarro, and Rossi 2011). Combined toxicogenomic investigations now allow investigators to study early changes in gene expression profiles. Since toxicogenomic profiling may precede clinical chemistry, histopathology, clinical, or even ultrastructural changes (Heinloth et al. 2007; Wang et al. 2011), it is possible to gain insight into the early signaling perturbations that presage toxicologic change (Ruepp et al. 2002).
The activation of nuclear hormone receptors and the induction of microsomal liver enzymes occur through well-recognized mechanisms that are amenable to toxicogenomic profiling. Since the majority of hepatotoxicants induce overwhelming responses in gene expression related to cell injury, degeneration, metabolism, DNA repair, and regeneration as the liver begins the healing process (Gerrish and Malarkey 2007), it seems likely that a toxicogenomic approach may provide a useful tool to understand the mechanistic basis of these changes. The challenge therefore now remains to find the key genetic and molecular events that link drug induced liver damage and, specifically, drug-induced enzyme induction with hepatocarcinogenesis in rodents and man (Figure 8 and Table 4). To that end, there have been about a dozen studies that profile known non-genotoxic or genotoxic carcinogens in the rat or mouse in order to identify a common toxicogenomic signature that may be used to classify chemicals with unknown carcinogenic activity (Auerbach et al. 2010; Ellinger-Ziegelbauer et al. 2005).
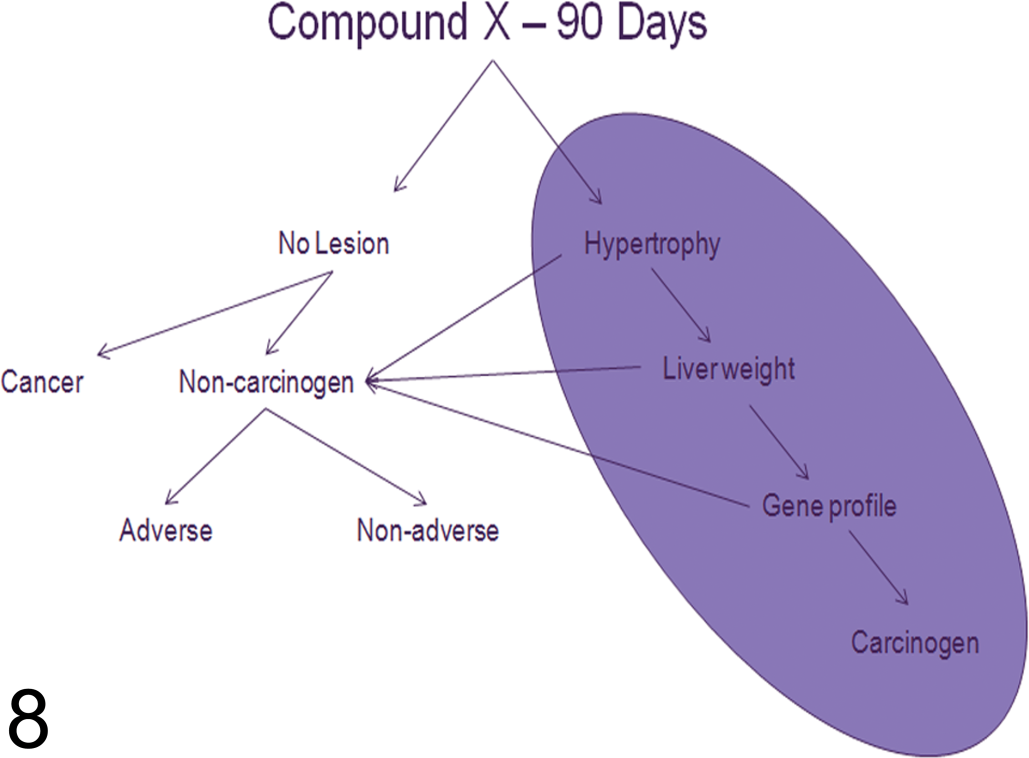
Flow-chart diagram demonstrating the NTP strategy of incorporating predictive hepatic transcriptomic data sets that may be useful in classifying compounds as non-genotoxic carcinogens after 90 days exposure. Currently, the presence of hepatocyte hypertrophy and increased liver weights have a low predictive value that will soon in combination with gene array profiles of genotoxicity and carcinogenicity make identifying and classifying carcinogenic agents more sensitive and specific.
Proposed mechanisms involved in tumorigenesis in rat liver according to observed gene expression changes.
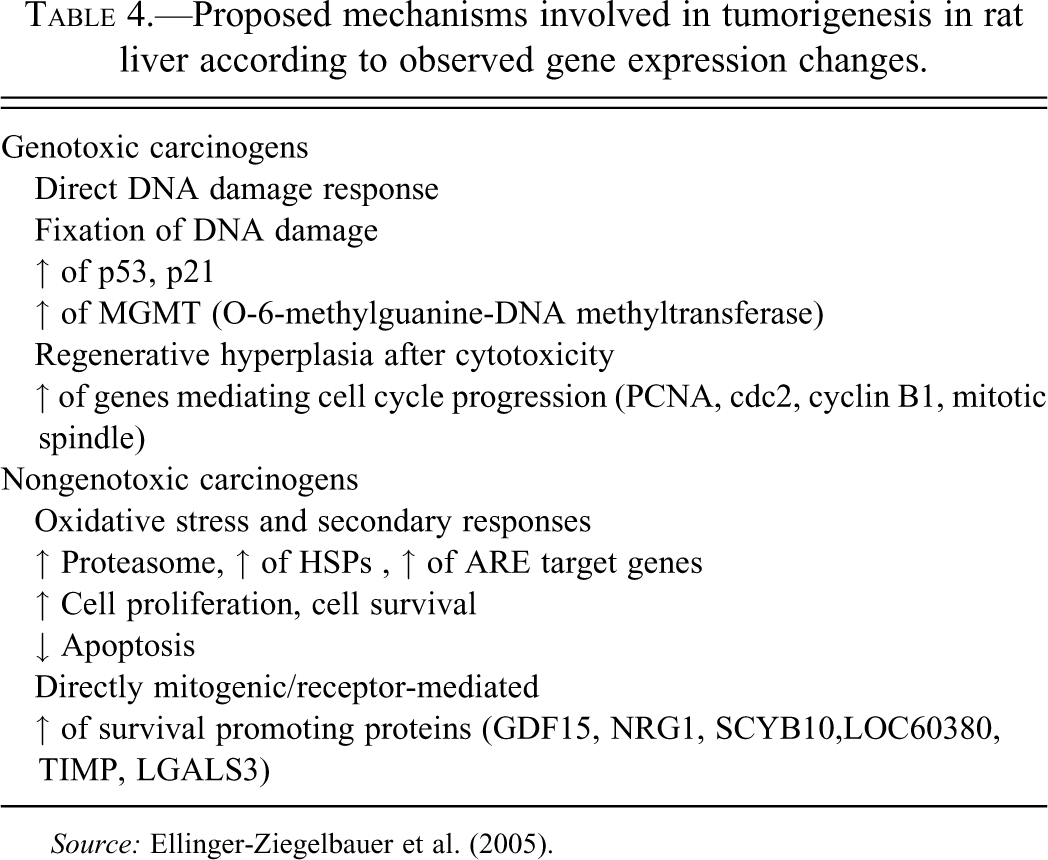
Source: Ellinger-Ziegelbauer et al. (2005).
Hepatic Hypertrophy: Adverse versus Non-Adverse
An adverse change is one “that . . . affects the performance of the whole organism’s ability to respond to an additional environmental challenge” (Lewis et al. 2002). Another widely accepted definition is that an adverse effect is “a change in morphology, physiology, growth, reproduction, development, or life span of an organism which results in impairment of functional capacity or impairment of capacity to compensate for additional stress or increased susceptibility to the harmful effects of other environmental influences” (WHO/IPCS [International Programme on Chemical Safety] 2004).
While such definitions may help summarize the broad category of changes that might be considered adverse, it was clear from the discussions arising from the expert panel that the operational use of the term adverse in the context of liver hypertrophy would benefit from some clarification. For example, a xenobiotic that induces an increase in liver weight of 150% in a 3-month study might be considered “adverse” in the context of dose setting for longer term studies but would not be considered adverse in the context of safety evaluation. In this situation, the increase in liver weight should be considered the maximum tolerated dose (MTD) for longer term dose setting (Carmichael et al. 1997; Maronpot and Malarkey 2011). In addition, while the initial effects of chemicals that induce hepatic metabolism may be regarded as adaptive and noninjurious, i.e., non-adverse (Greaves 2007; Schulte-Hermann 1974), it is clear that at higher dose levels, or following prolonged exposure, these adaptive responses can fail leading to degenerative hepatocellular changes including necrosis with additional involvement of the biliary systems as compensatory metabolic systems are overcome or where novel cytotoxic metabolites are generated (Klaunig et al. 1998; Williams and Iatropoulos 2002). In extreme cases, hepatocyte hypertrophy may lead to compression of the sinusoidal blood circulation and anoxic necrosis (Farber 1980). In these circumstances, the use of the term non-adverse is only valid for the dose and duration of exposure of that chemical as defined by the study in question.
Furthermore, the use of humanized mice has now shown that the rodent liver is primed toward proliferation in response to CAR/PXR/PPARα activation whereas the human liver shows considerable resistance to this mechanism of hepatocarcinogenesis. Therefore, the induction of a proliferative or even neoplastic response in the rodent liver through enzyme induction would be considered to have little relevance to man in the context of estimating the risk of human hepatocarcinogenesis.
The development of novel pharmaceuticals for the treatment of human disease often results in adverse events (AEs) in man. However, the identification of adverse effects in preclinical toxicity studies or even in patients does not necessarily preclude the development of these drugs into safe and effective treatments. Clearly, the risk evaluation of new therapeutic drugs requires careful consideration of the cost–benefit analysis. More tolerance to adverse effects noted in preclinical toxicity studies is given to drugs that treat serious disease and where the AEs are easily monitorable, have a good therapeutic margin, or the effect is fully reversible upon cessation of dosing without permanent loss of function.
Hence one of the underlying considerations when considering the adverse effects of a new chemical entity is that the exposure dose and duration in the pivotal laboratory animal toxicology studies should equal or, ideally, exceed that in the intended human population (IHC Guidance for Industry M3(R2), 2010).
Conversely, agrochemicals are exposed to healthy humans and therefore the risk-benefit assessment is different to drugs exposed to patients.
Non-adverse Effects in the Liver for Setting Dose Levels for Longer Term Studies
The highest dose level used on long-term animal studies generally requires that it satisfies the regulatory conditions of achieving a MTD level that is high enough to adequately assess the toxicity while preventing unnecessarily high dose levels that would induce chronic physiological dysfunction or reduce overall survival (OECD Guidance Notes 2002; ICH Guidance for Industry S1C(R2) 2008).
These requirements therefore dictate that for the purposes of dose setting in long-term toxicity studies, any change that results in structural or biochemical alterations indicative of liver cell damage is likely to result in increased turnover of hepatocytes, somatic mutation, and liver neoplasia in chronic 2-year rodent bioassays, and therefore exceed the MTD. In addition, increases in liver weight in short-term experiments (3 months to 1 year) of approx. ≥150% (although considered non-adverse for the purpose of risk assessment) even in the absence of all other adverse findings is also likely to result in liver tumors in chronic carcinogencity studies in rats and mice (Carmichael et al. 1997; Maronpot and Malarkey 2011). Therefore, a dose level of a xenobiotic that in short-term tests induced either structural or biochemical evidence of hepatocellular damage, or produced increases in liver weight of approx. ≥150% would be considered adverse in the context of dose setting and exceed the MTD.
When considering dose levels below the MTD, various formulae are applied but generally the bottom dose level needs to be a NOEL or NOAEL, as these will be used in estimating risk assessment for occupational or long-term unintentional exposure (OECD Draft Guidance Document No. 116
Non-adverse Effects in the Liver in Terms of the Hepatic Enzyme–inducing Agents
In the context of liver effects induced by hepatic enzyme–inducing agents, a dose level at which a non-adverse toxicity is present would, with every expectation, produce morphological changes in the liver characterized by liver weight increases, microscopic evidence of hepatocellular enlargement/hypertrophy, and generally no increases in plasma enzymes such as alanine and aspartate aminotransferase (Ennulat, Walker, et al. 2010). The exception to this rule is the dog where treatment with phenobarbital or corticosteroids results in induction of liver ALP and γGT in the absence of liver injury and independently of enzyme induction (Ennulat, Walker, et al. 2010).
In terms of the hierarchy of effects on the liver, the general consensus of the panel discussion was that liver enlargement, as characterized by increases in liver weight, was the most sensitive indicator of hypertrophy. This change precedes morphological effects, although measurable increases in mRNA (indicative of enzyme induction) are likely to occur prior to increases in liver weight. Considerable discussion occurred regarding the extent of liver weight increase that could be expected to be non-adverse in the long term. While empirical relationships of certain degrees of liver weight increase appeared to be correlated with the subsequent development of irreversible toxicity, such as fibrosis, necrosis, vacuolization, fatty degeneration, and even neoplasia, it was considered that a more scientific approach to the problem, taking into account proposed mode of action analysis, would be of use to guide the setting of better tolerated dose levels and a more systematic risk assessment process (Carmichael et al. 2011; Felter et al. 2011). The general opinion of the group was that liver weight increase through hepatocyte enzyme induction, in the absence of histopathologically demonstrated degenerative or necrotic changes and without significant changes in hepatic-derived plasma enzymes, would not be considered adverse and would have little relevance to man in terms of risk assessment and the development of liver tumors.
Assessment of Liver Weight Increases
When assessing a histological change caused by an increase in liver weight, in order to conclude whether the change is adverse or not, a number of steps must be carefully considered, summarized below (and in Figure 9):
Flow-chart diagram showing a decision tree for adverse versus non-adverse effects induced by compounds which increase liver weight. (Modified from Andrew 2005.) Is there histological evidence of structural degenerative or necrotic changes such as: hepatocyte necrosis, fibrosis, inflammation, and *steatotic vacuolar degeneration biliary/oval cell proliferation, degeneration, fibrosis, and cholestasis necrosis and degeneration of other resident cells within the liver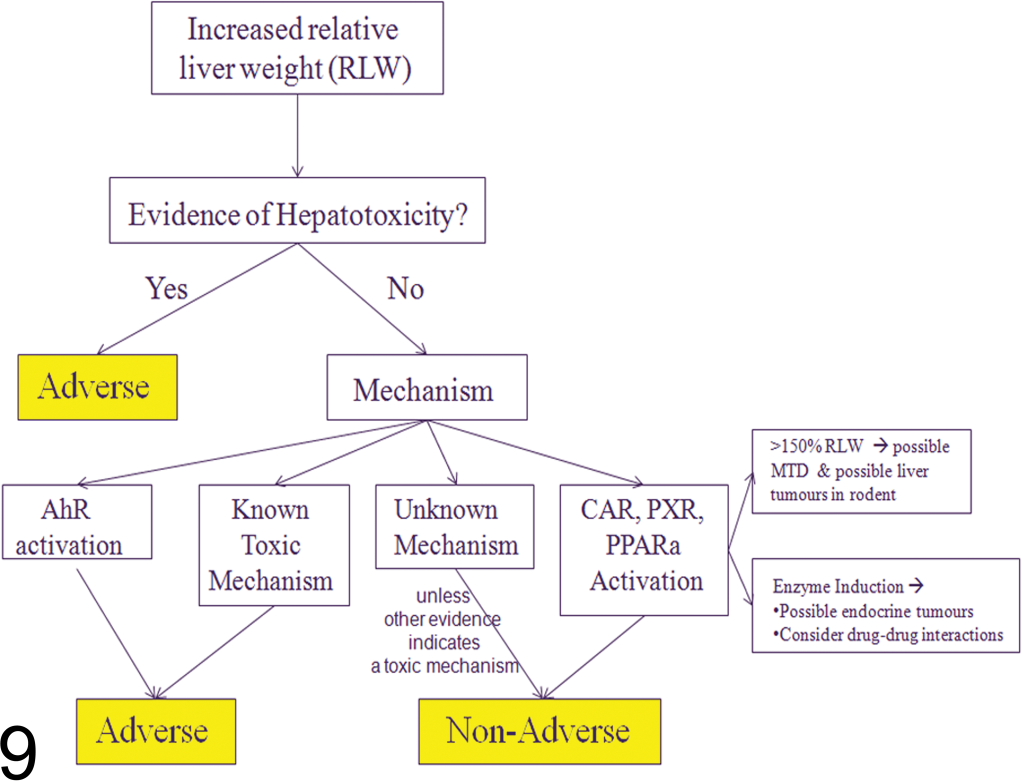
(*Minimal to mild increases in steatotic macro-vesicular vacuolation without other changes indicating cellular damage should be distinguished from micro-vesicular vacuolation and considered non-adverse since this is a common change induced by feeding high-fat diets.)
(Of note, transient increases in proliferative indices together with changes in hepatocyte ploidy, if induced through CAR/PXR/PPARα activation, is likely to be a rodent-specific phenomenon and therefore of little relevance to man even if this results in altered hepatic foci and/or primary liver tumors in chronic studies.) In the absence of histological changes, using a weight-of-evidence approach, is there clinical pathology evidence of hepatocyte damage characterized by a dose dependent and biologically significant and consistent increase in at least two liver parameters: at least ×2 to ×3 increase in ALT (EMEA 2010, FDA 2009; HED Guidance Document 2002) or a biologically significant change in other biomarkers of hepatobiliary damage (ALP, AST, γGT, GLDH, etc.) a biologically significant change in another clinical pathology marker indicating liver dysfunction (albumin, bilirubin, bile acids, coagulation factors, cholesterol, triglycerides etc.).
Clinical pathology changes should corroborate each other, be consistent with the expected species-specific patterns of change resulting from hepatobiliary injury, and take into account target pharmacology that may non-adversely alter one or more of these biomarkers. It should also be noted that statistical significance alone is not a reliable indicator of hepatic toxicity (HED guidance document 2002).
If the above mentioned adverse criteria are not observed, then increases in liver organ weight and liver cell hypertrophy due to enzyme induction can be considered as an adaptive response to a xenobiotic and of little relevance to man.
However, prolonged exposure to a xenobiotic at levels that have previously been shown to be adaptive may eventually result in liver cell injury due to a failure of adaptive mechanisms. In this case, the combination of dose level and duration of exposure to the xenobiotic under the terms and conditions of the new experiment would now be considered adverse.
Summary and Conclusions
The purpose of the 3rd International ESTP workshop was to discuss the significance of hepatocellular hypertrophy in rodents and define more clearly which responses were considered to be adaptive in nature so as to help guide scientific opinion. The consensus of opinion was that hepatomegaly as a consequence of hepatocellular hypertrophy without histologic or clinical pathology alterations indicative of liver toxicity was considered to be an adaptive non-adverse change. This conclusion should normally be reached by an integrative weight of evidence approach. Under these circumstances, hepatocellular hypertrophy through enzyme induction may therefore be considered a fully reversible change that does not compromise viability or functional integrity and potentially benefits the organism through increased metabolic capability.
During the assessment of increased liver weight due to enzyme induction, one should consider for long-term dose setting that non-adverse adaptive changes at low exposures may very well become adverse at higher/longer dosages. In addition, increases in liver organ weight ≥150% in 3-month to 1-year studies may very well lead to hepatocarcinogenesis and/or endocrine tumors (though increased degradation of thyroid and possibly reproductive hormones) in longer term studies and appropriate measures should be put in place to ensure adequate survival of the top dose animals on the study.
Finally, the development of newer technologies and the use of genetically modified mice have considerably advanced our understanding of the underlying mechanisms involved in nuclear hormone receptor (CAR, PXR, and PPARα) induced hepatocellular hypertrophy. The imminent generation of triple humanized mice are likely to further increase our understanding of the response of the humanized receptor to these types of activators by removing the potentially confounding effects of “off-target” nuclear hormone receptor signaling by “promiscuous” activators and thus increase the confidence that hepatocarcinogenesis in response to these types of compounds is a rodent-specific phenomenon.
Footnotes
Authors’ Note
Workshop Participants—Clifford Elcombe, CXRBiosciences, Great Britain, BSTP; John Foster, AstraZeneca, Great Britain, BSTP; Peter Hall, AstraZeneca, Great Britain, BSTP; Takanori Harada, IET (Institute of Environmental Toxicology), Japan, JSTP; Wolfgang Kaufmann, Merck KGaA, Germany, ESTP (organizer of the 3rd International ESTP expert workshop); Anja Knippel, Merck KGaA, Germany, ESTP; Karin Küttler, BASF SE, Germany, ESTP; David E Malarkey, NIEHS (National Institute for Environmental Health Science), USA, STP; Robert R. Maronpot, Consultant, USA, STP; Akiyoshi Nishikawa, NIHS (National Institute of Health Sciences), Japan, JSTP; Thomas Nolte, Boehringer Ingelheim Pharma GmbH & Co. KG, Germany, ESTP; Agnes Schulte, BfR (German Federal Institute for Risk Assessment), Germany, ESTP; Volker Strauss, BASF SE, Germany, ESVCP/ESTP; Malcolm York, GSK, Great Britain, ESVCP.
Acknowledgments
The authors wish to thank ESTP for organizing the workshop and MerckSerono for giving logistical support at the venue.
The author(s) declared a potential conflict of interest with respect to the research, authorship, and/or publication of this article: Financial support for this workshop was provided by the ESTP. This article may be the work product of an employee or group of employees of the National Institute of Environmental Health Sciences (NIEHS), National Institutes of Health (NIH); however, the statements, opinions or conclusions contained therein do not necessarily represent the statements, opinions or conclusions of NIEHS, NIH, or the United States government.
The author(s) received no financial support for the research, authorship, and/or publication of this article.