
Abstract
Fenoldopam, a dopaminergic DA1 agonist, induces vasodilatation via nitric oxide (NO), and this may be associated with mesenteric arterial injury. NO is produced from the enzymatic action of nitric oxide synthase (NOS), which is regulated by the shear-stress mediating protein caveolin-1. Profound vasodilatation and accompanied decreased shear are early events that could initiate vascular injury. Therefore, it is of interest to determine the role of caveolin-1 and the NO pathway in fenoldopam-induced vascular injury. At sites of fenoldopam-induced mesenteric arterial injury, decreased caveolin-1 expression and apoptosis were prominent immunohistochemical findings. An additional finding at these sites of injury were loss and/or reduced expression of caveolin-1 regulated structural proteins, connexin-43, (gap junction) ZO-1, and claudin (tight junctions). Because functional loss of caveolin-1 is associated with increased NOS activity and vasodilatation via NO, studies were conducted to show a NO donor produced vascular lesions in the mesenteric arteries morphologically similar to those induced by fenoldopam. Moreover, the incidence and severity of fenoldopam-induced vascular injury were reduced when an NOS inhibitor or a scavenger of NO-generated free radicals were coadministered with fenoldopam. Collectively, these data suggest that caveolin-1 and its regulated NO pathway may play an important role in vasodilatory drug-induced vascular injury.
Keywords
Introduction
Clinical development of novel life-saving therapies is often hindered because the nonclinical safety profiles of candidate drugs are associated with occult pathology that are not detected by noninvasive, routine clinical monitoring. In nonclinical safety studies, drug-induced vascular injury (DIVI) is an example of such a profile, because there are no biochemical markers for monitoring this lesion clinically and our understanding of the pathophysiology is limited. This type of vascular toxicity is most often associated with candidate drugs that are pharmacologically active in the vascular bed. There are a number of drugs from different pharmacologic classes and chemical structures that have been approved for human use but are known to cause occult mesenteric or coronary arterial vascular lesions in animal studies (Albassam et al. 1999; Chelly et al. 1986; Gans et al. 1980; Greaves 1998; Joseph 2000; Kerns, Arena, Macia, et al. 1989; Kerns, Arena, and Morgan 1989; Kerns et al. 2005; Louden et al. 2000; Louden and Morgan 2001; Mesfin et al. 1989). In dogs, understanding DIVI pathophysiology and potential mode of action has been aided by data describing a relationship between predisposed toxicity site/sites and the distribution, density, type, and ratio of vasoactive endothelin receptors in the heart and coronary arteries (Louden et al. 2000). Additionally, the composite analysis of receptor subtype profiles, messenger RNA (mRNA) expression and regional blood flow measurements clearly supported the hypothesis that a disproportionate receptor distribution was responsible for the marked functional differences in regional blood flow at the affected sites of injury, the right atrium and right coronary arteries (Chelly et al. 1986; Louden et al. 2000). Collectively, these data in conjunction with other published reports strongly suggest that mechanistically, exaggerated pharmacology causing profound regional vasodilatation, increased regional blood flow and dysregulation of arterial tone are the basis for selective, site-specific, mesenteric, or coronary arterial damage in rats and dogs (Chelly et al. 1986; Joseph 2000; Louden et al. 2000).
Fenoldopam, a dopaminergic-1 agonist, mediates renal vasodilatation through the nitric oxide (NO) pathway (Venkatakrishnan et al. 2000) and induces renal and mesenteric arterial lesion in rats (Bugelski et al. 1989; Yuhas et al. 1985). NO mediates the vasodilatation that can be induced by antagonism of endothelin receptors and/or opening of potassium ion channels (Kuhlmann et al. 2004; Tirapelli et al. 2005; Yamaguchi et al. 2003). There is growing evidence that vascular injury may be a consequence of exaggerated pharmacology of receptors or ion channels that have downstream signaling activity in the nitric oxide synthase (NOS) pathway either directly or indirectly (Kerns et al. 2005; Louden and Morgan 2001). It is possible that in drug-induced vascular injury, NO mediates the prolonged and sustained vasodilatation that causes dysregulation of arterial tone leading to arterial damage (Joseph et al. 1997; Louden et al. 2000). Concomitantly, there is loss of regulation and control of key biochemical pathways in endothelial cells (EC) and smooth muscle cells (SMC), breakdown in cell-to-cell communications and ultimately, arterial wall damage.
Cav-1, the major structural protein of caveolae, is expressed within the vascular wall, specifically on the phenotypically anchored primary targets of drug-induced vascular injury EC and SMC (Drab et al. 2001; Zhao et al. 2002). Within these cells, cav-1 is an important mediator of many signal transduction pathways including regulation of vascular tone (Liu et al. 2002; Martens et al. 2001; Shaul and Anderson 1998; Yu et al. 2004). Functionally, caveolae appear to be the focal point for compartmentalizing, organizing, and modulating signal transduction activities (Linder et al. 2005; Liu et al. 2002; Martens et al. 2001; Shaul and Anderson 1998; Yu et al. 2004) for many receptors such as adenosine, dopaminergic-1, and endothelin, and enzymes including phosphodiesterase (PDE), NOS, and adenylyl cyclase. These and other proteins are regulated by cav-1. An example of this regulation is seen in mice with targeted gene disruption of cav-1. This induced genetic defect causes impairment of NOS, NO, and calcium signaling in the cardiovascular system causing aberrations in endothelium-dependent relaxation, contractility, and maintenance of vasculomyogenic tone (Drab et al. 2001). Functionally, genetic loss of cav-1 was also associated with a 5-fold increase in systemic NO but no major changes in NOS expression (Drab et al. 2001; Zhao et al. 2002), suggesting that deregulation and/or hyperactivity of NOS was responsible for the massive increase in NO production. Collectively, these data suggest that cav-1 plays an important role in regulating NOS activity and potentially vascular tone.
Data from our previous studies reported loss of cav-1 in SMC and/or EC in DIVI (Brott, Jones, et al. 2005; Brott, Gould et al. 2005). Furthermore, studies in vitro showed loss of cav-1 was an early event that preceded SMC apoptosis/necrosis (Brott, Jones, et al. 2005). Based on these data, we hypothesized that DIVI is mediated in part through the cav-1 regulated NO pathway and the principal objective of this study was to define and characterize the potential role of NO and components of the NO pathway in the development of DIVI.
Materials and Methods
Animals
Studies were approved by the institutional animal care and use committee (AstraZeneca Pharmaceuticals, Wilmington, DE) and were designed to minimize clinical signs of toxicity. All studies were conducted in 8- to 10-week-old male Han Wistar rats (Charles River Laboratories Wilmington, MA). Rats were single housed, had free access to water, and fed a complete pelleted rodent diet. The testing facility is illuminated with a 12-hr light/dark cycle, and temperature, humidity, and air changes were controlled.
Test Preparation and Administration
Fenoldopam (Sigma, St. Louis, MO) as the mesylate salt was prepared as a 6 mg/ml solution in sterile 0.9% aqueous sodium chloride. Sodium nitroferricyanide (III) dihydrate (sodium nitroprusside; SNP; nitric oxide donor; Sigma), L-NAME (NOS inhibitor; Cayman Chemical, Ann Arbor, MI), and Tempol (extracellular superoxide dismutase [ecSOD] mimetic; EMD Biosciences, La Jolla, CA) were dissolved in sterile 20% (v/v) propylene glycol:80% (v/v) of 0.9% (w/v) sodium chloride. Saline (0.9% sodium chloride) was used as the test article vehicle control. All compounds were administered as continuous infusions using Harvard® minipumps.
Study Design, Treatments (Infusion Studies)
Briefly, groups of rats (3–5/group) were administered fenoldopam, sodium nitroprusside, L-NAME or tempol at a rate of 0.3 mL/hr. In a pilot study, fenoldopam dosages of 1, 10, or 100 µg/kg/min were infused for 3, 6, 12, or 24 hr. From the pilot study, the dose (100 µg/kg/min) and infusion duration (24 hr) were selected for fenoldopam. For the NO donor sodium nitroprusside, dosages evaluated were 20, 50, 100, and 200 µg/kg/min; for L-NAME 50 and 200 µg/kg/min and for Tempol 500 µg/kg/min. In combination studies (fenoldopam +L-NAME or fenoldopam+Tempol), rats were pretreated with L-NAME and Tempol for at least 90 min before co-infusion with fenoldopam was initiated. In all studies, rats were subjected to a complete necropsy ∼26 hr post dose initiation.
Necropsy, Tissue Preparation
At necropsy the entire mesentery was dissected free from the viscera and prepared using the methods described by Newsholme et al. (2000). The tissues were fixed in NBF (neutral buffered formalin, 10%) for all studies or ZF (zinc-buffered formalin fixative, Anatech Ltd., Battle Creek, MI) in the fenoldopam only study to allow immunohistochemistry staining. The NBF-fixed tissues were processed and stained with hematoxylin and eosin (H&E). This method of preparation enabled microscopic evaluation to be standardized and optimized so that numerous profiles of variable sized mesenteric arteries could be examined. The severity of arterial lesions within the profiles were graded and scored as previously described (Slim et al. 2003).
Immunohistochemistrty
The immunohistochemistry staining procedures were done using the Discovery XT system (Ventana Medical Systems Inc., Tucson, AZ). ZF-fixed tissues were processed and paraffin sections (5 µm) of mesenteric arteries from control and treated rats were immunostained for caveolin-1, activated caspase-3, tight junctions (ZO-1 and claudin), gap junctions (connexin 43), plasma proteins (von Willebrand Factor [VWF] and fibrinogen), nitrotyrosine (NT), and ecSOD. The staining procedures were optimized for each protein according to the manufacturers’ specifications and recommendations.
Caveolin-1
Sections were pretreated with citrate for 15 min, blocked for 4 min using 5% (w/v) bovine serum albumin (BSA; Sigma), 2% NGS (Vector, Burlingame, CA) in triton x-100 (Sigma) and washed with approximately 100 mL triton x-100. Following blocking and subsequent washings, tissues were subjected to 30 min incubation with anti-mouse cav-1 (1:50, Research Diagnostics, Concord, MA). Sections were then incubated for 24 min with goat anti-mouse biotin (Jackson ImmunoResearch).
Activated caspase-3
Following treatment with EDTA for 30 min and an 8-min protease 1 (Vantana) treatment, sections were incubated for 2.5 hr with anti-rabbit cleaved caspase-3 antibody (Biocare, Concord, CA; 3:20 with antibody diluent). Sections were then treated for 50 min with goat anti-rabbit biotin (Jackson ImmunoResearch 1:25 with antibody diluent).
ZO-1
Sections were pretreated with citrate for 8 min, incubated for 2 hr with anti-rabbit ZO-1 (1:5 diluted; Zymed, Carlsbad, CA). Sections were then incubated with goat anti-rabbit biotin for 30 min (1:25 diluted, Jackson ImmunoResearch).
Claudin
Sections were incubated 1 hr with anti-mouse Claudin (1:500 diluted; Zymed), followed by a 30-min incubation with horse anti-mouse-biotin (1:100 diluted; Jackson ImmunoResearch).
Connexin 43
Sections were pretreated with citrate for 16 min and then incubated for 3 hr with anti-rabbit connexin 43 (1:5 diluted, Zymed). Sections were then incubated 30 min with goat anti-rabbit biotin (1:25 diluted, Jackson ImmunoResearch).
VWF
Sections were incubated for 10 min with 0.4% trypsin containing 1% (w/v) calcium chloride, blocked for 2 min with 3% hydrogen peroxide followed by a 10-min protein block (Dako, Carpinteria, CA). Sections were then incubated 1 hr with anti-rabbit vWF (1:600 diluted; Dako) and then 30 min with goat anti-rabbit biotin (1:50 diluted; Jackson ImmunoResearch).
Fibrinogen
Sections were pretreated with citrate for 8 min, followed by a protease treatment for 6 min. Sections were then incubated 1 hr with goat anti-fibrinogen (1:500 diluted; Nordic Immunology), followed with a 30-min incubation with horse anti-goat-biotin (Jackson ImmunoResearch).
NT
Sections were blocked in 3% (v/v) hydrogen peroxide for 15 min, incubated for 2 hr with anti-mouse nitrotyrosine antibody (diluted 3:20; Zymed), followed by incubation for 30 min with goat anti-mouse-biotin (diluted 1:25; Jackson ImmunoResearch.
ecSOD
Sections were incubated in 0.4% (w/v) trypsin containing 1% calcium chloride for 10 min, blocked in 3% (v/v) hydrogen peroxide for 15 min, blocked with protein block (Dako) for 20 min, and then incubated for 30 min with anti-rabbit ecSOD (1:500 diluted; Stressgen, San Diego, CA). Sections were then incubated for 10 min with goat anti-rabbit-biotin primary (1:50 diluted; Jackson ImmunoResearch).
In Vitro Studies
Test Articles and Controls
IBMX (3-isobutyl-1-methylxanthine)/dimethyl sulfoxide (DMSO) was dissolved in fenoldopam was dissolved in A10 culture media. The final concentration of ethanol or DMSO in culture was 0.1%.
Experimental Design
Semi-confluent A10 cells (rat embryonic aortic smooth muscle cells; ATCC, Manassas, VA), were cultured according to manufacturers recommendations. The cells were treated for 6 or 24 hr with IBMX or the appropriate vehicle controls. The cultured A10 cells were evaluated for relative cell number (CellTiter Glo), activated caspase-3 (as a biomarker of apoptosis), and/or caveolin-1 expression (by Enzyme-linked immunosorbent assay [ELISA]). CellTiter Glo (Promega, Madison, WI) and activated caspase-3 (Promega) measurements were determined according to the manufacturer’s recommendations.
Caveolin-1 ELISA
A10 cell lysates were incubated overnight in antibody (BD Transduction Labs, San Diego, CA) coated caveolin-1 wells, then blocked for 2 hr with Tris-buffered saline, 3% BSA, and 0.05% tween-20. Wells were washed with blocking buffer, incubated with antibody for 90 min, washed with Tris-buffered saline and developed using 3,3’,5,5’-tetramethylbenzidine (Sigma) and sulfuric acid as the stop solution.
Statistical Analysis
Statistical analyses were performed using analysis of variance (ANOVA) followed by post hoc comparison. A value of p < .05 and p < .01 determined statistical significance.
Results
Fenoldopam-Induced Vascular Injury
Continuous intravenous infusion of fenoldopam was associated with macroscopic and microscopic vascular lesions 26 hr post dosing. The dose of 100 µg/kg/min consistently induced severe lesions in all rats (Table 1). Additionally, there was a dose–response in incidence and severity of vascular toxicity in rats administered 1, 10, and 100 µg/kg/min. Regardless of the dose, vascular lesions were only evident when fenoldopam was infused for more than 12 hr (data not shown). Briefly, the typical histologic appearance was segmental medial SMC necrosis/apoptosis with multifocal hemorrhage (Figure 1). Positive staining for activated caspase-3 confirmed apoptosis of SMC at these sites of injury (Figure 2). Based on these data, continuous intravenous infusion of 100 µg/kg/min of fenoldopam for 24 hr was used to assess the potential role of the NO pathway in the initiation and development of fenoldopam-induced vascular injury.
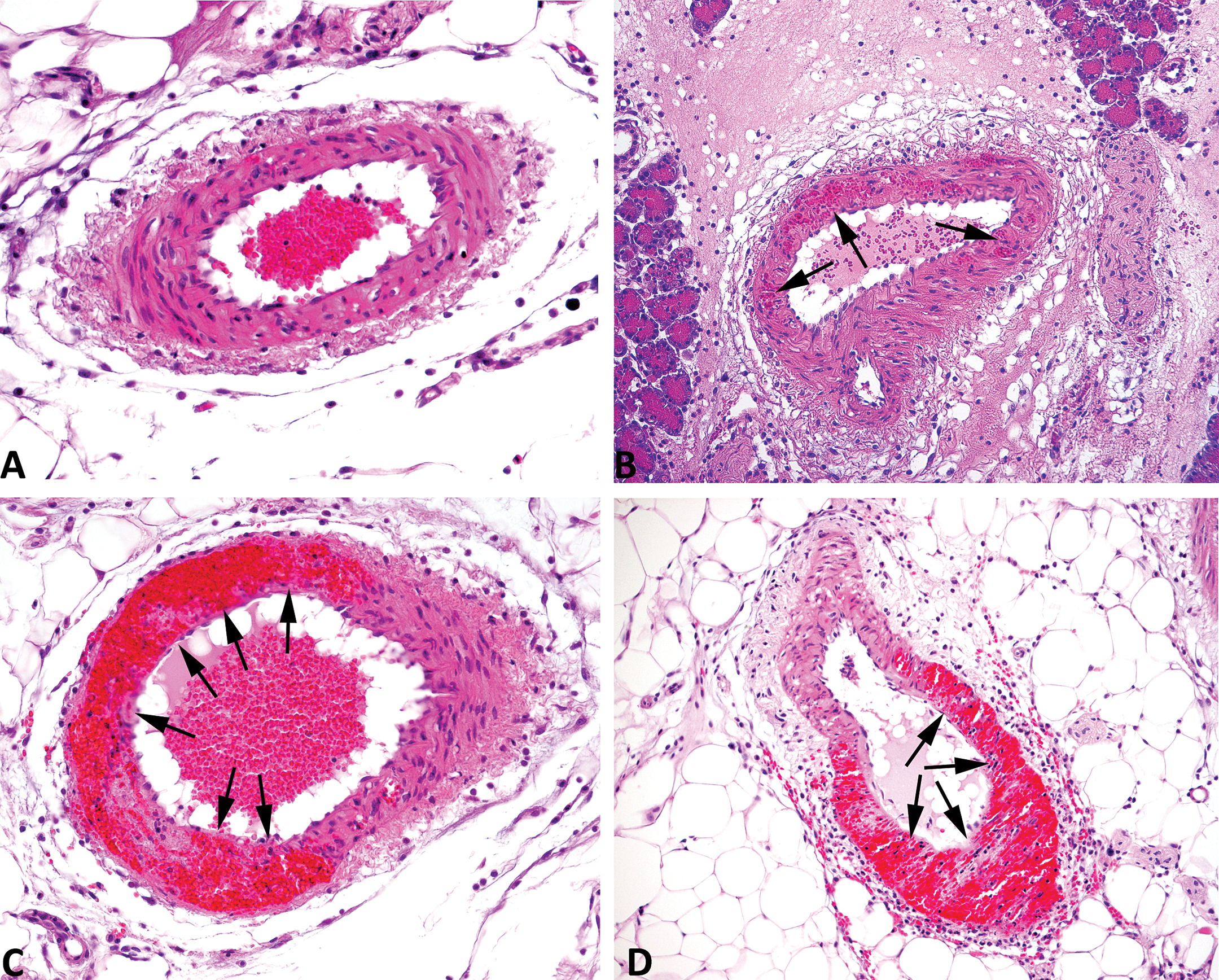
Hematoxylin and eosin (H&E) staining of mesenteric arteries from rats treated 24 hr with, (A) saline, (B) 1 µg/kg/min fenoldopam, (C) 100 µg/kg/min fenoldopam, and (D) 50 µg/kg/min sodium nitroprusside. Arrows indicate areas of drug-induced vascular injury. H & E, 200×.
Apoptosis of smooth muscle cells in fenoldopam-induced vascular injury. Mesenteric arteries from control (0 µg/kg/min) and fenoldopam (100 µg/kg/min) treated rats were excised, zinc fixed, processed, and stained for activated caspase-3. Smooth muscle cells were positive for the apoptosis marker activated caspase-3. Arrows indicate areas of drug-induced vascular injury (200×).
Incidence and severity of mesenteric arterial lesions induced by fenoldopam.

Fnd = fenoldopam.
Groups of rats (3 males/group) were treated for 24 hr, necropsied at 24 hr, mesenteric arteries excised, formaldehyde fixed, processed, and hematoxylin and eosin (H&E) stained. The incidence and severity of vascular lesions were recorded by a pathologist, as previously described (Slim et al. 2003).
Reduced Vascular Smooth Muscle Cell cav-1 in Vivo and in Vitro
From rats treated with vasculotoxic doses of fenoldopam, mesenteric arteries with histologic evidence of segmental medial hemorrhage and necrosis were evaluated immunohistochemically, to determine whether caveolin-1 was reduced at the specific sites of SMC damage (Figure 3). Within damaged mesenteric arteries, caveolin-1 immunoreactivity, used as an indicator of expression, was markedly reduced or absent only at the localized sites of SMC damage. Loss of immunoreactivity was segmental and confined to the focal areas of damage, while histologically normal arterial segments of SMC expressed caveolin-1 similar to unaffected or vehicle control arteries. Furthermore, caveolin-1 immunoreactivity in affected mesenteric arteries was linked to the severity of vascular damage with some variation in the intensity of immunoreactivity. Interestingly, in some damaged areas where necrosis was not apparent, there was reduced but not complete loss caveolin-1 immunoreactivity.
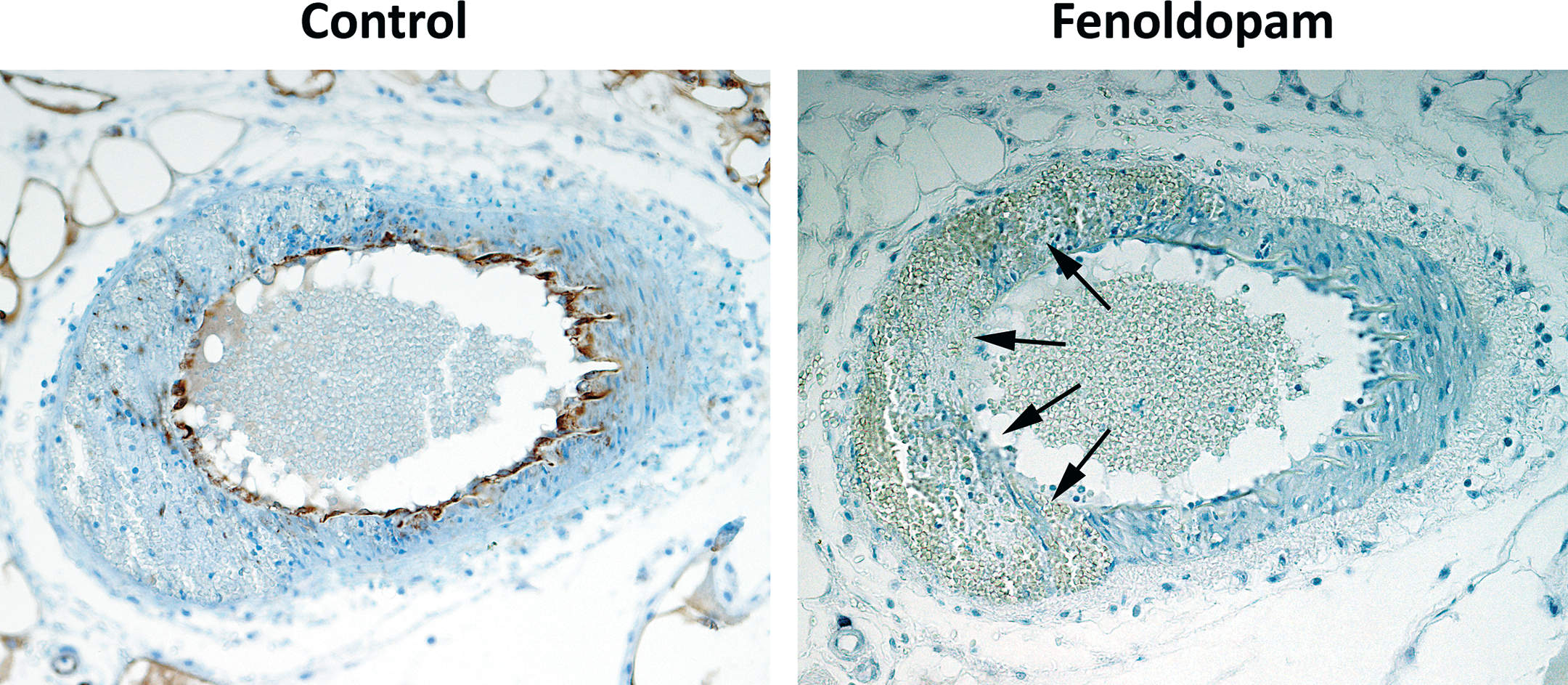
Expression of Cav-1 in fenoldopam-induced vascular injury. Immunohistochemistry of cav-1 expression in mesenteric arteries from control and fenoldopam (100 µg/kg/min) treated rats. Note positive immunoreactivity for cav-1 in SMC and EC in controls and the absence of immunoreactivy in EC and SMC damaged from fenoldopam treatment. Arrows indicate areas of drug-induced vascular injury (200×).
While these data suggest an association between DIVI and changes in SMC caveolin-1 expression, it is unknown if caveolin-1 loss is an early event that precedes SMC damage induced by fenoldopam and other in vivo vasculotoxicants. This was assessed using an in vitro cell culture system of embryonic rat aortic SMC (A10 cells) and varying concentrations of IBMX a broad spectrum PDE inhibitor that belongs to a class of well-known in vivo vascular toxicants. Cell viability, caveolin-1, and caspase-3 activation as biomarker of apoptosis were assessed.
In vitro treatment of rat aortic SMC with the broad spectrum PDE inhibitor IBMX caused reduced and/or loss of caveolin-1 expression. Because PDE inhibitors are known to cause vascular lesions in vivo IBMX was used to better define the temporal relationship between caveolin-1 loss, cell death, and apoptosis (Figure 4). Based on caveolin-1 ELISA analyses, there was a significant dose-related reduction in SMC caveolin-1 expression 6 hr post-treatment with IBMX and at this time point cell viability was not affected and caspase-3 activation was similar to untreated or vehicle control SMC. However, at 24 hr posttreatment, at high doses, reduced cell viability and caveolin-1 expression was accompanied by a significant increase in caspase-3 activation indicating SMC apoptosis and possible necrosis. At the intermediate dose at this time point, there was significant reduction in caveolin-1 with increased caspase-3 activation and no loss in cell viability. Interestingly, after 24 hr of treatment, the low dose of the PDE inhibitor was associated with a slight increase in caveolin-1 (not statistically significant) and caspase activation but with no loss of cell viability. Both fenoldopam and minoxidil® caused a similar response in caveolin-1 expression and apoptosis (Brott, Jones, et al. 2005).
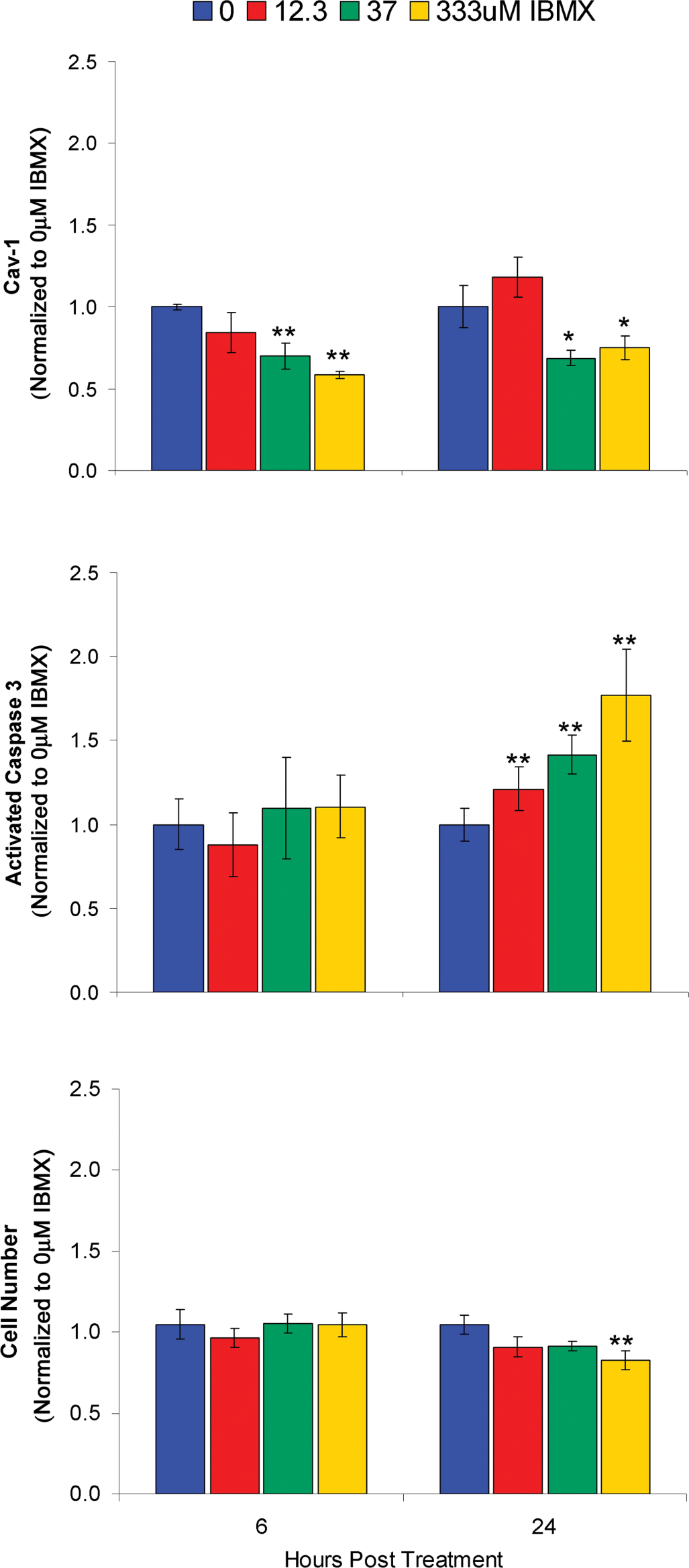
Relationship between cell viability, caveolin-1 expression, and caspase-3 activation as a biomarker of apoptosis. A10 cells were treated with the broad spectrum phosphodiesterase inhibitor IBMX for 6 and 24 hr and then the cells were analyzed. Note the dose and time-related reduction in caveolin-1 expression, caspase-3 activation, and cell viability (*p < .05, **p < .01).
Caveolin-1 Loss Correlates with the Breakdown of Vascular Wall Integrity
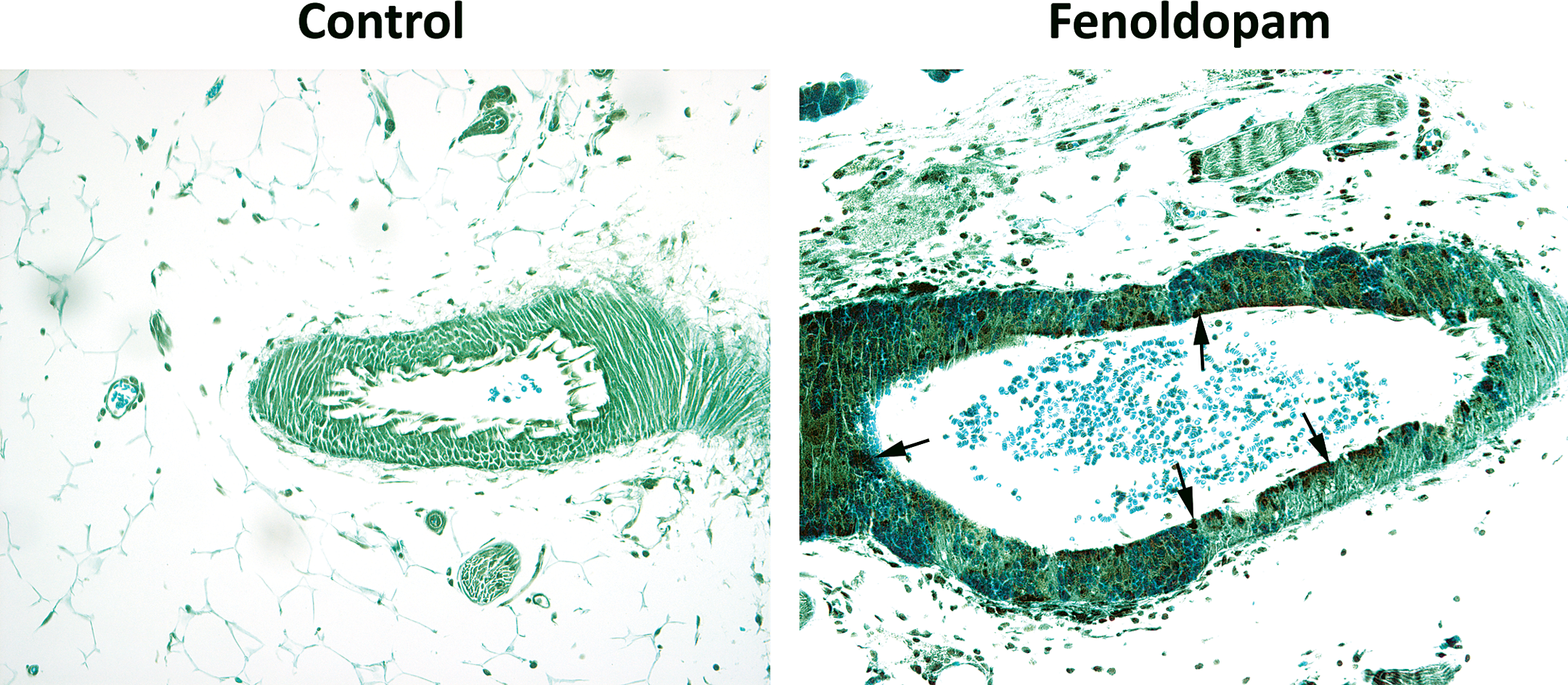
Fenoldopam-induced mesenteric arterial injury is associated with excessive vasodilatation, extensive medial hemorrhage, and edema that indicates there is a breakdown in the structural integrity of the vessel wall (Yuhas et al. 1985). Tight and gap junction proteins are important in maintenance of the structural integrity of the vascular wall and are regulated by cav-1 (Nusrat et al. 2000). The status of ZO-1, Claudin (tight junction proteins), and connexin 43 (a gap junction protein) were evaluated using immunohistochemistry to determine if there was concomitant reduction in the expression pattern of these proteins similar to cav-1 at the specific sites of injury. ZO-1, claudin, and conexin 43 immunoreactivity were markedly reduced at the sites of injury in mesenteric arteries (Figure 5).
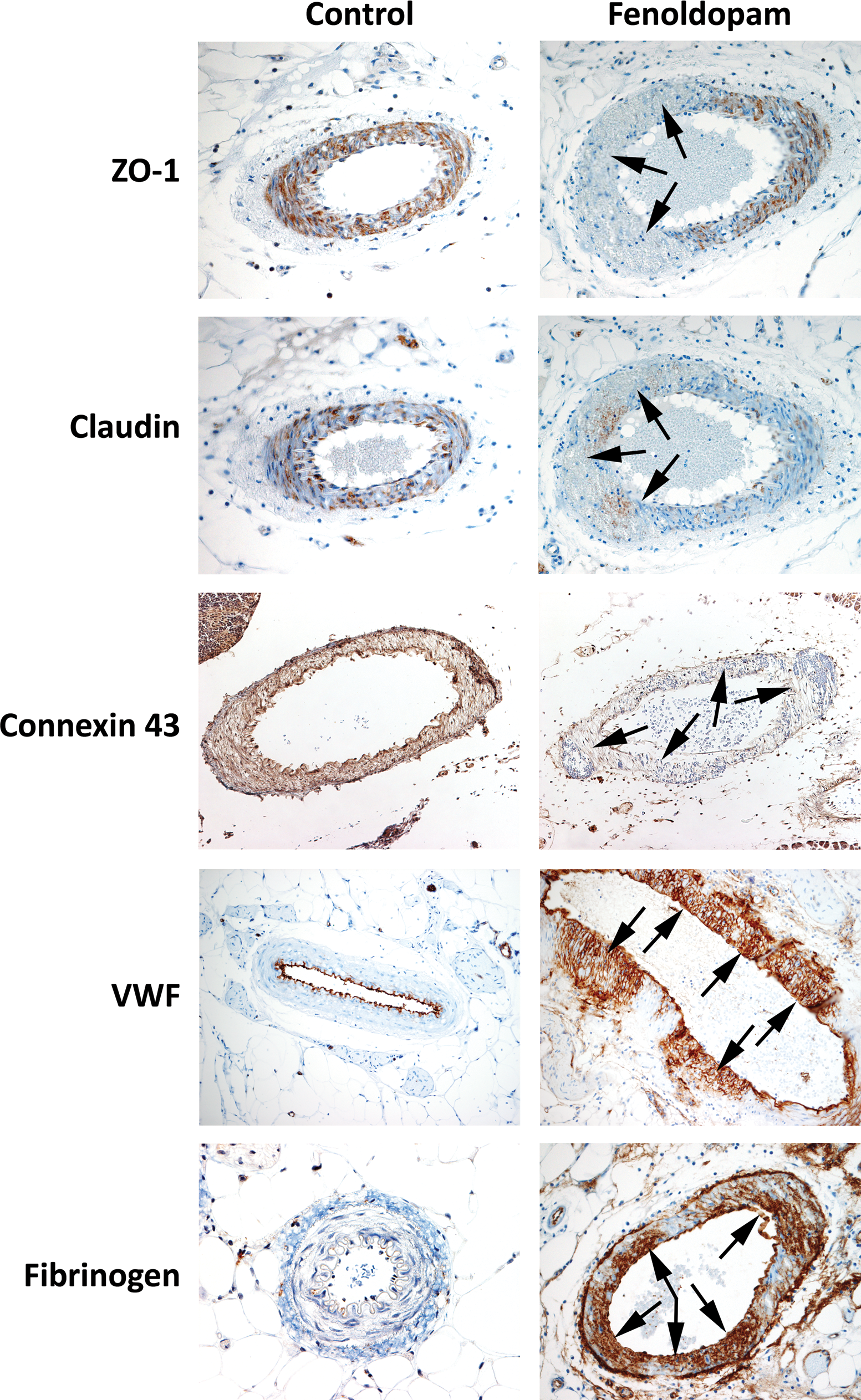
Immunohistochemcal staining for tight junctions (ZO-1 and claudin), gap junctions (connexin 43), and plasma proteins (von willibrand factor [vWF] and fibrinogen) in mesenteric arteries from control (0 µg/kg/min) and fenoldopam (100 µg/kg/min) treated rats. Fenoldopam-induced vascular injury was associated with gap and tight junction loss within the region of SMC necrosis and with extravasation of plasma proteins fibrinogen and vWF. Arrows indicate areas of drug-induced vascular injury (200×).
Because gap and tight junctions are vital structures in the maintenance of vascular wall integrity, breakdown and/or functional loss of these proteins would lead to changes in the spatial relationship of the phenotypical anchoring cells within the vascular wall. As a result, there would be extravasation of red blood cells, edema, and leakage of plasma proteins all of which are early morphologic features of fenoldopam-induced vascular injury. This was confirmed when increased immunoreactivity to the plasma proteins von Willebrand Factor (VWF and fibrinogen were observed within the damaged vascular wall of mesenteric arteries from fenoldopam treated rats (Figure 5). Taken together these data could suggest that the pathophysiological mode of action for fendoldopam-induced vascular injury may involve an interrelationship between caveolin-1 loss, sustained and prolonged NO mediated vasodilatation, functional loss of tight and gap junctions that results in subsequent breakdown in vascular wall integrity.
SNP, a Nitric Oxide Donor, Induces Fenoldopam-Like Mesenteric Arterial Injury
Because fenoldopam causes vasodilatation that is mediated by NO (Venkatakrishnan et al. 2000) and genetic functional loss of caveolin-1 is associated with sustained NOS activity and NO production (Drab et al. 2001; Zhao et al. 2002), in vivo studies were conducted to show that in the rat, continuous intravenous infusion of the NO donor SNP produced vascular lesions morphologically similar to those induced by fenoldopam. SNP induced damage in the mesenteric arteries when infused continually for 24 hr with 50 µg/kg/min (Table 2). The vascular lesions were morphologically indistinguishable from fenoldopam-induced lesions (Figure 1). Briefly, SNP-induced vascular damage was characterized by mild-to-moderate medial hemorrhage, medial necrosis, degeneration, and apoptosis of vascular SMC. Apoptosis was confirmed with positive immunostaining for activated caspase-3 within SMC at the site of vascular damage (data not shown). Dosages lower than 50 µg/kg/min of SNP were not associated with mesenteric vascular damage 24 hr after administration. Dosages of 100 and 200 µg/kg/min were associated with severe hypotension and prostration within 6 hr of infusion. These rats were terminated prematurely from the study and histopathology did not reveal any evidence of vascular lesions.
Incidence and severity of mesenteric arterial lesions induced by SNP.

SNP = Sodium nitroprusside. Groups of rats (3 males/group) were treated for 24 hr, necropsied at 24 hr, mesenteric arteries excised, formaldehyde fixed, processed, and hematoxylin and eosin (H&E) stained. The incidence and severity of vascular lesions were recorded by a pathologist, as previously described (Slim et al. 2003). Rats dosed with 100 or 200 µg/kg/min were euthanized within 6 hr of dose initiation due to adverse clinical observations.
Based on the data showing that SNP, a known vasodilatator and NO donor, induced mesenteric vascular injury, studies were initiated to determine if pre- and co-treatment of the NOS inhibitor L-NAME (50 µg/kg/min) with fenoldopam reduced the incidence and/or severity of mesenteric arterial injury. L-NAME reduced the incidence and severity of fenoldopam-induced mesenteric vascular injury (Table 3). Collectively, these data suggests that NO plays an important role in vasodilatatory drug-induced mesenteric arteriopathy in rats.
Incidence and severity of mesenteric arterial lesions induced by fenoldopam, fenoldopam in comination with L-NAME and fenoldopam in combinatuion with tempol.

Fnd = fenoldopam; L-NAME— a nitric oxide synthase inhibitor; and Tempol—a membrane permeable extracellular superoxide dismutase mimetic.
Groups (3 male/group) of rats were pretreated for 1.5 hr with Tempol or L-NAME alone; or in combination with fenoldopam as a continuous intravenous infusion for 24 hr and necropsied at the end of the infusion period. The incidence and severity of vascular lesions were recorded by a pathologist, as previously described (Slim et al. 2003).
Evidence for Peroxynitrite as a Mediator of Fenoldopam-Induced Vascular Injury
Increased vascular NO can cause excess production of NO free radicals that leads to vascular SMC apoptosis, tissue injury, and vascular damage. Because fenoldopam-induced vasodilatation and potential vascular damage could be mediated through NO-derived oxidizing free radicals, it was of interest to determine the presence and spatial relationship of NT residues as a footprint of peroxynitrite at the sites of vascular injury induced by fenoldopam. Peroxynitrite, a NO-derived free radical, is produced when NO binds to superoxide, an endogenously produced oxidant.
Very weak NT immunoreactivity was detected in mesenteric arteries from control rats (Figure 6). Whereas, focal, segmental, intense positive NT immunoreactivity was localized to the area around the sites of SMC damage in mesenteric arteries from fenoldopam or SNP-treated rats. Within damaged mesenteric arteries, positive NT immunoreactivity was seen around similar sites where there was evidence of caspase-3 activation, loss of caveolin-1, ZO-1, Claudin, and Connexin 43. Positive NT immunoreactivity was also seen at sites of SMC necrosis. Weak NT immunoreactivity was observed in the endothelial layer.
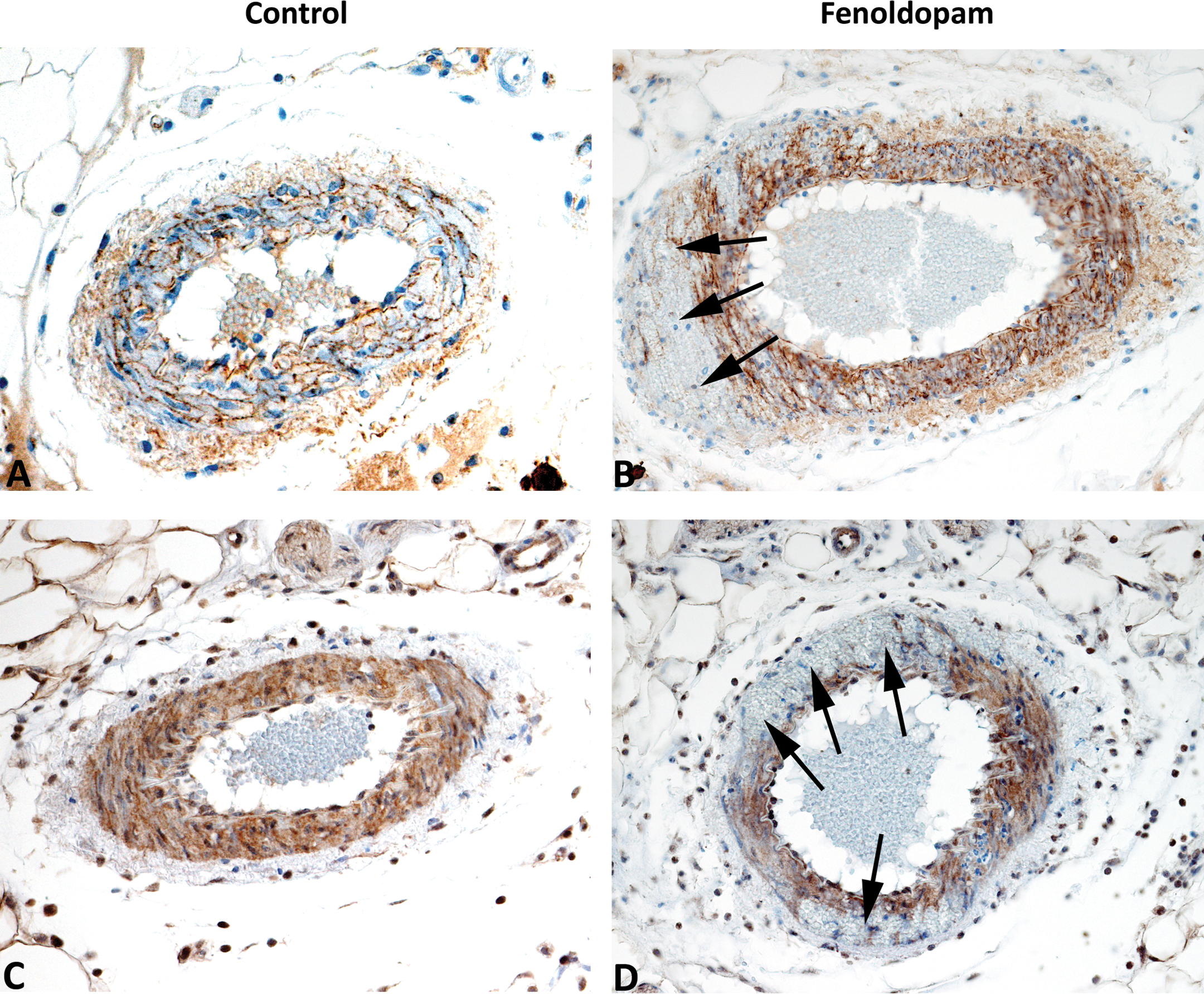
Immunohistochemcal staining for nitrotyrosine residues (A&B) and ecSOD (C&D) in mesenteric arteries from control (0 µg/kg/minute) and fenoldopam (100 µg/kg/minute) treated rats. Note the strong positive immunoreactivity for nitrotyrosine in mesenteric arteries from fenoldopam treated rats around the injury (B) with only a weakly positive immunoreactivity in the controls (A). Conversely, ecSOD activity is abundantly expressed in controls (C) and markedly reduced with fenoldopam treatment (D). Arrows indicate areas of drug-induced vascular injury. x200
Based on the strong positive NT immunoreactivity, it was of interest to determine ecSOD activity in mesenteric arteries damaged by fenoldopam treatment. Extracellular SOD activity limits the amount of NT formed because ecSOD inactivates superoxide thus reducing its availability to react with NO and form the cell destroying peroxynitrite free radicals. In normal uninjured mesenteric arteries, ecSOD is expressed abundantly within the SMC layer, in contrast to fenoldopam-damaged mesenteric arterial segments (Figure 6). Specifically, at the sites of SMC damage, there was loss of ecSOD immunoreactivity suggesting that this loss and/or reduced activity enabled NO to bind with superoxide, form peroxynitrite, and induce free radical damage in SMC, resulting in necrosis and apoptosis (Figure 6). Because the level of ecSOD could affect the development and/or severity of vascular damage, the cell permeable, ecSOD mimetic Tempol was evaluated to determine the effect of pre- and co-treatment with fenoldopam to induce vascular injury. When fenoldopam was infused for 24 hr at a rate of 100 µg/kg/min, Tempol had no effect on fenoldopam-induced vascular injury (Table 3). In a dose–response study, however, Tempol reduced the severity and incidence of vascular injury induced by a lower dose (1 µg/kg/min) of fenoldopam.
Discussion
Data from this series of experiments demonstrates that the NO pathway and its regulatory protein cav-1 may play an important causal role in the development of fenoldopam-induced vascular injury seen in nonclinical safety studies. Mesenteric arterial lesions were macroscopically and microscopically identical to those described in previous reports (Joseph et al. 1997; Joseph 2000; Kerns, Arena, Macia, et al. 1989; Kerns, Arena, and Morgan 1989; Yuhas et al. 1985). Our in vivo and in vitro data clearly shows that vascular injury mediated by prolonged vasodilatation induced by NO is associated with loss of cav-1 that precedes apoptosis and necrosis of EC and SMC. Furthermore, at these sites of SMC damage, breakdown in the structural integrity of the vascular wall, hemorrhage, and extravasations of plasma proteins such as vWF and fibrinogen were noted. Fenoldopam is a well-recognized vasodilatator in multiple species (Kerns, Arena, and Morgan 1989; Ventura et al. 1984) and there is strong evidence that binding of fenoldopam to specific dopaminergic receptor subtypes (DA1) leads to activation of the NO pathway and increased vascular NO that causes marked vasodilatation (Venkatakrishnan et al. 2000). Data from our series of experiments provide the in situ evidence that fenoldopam-induced vascular injury may be mediated in part by excess production of NO-derived oxidizing radicals that can cause apoptosis and necrosis. In support of our findings, data from other studies show that NO-derived oxidizing radicals can induce apoptosis and necrosis of vascular SMC (Lau 2003; Li et al. 2004). Additionally, this report also describes the spatial relationship between the signature footprint of peroxynitrite, apoptosis, and loss of cav-1 in EC and SMC of mesenteric arteries damaged by fenoldopam administration. A relatively novel finding in our studies is the in situ expression of NT residues in areas localized to loss of ecSOD and the ability of an exogenous ecSOD mimetic to confer partial protection from the vascular injury induced by fenoldopam. NT levels were not measured in our studies, but evaluation of this parameter in plasma could serve as a potential diagnostic as well as mechanistic biomarker of NO-mediated vascular injury (Radabaugh et al. 2008).
Extracellular superoxide dismutase (ecSOD) is the most prevalent antioxidant within the vascular wall and its activity reduces the availability of endogenous superoxide to react with NO thus preventing the formation of the toxic peroxynitrite (Fukai et al. 2002; Marklund 1984; Strälin et al. 1995). In our studies, we provide evidence showing that fenoldopam treatment resulted in loss of vascular ecSOD specifically at the sites of injury and these data suggest that the loss of ecSOD enabled NO to bind with superoxide and form peroxynitrite that induced free radical SMC damage and apoptosis. Therefore, the presence of peroxynitrite fingerprints and the inhibitory effects of an ecSOD mimetic suggest that vascular damage is mediated in part through NO-derived oxidizing free radicals.
Explanations for the loss of vascular ecSOD during drug-induced injury are complex and multifactorial. Biologically, the major isoform of SOD within the vessel wall is ecSOD and this potent antioxidant comprises one-third to one-half of the total vascular SOD activity, and in comparison to other tissues, ecSOD activity is ∼10-fold higher in the vessel wall (Fukai et al. 2002; Strälin et al. 1995). Based on immunohistochemical studies, ecSOD is expressed in high concentrations between the endothelium and smooth muscle cells (Oury et al. 1996). This enzyme is made predominantly by vascular SMCs but it binds to heparin sulfates on the EC surface where it undergoes internalization and so the protein can be detected by immunohistochemistry (IHC) (Ohata et al. 1994; Oury et al. 1996). Because of its location, it regulates the vascular redox state in the extracellular space and is also considered the principal regulator of endothelium-derived NO (Fukai et al. 2002; Oury et al. 1996). Therefore, ecSOD provides antioxidant activity to endothelial and vascular SMCs as well as the vascular extracellular matrix. Given this complexity in biology, location and biochemical regulation, loss and/or decreased expression of vascular ecSOD can be attributed in part to (1) exhaustion of the enzyme because of the high levels of peroxynitrite formed during NO-mediated DIVI, (2) decreased synthesis and production of ecSOD as a result of injury to SMC, (3) displacement of extracellular matrix-bound ecSOD, and/or (4) proteolytic degradation of ecSOSD by cellular and plasma proteins that are released during injury within the vascular wall.
As a result of tissue damage, necrosis, and subsequent vascular injury, there is release of oxygen-derived radicals that bind with NO to form nitrosating products including peroxynitrite (Fukai et al. 2002). Peroxynitrite is a potent oxidant and within the vascular wall ecSOD is the primary scavenger and antioxidant, so, it should not be surprising that expression of this enzyme is reduced at the site of vascular damage mediated by NO. Additionally, because SMCs are the primary site of production, injury, necrosis, and/or apoptosis could reduce the ability and/or number of viable SMCs to synthesize ecSOD. Therefore, this lack of production could contribute to reduced ecSOD expression at the sites of vascular injury.
A small portion of the extracellular matrix-bound ecSOD can be displaced, thus increasing circulating levels of this enzyme in plasma while decreasing vascular wall expression (Fukai et al. 2002; Karlsson and Marklund 1987; Oury et al. 1996). In our studies, it is possible that as a result of increased vascular oxidative stress and/or SMC damage and necrosis, there is a release of cellular and plasma proteins that cleave matrix-bound ecSOD releasing it into circulation and therefore reducing expression at the site of injury (Karlsson and Marklund 1987; Landmesser et al. 2002). Unfortunately, plasma ecSOD was not measured in our studies but evaluation of this protein in circulation could add significant value in determining if it is a suitable mechanistic biomarker of NO-mediated vasodilatatory injury. It is also possible that within the vascular wall proteolytic cleavage destroys ecSOD that ultimately reduces expression (Fukai et al. 2002). Because the enzyme would be destroyed in this situation, measurement of plasma ecSOD would not be beneficial.
It is worthy to note, that compared to other species, dog and rats have the lowest level of ecSOD, (Marklund 1984) an observation that may explain the unique susceptibility of these species to vascular injury seen in nonclinical safety studies when large doses of vasodilatators are administered. It is clear that the ecSOD mimetic did not prevent the vascular lesions induced by high-dose fenoldopam (100 µg/kg/min). This could be due to insufficient ecSOD reaching the tissues, or because fenoldopam can exert vasodilatation via poly pharmacologic action through non-NOS pathways (Bugelski et al. 1989; Joseph 2000; Yuhas et al. 1985).
Excessive vasodilatation, loss of vascular tone regulation, and changes in shear stress have been identified as the critical early events of vascular injury induced by vasodilators with a wide range of pharmacologic activity in rats, dogs, and monkeys (Albassam et al. 1999; Chelly et al. 1986; Dogterom et al. 1992; Gans et al. 1980; Greaves 1998; Joseph 2000; Kerns, Arena, Macia, et al. 1989; Kerns, Arena, and Morgan 1989; Kerns et al. 2005; Louden et al. 2000; Louden and Morgan 2001; Mesfin et al. 1989; Nyska et al. 1998). Inter-endothelial breaks, gradual breakdown of vessel wall integrity, breaks in the internal elastic lamina, and hemorrhage (Bugelski et al. 1989; Yuhas et al. 1985) are the morphologic manifestations and consequence of excessive vasodilatation that also includes markedly increased blood flow, decreased shear stress (EC), and increased hoop stress on the vessel wall (Chelly et al. 1986; Humphrey and Zins 1984; Kerns et al. 2005; Louden et al. 2000). Data from our studies show that there is a breakdown in the structural integrity of the vascular wall causing increased permeability and leakage of plasma proteins such as vWF and fibrinogen. In addition, we report that at these localized sites of SMC vascular damage, there was concomitant reduction in the expression pattern of caveolin-1, tight junctions (ZO-1 and claudin), and gap junction (connexin 43).
An alternative interpretation to the loss and/or reduced immunoreactivity is that within the injured vascular wall the antigenic epitopes of caveolin-1 and related structural proteins were destroyed by the presence of red blood cells, plasma proteins, and cellular enzymes released during tissue damage and inflammation. The presence of a large number of red blood cells within the vascular wall could potentially have an adverse effect on caveolin-1 immunoreactivity and this must be at least considered as a contributory factor. In order to address the effects of inflammation, multiple sections from the mesenteric vascular bed were evaluated histologically and immunohistochemically to characterize any adverse effects on caveolin-1 and gap junction protein immunoreactivity. In the perivascular space and surrounding tissues, inflammation and edema did not adversely affect caveolin-1 and gap junction protein immunoreactivity in SMC of adjacent arteries, arterioles, and veins with perivascular inflammation. Therefore, loss and/or reduced expression were consequences of the pathological process and not a secondary event or a bystander phenomenon related to inflammation. In support of this, we found that in vitro, caveolin-1 loss was an early (6 hr) dose-dependent event that preceded apoptosis of SMC when they were treated with a broad spectrum PDE inhibitor (IBMX) that belongs to a pharmacologic class of known in vivo vascular toxicants. This corroborates our previous finding in that, when SMC were treated with fenoldopam or minoxidil and caveolin-1 was evaluated using fluorescent microscopy, western blot analysis and flow cytometry, decreased expression was observed prior to any increases in annexinV that was used as marker of apoptosis (Brott, Jones, et al. 2005). However, in our current study, at the 24-hr time point, the low dose of this PDE inhibitor was associated with slight, increase (nonstatistically significant) in caveolin-1 and a statistically significant increase in activated caspase-3. A possible explanation for this paradox is that the increase in caveolin-1 is a physiological, compensatory, adaptive response to regulate NOS and reduce the magnitude of apoptosis and sustain cell viability. This explanation may be plausible because cell viability was not affected significantly at the low dose at 24 hr.
Because gap and tight junctions proteins as well as the NO pathway are regulated in part by caveolin-1 (Feron and Kelly 2001; Garcia-Cardena et al. 1997; Linder et al. 2005; Liu et al. 2002; Nusrat et al. 2000; Shaul and Anderson 1998) these data suggest that in EC and SMC, loss of caveolin-1 could be an obligatory step that initiates a series of biochemical events culminating in excessive vasodilatation, loss of vascular wall integrity, and cell death.
Collectively, the in vivo and in vitro data suggest that loss or reduced caveolin-1 expression occurs early, precedes apoptosis/necrosis, and could be the critical event in DIVI mediated by NO-induced excessive vasodilatation in sensitive arterial beds.
Functionally, caveolin-1 is a cell surface, shear stress mediating protein whose signaling activities include both vascular tone regulation and maintenance of vascular wall structural integrity through gap and tight junctions. Recently, it has been shown that through genetic disruption leading to functional deficiency of cavolin-1, there is impaired NO and calcium signaling, aberrations in endothelium-dependent relaxation, contractility, and maintenance of myogenic vascular tone (Drab et al. 2001). Additionally, vascular shear stress response, flow-mediated mechanotransduction, and vascular remodeling are reportedly cav-1 regulated (Frank and Lisanti 2006; Yu et al. 2006). In cav-1 KO mice, chronic changes in blood flow and shear stress caused vascular remodeling characterized by increased wall thickness and EC proliferation in rats (Drab et al. 2001; Zhao et al. 2002). Similar morphologic descriptions of vascular lesions are associated with long-term administration of low doses of vasodilatators such as theophylline, fenoldopam, and caffeine (Collins et al. 1988; Johansson 1981; Kerns, Arena, and Morgan 1989; Nyska et al. 1998).
Because functional loss of cav-1 is associated with increased NOS activity and vasodilatation via NO, in vivo studies were conducted to show that in the rat mesentery, continuous intravenous administration of SNP (NO donor) caused vascular lesions, morphologically and immunohistochemically similar to vascular lesions induced by fenoldopam. The SNP vascular lesions, however, were not as severe as those induced by fenoldopam and occurred at a lower incidence. In our studies, higher doses of SNP caused severe prostration, systemic hypotension, and cardiovascular collapse, while 50 µg/kg/min was associated with mesenteric vascular injury. In concordance with a previous report (Kerns, Arena, and Morgan 1989), our data confirm that when 20 µg/kg/min of SNP is infused, mesenteric vascular lesions do not develop. The reason/reasons for these observations are not clear but may be attributable to the level of vasodilatation in the rat mesenteric arteries induced by SNP at respective doses versus fenoldopam. This assumption is supported by data that report an additive effect when a non-vasculotoxic dose of SNP (20 µg/kg/min) is coadministered with a low dose of fenoldopam (Kerns, Arena, and Morgan 1989). This combination caused an increased incidence and severity of vascular lesions when compared to low, vasculotoxic doses of fenoldopam. Another possible reason for this difference between SNP and fenoldopam may be attributed to the synergistic poly pharmacologic action of fenoldopam via dopamine receptors, alpha-adreno receptors, NO, and other cellular mediators of vasodilatation. Our data support this because using two different approaches, inhibiting the NO pathway was only effective in attenuating, but not preventing, vascular lesions when coadministered with fenoldopam at low doses. These data suggest that the NOS pathway does contribute to the initiation and development of fenoldopam-induced vascular injury, but it may not be the sole mode of action. In rats, morphologically similar lesions involving the mesenteric arteries have been described following oral administration of PDE4s including the prototypic inhibitor rolipram (Larson et al. 1996; Slim et al. 2002). However, oral administration of IC542, a selective PDE4 inhibitor, was associated with a pro-inflammatory response in the rat mesentery with minimal vascular injury (Dietsch et al. 2006). Coadministration of IC542 with a potent anti-inflammatory agent dexamethasone completely blocked the inflammation in the rat mesentery (Dietsch et al. 2006). These results were in contrast to other published studies using a similar study design (Slim et al. 2002). The differences in response may be related to the selectivity of various molecules for members within the PDE family. Based on these data, one cannot exclude the possibility that inflammatory cytokines may be a contributing factor to the mode of action of mesenteric arterial injury induced by vasodilators.
In conclusion, the events of gap and tight junction loss, increased expression of NT residues, and early loss of the NOS regulated protein caveolin-1 suggest that the vasodilatator NO in part mediates fenoldopam-induced vascular injury. Moreover, infusion of a NO donor caused mesenteric vascular injury similar to fenoldopam and direct or indirect interference of NO or its toxic radicals attenuated this vascular injury. Collectively, these data suggest that the NO pathway may play a crucial role in vasodilatatory DIVI. The mechanistic data derived from our studies clearly suggest that caveolin-1, NT, and ecSOD activities are potentially mechanistically linked, candidate biomarkers of vasodilatatory induced vascular injury mediated through the NO pathway. Therefore, in nonclinical safety studies, measurement of these biomarkers in plasma could improve our overall assessment of DIVI and this could lead to the development of a clinically translatable marker.
Further in vivo and in vitro studies are necessary to determine if other well-recognized vascular toxicants of different pharmacologic classes induce injury to the phenotypically altered cells through cav-1 and the NO pathway. This will be of great interest because (1) the regulatory role of caveolin-1, (2) co-localization of caveolin-1 with various receptors and ion channels that increase cAMP levels as a consequence of pharmacology, and (3) exaggerated pharmacology of these receptors and ion channels is associated with vascular injury in rodents and non-rodents. (Kuhlmann et al. 2004; Linder et al. 2005; Martens et al. 2001; Nusrat et al. 2000; Shaul and Anderson 1998; Tirapelli et al. 2005; Yamaguchi et al. 2003; Yamamoto et al. 1999; Yu et al. 2004). Data from studies of this nature could also identify additional candidate biomarkers with clinically translatable application for monitoring DIVI.
Footnotes
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
The authors received no financial support for the research, authorship, and/or publication of this article.