
Abstract
In this study, the authors evaluated the ability of diphenyl diselenide (PhSe)2 to reverse acute hepatic failure induced by acetaminophen (APAP) in mice. The animals received an APAP dose of 600 mg/kg intraperitoneally (i.p.), and then 1 hour later, they received 15.6 mg/kg i.p. of (PhSe)2. Three hours after (PhSe)2 administration, the animals were sacrificed and blood and liver samples were collected for analysis. The serum alanine aminotransferase (ALT) and aspartate aminotransferase (AST) levels were measured. The levels of reduced glutathione (GSH) and oxidized glutathione (GSSG), thiobarbituric acid-reactive substances (TBARS), 2’,7’-dichlorofluorescein (DFC), catalase activity (CAT), and myeloperoxidase (MPO) activity were determined in the liver. A methyl-tetrazolium reduction (MTT) assay was also performed on the liver. Histopathological studies were conducted in all groups. Exposure of animals to APAP induced oxidative stress, increased lipid peroxidation (LPO), and the generation of reactive species, reduced the levels of GSH, and caused an increase in the MPO activity. Treatment with (PhSe)2 reduced LPO and the formation of reactive species and inhibited the processes of inflammation, reducing the hepatic damage induced by APAP. The results of this study show that (PhSe)2 is a promising therapeutic option for the treatment of acute hepatic failure.
Introduction
Acetaminophen (N-acetyl-p-aminophenol; APAP), a commonly used analgesic and antipyretic drug, can induce severe hepatotoxicity following accidental or intentional overdose in both experimental animals and humans (Kaufman et al. 2002). APAP overdose is the most frequent cause of drug-induced liver failure in the United States and Great Britain and the second leading cause of liver transplants, both of which are associated with considerable morbidity and mortality (Lee 2004; Ostapowicz et al. 2002). The characteristic dose-dependent hepatotoxicity of an APAP overdose results from the cytochrome P450-mediated oxidative metabolism of APAP, yielding the highly reactive metabolite N-acetyl-p-benzoquinone imine (NAPQI). At therapeutic doses, NAPQI is efficiently detoxified by glutathione (GSH) and eliminated in the urine or bile (Manyike et al. 2000; Nelson 1990). After an overdose or chronic, long-term use of APAP, the glucuronidation and sulfation routes become saturated, and more extensive bioactivation of APAP occurs, leading to the rapid depletion of the hepatic GSH pool. Subsequently, NAPQI binds to cellular proteins, including a number of mitochondrial proteins, which in turn causes oxidative stress that triggers cell death and tissue necrosis (Jaeschke, Knight, and Bajt 2003; James, Mayeux, and Hinson 2003). The cytotoxicity of APAP is also related to inflammation; the accumulation of neutrophils; and the release of proinflammatory and cytotoxic mediators, such as reactive oxygen and nitrogen intermediates and hydrolytic enzymes (Laskin and Laskin 2001; Luster et al. 2001).
Several previously published studies have shown that oxidative stress plays an important role in the development of hepatic failure from hepatotoxic doses of APAP, as evidenced by the elevated levels of reactive species and increased lipid peroxidation (LPO) (Bajt et al. 2004; Bessems and Vermeulen 2001; Schnellmann et al. 1999). Because of this role of oxidative stress, several substances with antioxidant properties have been tested as protectants against the hepatotoxicity of APAP (Ghosh and Sil 2007; Ghosh et al. 2010; Reisman et al. 2009).
The interest in organoselenium compounds has intensified because of their pharmacological potential—particularly their antioxidant potential—because oxidative stress has been implicated in the etiology of various diseases (Commandeur, Rooseboom, and Vermeulen 2001; Klotz and Sies 2003). Of these compounds, diphenyl diselenide [(PhSe)2] has been shown to have neuroprotective, hepatoprotective, and anti-inflammatory properties, in addition to being less toxic than other selenium compounds (Borges, Borges, et al. 2005; Nogueira, Quinhones, et al. 2003; Nogueira, Zeni, and Rocha 2004). (PhSe)2 has the ability to reduce oxidative damage induced by glycerol in the kidneys of rats, LPO induced by nitroprusside and quinolinic acid in brain homogenates, and acute hepatic damage induced by 2-nitropropane in rats (Borges, Borges, et al. 2005; Brandão et al. 2009; Rossato et al. 2002). The antioxidant capacity of (PhSe)2 is related in part to its glutathione peroxidase (GPx)-mimetic action. GPx enzymes are selenoenzymes that protect against oxidative stress by catalyzing the reduction of hydroperoxides; however, the mechanisms involved in this antioxidant action are not entirely clear (Nogueira, Zeni, and Rocha 2004; Ogunmoyole et al. 2009; Straliotto et al. 2010). The toxicity of (PhSe)2 depends on the route of administration and the species; the LD50 calculated for intraperitoneal (i.p.) administration in mice is 655 mg/kg (210 µmol/kg) (Nogueira, Meotti, et al. 2003). In contrast, high doses of (PhSe)2 inhibit δ-ALA-D activity in vitro in human blood (Nogueira, Borges, et al. 2003) and cerebral Na+, K+-ATPase activity (Borges, Rocha, and Nogueira 2005) by interacting with the SH groups of these enzymes (Brandão, de Oliveira, and Nogueira 2010). However, studies have shown that (PhSe)2 at a low concentration does not have a toxic effect (Rosa et al. 2007; Straliotto et al. 2010).
A previous study using (PhSe)2 (31 mg/kg of body weight given orally for 2 days) as a pretreatment for rats exposed to APAP demonstrated beneficial effects by preventing APAP-associated hepatotoxicity, reducing LPO and preventing a decrease in the ascorbic acid levels (Wilhelm et al. 2009).
In cases of APAP overdose, treatment primarily consists of the administration of N-acetylcysteine (NAC), a precursor of GSH. NAC reduces hepatic damage by increasing GSH levels and antagonizing the oxidative stress induced by NAPQI (Bajt et al. 2004; Kelly 1998; Mitchell et al. 1973; Terneus et al. 2007). However, for NAC to be effective, it must be administered quickly before the elevation of the alanine transaminase (ALT) level is seen (Latchoumycandane et al. 2007). Therefore, the search for alternative treatments that can be effective in patients intoxicated with APAP is necessary. In view of these facts, we evaluated the effects of (PhSe)2 on mice given an overdose of APAP.
Materials and Methods
Chemicals
Diphenyl diselenide (PhSe)2 was prepared as previously described Paulmier (1986) and was dissolved in canola oil. Analysis of the 1HNMR and 13CNMR spectra showed that (PhSe)2 presented analytical and spectroscopic data in full agreement with its assigned structure. The chemical purity of (PhSe)2 (99.9%) was determined by GC/HPLC. This drug was dissolved in canola oil, which was obtained from a standard commercial supplier. All other chemicals were of analytical grade and obtained from standard commercial suppliers.
Animals
Male adult Swiss albino mice (2 months old, 25–35 g) from our own breeding colony were used. The animals were kept in a separate animal room, on a 12 h light/dark cycle, at temperature of 22 ± 2°C, with free access to food and water. This study was approved by the Ethics and Animal Welfare Committee of Federal University of Santa Maria, Brazil.
Experimental Protocol
The animals were divided into four groups, with 6 mice each. Groups 1 (control) and 2 [(PhSe)2] received the vehicle of APAP (saline 0.9%, 20 ml/kg i.p.) and 1 h after they both received vehicle of (PhSe)2 (canola oil 2.5 ml/kg i.p.) or (PhSe)2 (15.6 mg/kg i.p.) (Rosa et al. 2007), respectively. Groups 3 (APAP) and 4 (APAP + (PhSe)2) received APAP (600 mg/kg i.p.) (Chan, Han, and Kan 2001) dissolved in warm buffered saline, and 1 h after they received vehicle or (PhSe)2, respectively. The intraperitoneal route of administration was used, because it provides a higher effect of (PhSe)2 than the oral route at the same time, being more efficient for this kind of treatment. All animals were sacrificed 3 hours after receiving (PhSe)2 or vehicle. The animals were anesthetized with equitesin (3 ml/kg i.p.), and blood collection was done by heart puncture. The anesthesia does not cause interference in the experimental procedure (hepatotoxicity). Serum was obtained by centrifugation at 2,000 × g for 10 min (hemolyzed plasma was discarded) and used for biochemical assays. After this procedure, animals were sacrificed and liver was removed, dissected, and kept on ice until the time of assay. The liver samples were homogenized in 10 mM Tris–HCl, pH 7.4 (1/10, w/v), centrifuged at 2000 × g for 10 min, and the low-speed supernatants (S1) were separated and used for experiments except for GSH and GSSG levels.
Measurement of Serum ALT and AST Level
Serum activity of alanine aminotransferase (ALT) and aspartate aminotransferase (AST) was determined photometrically using commercial kit (Labtest®, Diagnostica S.A., Minas Gerais, Brazil).
Methyl-Tetrazolium (MTT) Reduction Levels
MTT reduction levels were determined as an index of the dehydrogenase enzymes functions, which are involved in the cellular viability (Bernas and Dobrucki 2002). Aliquots of liver S1 (200 µL) were added to a medium containing 0.5 mg/mL of MTT and were incubated in the dark for 1 h at 37ºC. The MTT reduction reaction was quenched by the addition of 1 mL of dimethylsulfoxide (DMSO). The formed formazan levels were determined spectrophotometrically at 570 nm, and the results were corrected by the protein content (Mosmann 1983).
Thiobarbituric Acid–Reactive Substances Levels
Lipid peroxidation was determined by measuring thiobarbituric acid reactive substances (TBARS) as described by Ohkawa et al. (1979). An aliquot (200 µL) of S1 of liver was mixed with 500 µL thiobarbituric acid (0.6%), 200 µL sodium dodecyl sulphate (SDS) 8.1%, and 500 µl acetic acid (pH 3.4) and incubated at 90°C for 1 h. TBARS levels were measured at 532 nm using a standard curve of MDA (malondialdehyde), and the results were reported as nmol MDA/mg protein.
Measurement of Intracellular Reactive Oxygen Species Production
2’-7’-Dichlorofluorescein (DCF) levels were determined as an index of the reactive species production by the cellular components (Myhre et al. 2003). Aliquots (20 µl) of liver S1 was added to a medium containing Tris–HCl buffer (10 mM; pH 7.4) and 2’-7’-dichlorofluorescein diacetate DCFH-DA (1 mM). After DCFH-DA addition, the medium was incubated in the dark for 1 h until fluorescence measurement procedure (excitation at 488 nm and emission at 525 nm, and both slit widths used were at 1.5 nm). DCF levels were determined using a standard curve of DCF, and results were corrected by the protein content.
Catalase (CAT) Activity
The measurement of the CAT activity was determined according to the method proposed by Aebi (1984). An aliquot of the S1 (40 µl) was added to a medium containing phosphate buffer (50 mM; pH 7.4) and H2O2 (10 mM). The kinetic analysis of catalase was started after H2O2 addition and the rate of H2O2 decomposition was measured spectrophotometrically at 240 nm during 120 seconds. One unit of the enzyme was considered as the amount of enzyme that decomposes 1 µmol H2O2/min at pH 7.
Myeloperoxidase (MPO) Activity
The MPO enzyme activity was determined in liver S1 according to the method proposed by Grisham et al. (1986), with some modifications. Briefly, a sample of the liver S1 preparation (20 µL) was added to a medium containing potassium phosphate buffer (50 mM; pH 6.0), hexadecyltrimethylammonium bromide (0.5%), and N, N, N’, N’-tetramethylbenzidine (1.5 mM). The kinetic analysis of MPO was started after H2O2 (0.01%) addition, and the color reaction was measured at 655 nm at 37ºC.
Fluorimetric Assay of Reduced (GSH) and Oxidized (GSSG) Glutathione
For the measurement of GSH and GSSG levels, we used a method previously described by Hissin and Hilf (1976). Briefly, 250 mg of liver were homogenized in 3.75 mL phosphate–EDTA buffer (pH 8) plus 1 mL HPO3 (25%). Homogenates were centrifuged at 4ºC at 100,000 × g for 30 min, and the supernatants (S2) were separated in two different aliquots of 500 µL each for measurement of GSH and GSSG. For GSH measurement, 500 µL of the supernatant (S2) was diluted in 4.5 mL of phosphate–EDTA buffer (pH 8) (sodium phosphate 100 mM and EDTA 5 mM). The final assay mixture (2.0 mL) contained 100 µL of the diluted tissue supernatant, 1.800 µL of phosphate–EDTA buffer and 100 µL of O-Phthalaldehyde (OPA) (1 µg/µL). The mixtures were incubated at room temperature for 15 min and their fluorescent signals were recorded in the RF-5301 PC Shimadzu spectrofluorometer (Kyoto, Japan) at 420 nm of emission and 350 nm of excitation wavelengths. For measurement of GSSG levels, a 250 µL of the supernatant (S2) was incubated at room temperature with 100 µL of N-ethylmaleimide (NEM) (0.04 M) for 30 min at room temperature, and after that, 140 µL of the mixture, were added to 1,760 µL of NaOH (0.1 N) buffer, following of added 100 µL OPT and incubated for 15 min, using the procedure outlined above for GSH assay.
Histopathology
Liver tissues of mice (n = 6 per each group) were fixed in 10% formalin. For light microscopy examination, tissues were embedded in paraffin, sectioned at 4 µm and stained with hematoxylin and eosin to analysis qualitative of alterations histopathological.
Protein Estimation
Protein content was measured in fraction S1 by Bradford’s (1976) method using bovine serum albumin as standard.
Statistical Analysis
Data were analyzed by one-way ANOVA, followed by multiple comparison test of Newman-Keuls. Differences between groups were considered significant when p < .05.
Results
Biochemical Assays
The serum concentrations of both AST (A) and ALT (B), used as markers of liver damage, were significantly greater in the APAP group. The AST level was 3.66-fold higher and the ALT level was 14.34-fold higher when compared with the control group (p < .0001), providing evidence of APAP-induced hepatotoxicity (Figure 1). The animals treated with (PhSe)2 exhibited ALT and AST levels similar to those of the control group and significantly lower levels of both AST and ALT when compared with the APAP group (p < .0001) (Figure 1), indicating a reduction in tissue damage.
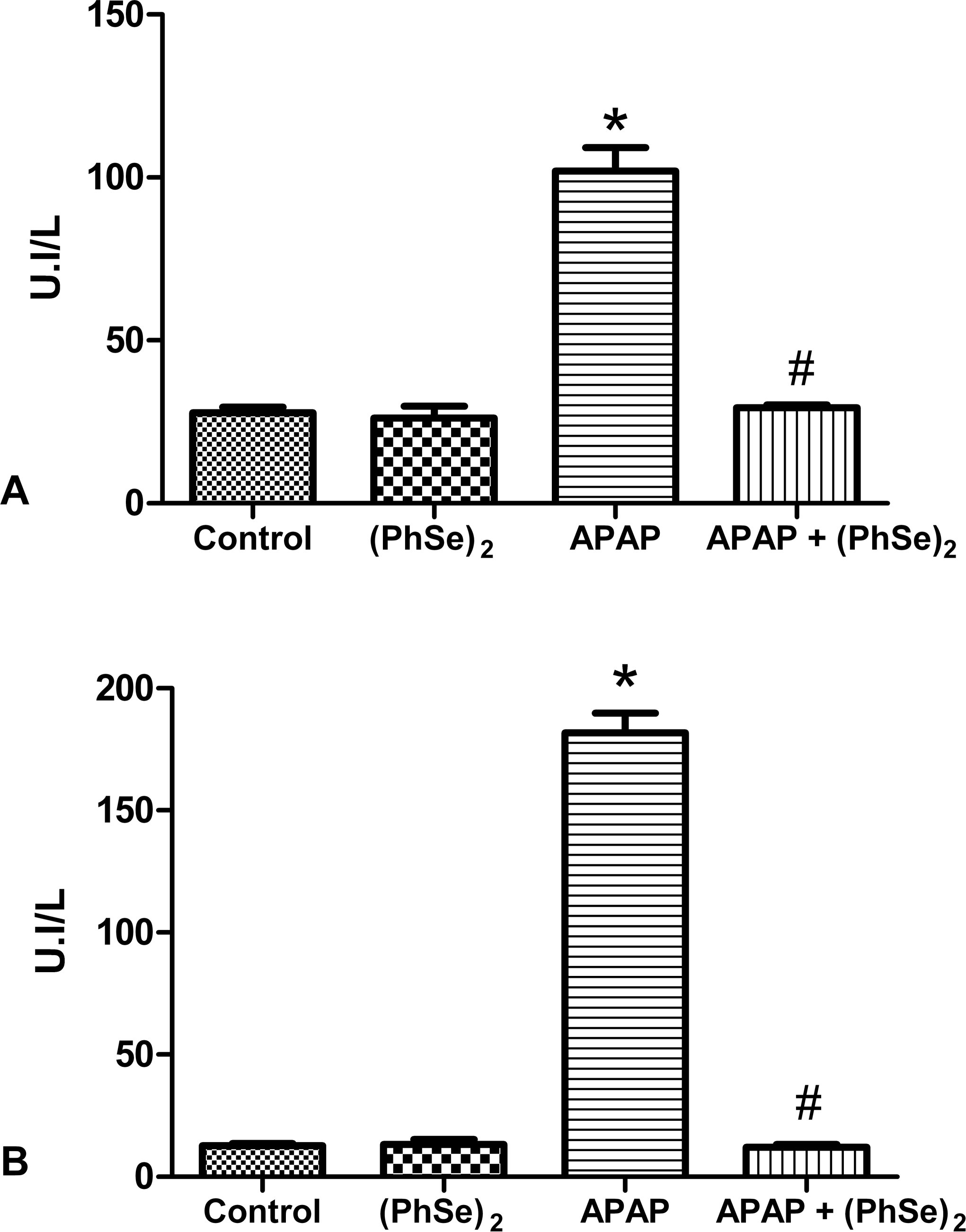
Effect of administration of APAP and treatment with (PhSe)2 in markers enzymatic of liver damage. Aspartate aminotransferase (A) and alanine aminotransferase (B). Data are reported as mean ± SD of 6 animals per group. Significance was assessed by one-way analysis of variance (ANOVA), followed by Newman-Keuls’s test for post hoc comparison. *Indicates p < .05 as compared to the control group. #Indicates p < .05 as compared to the APAP group.
Mitochondrial Dehydrogenases Activity Reduction Levels
The MTT reduction assay showed that APAP caused severe cellular injury, reducing cell viability by 50% compared to that of the control group (p < .0001) (Figure 2). Additionally, we observed that the treatment with (PhSe)2 minimized the toxic effects of APAP, with a significant increase in the cell viability when compared with the APAP group (p < .0001). There was no difference in cell viability between the (PhSe)2 group and the control group, demonstrating that the treatment was able to protect against the damage caused by APAP.
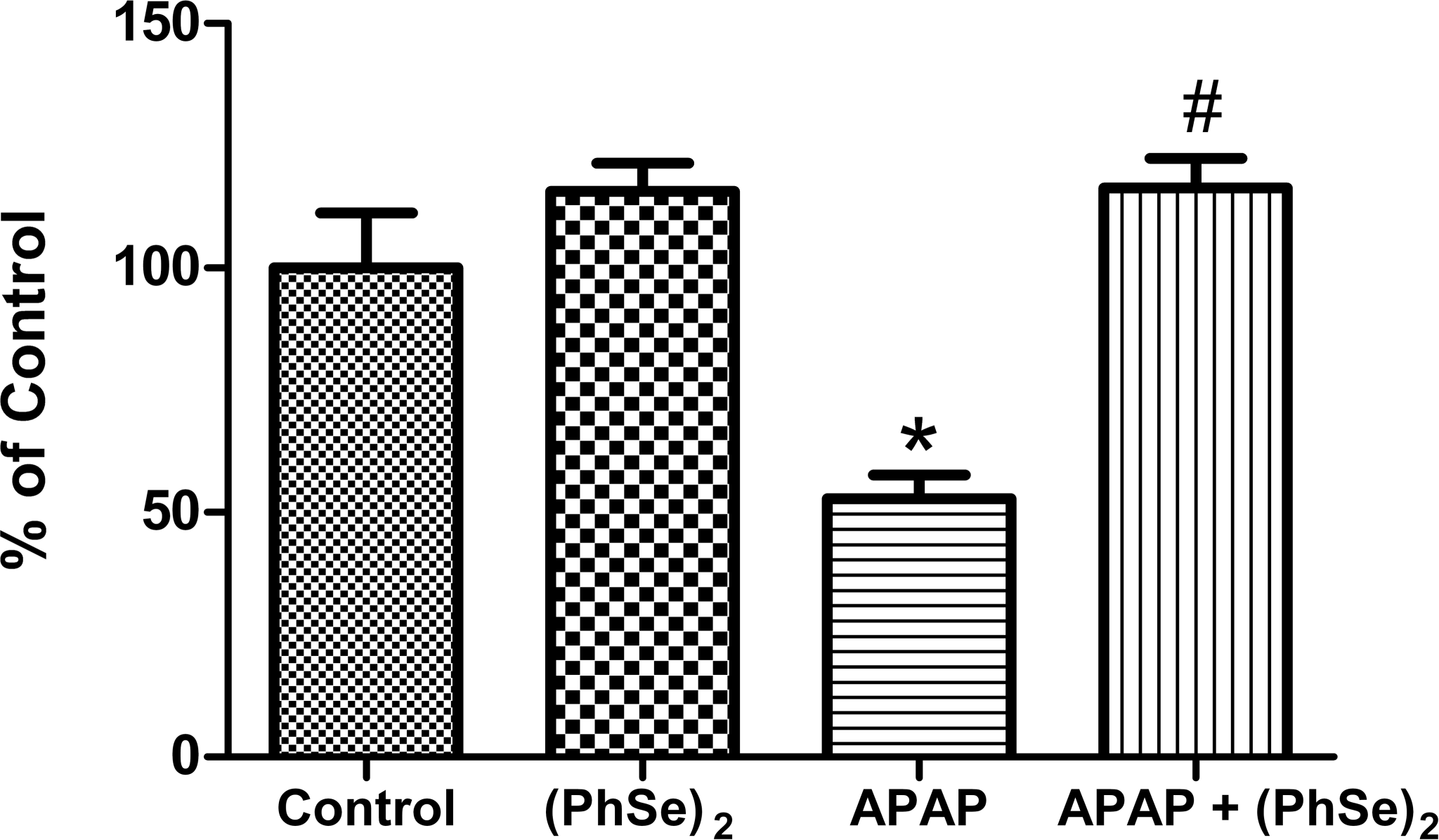
Effects of the administration of APAP and treatment with (PhSe)2 in the reduction of MTT. Data are reported as mean ± SD of 6 animals per group. Significance was assessed by one-way analysis of variance (ANOVA), followed by Newman-Keuls’s test for post hoc comparison. *Indicates p < .05 as compared to the control group. #Indicates p < .05 as compared to the APAP group.
Thiobarbituric Acid Reactive Substance Levels
The administration of a toxic dose of APAP caused a significant increase in LPO, as determined by the increase in TBARS level (3.8-fold higher than the control group) (p < .0001), indicating damage caused by oxidative stress (Figure 3). Treatment with (PhSe)2 significantly reduced the APAP-induced formation TBARS (p < .0001), keeping these levels at baseline and indicating a marked reduction in oxidative stress.
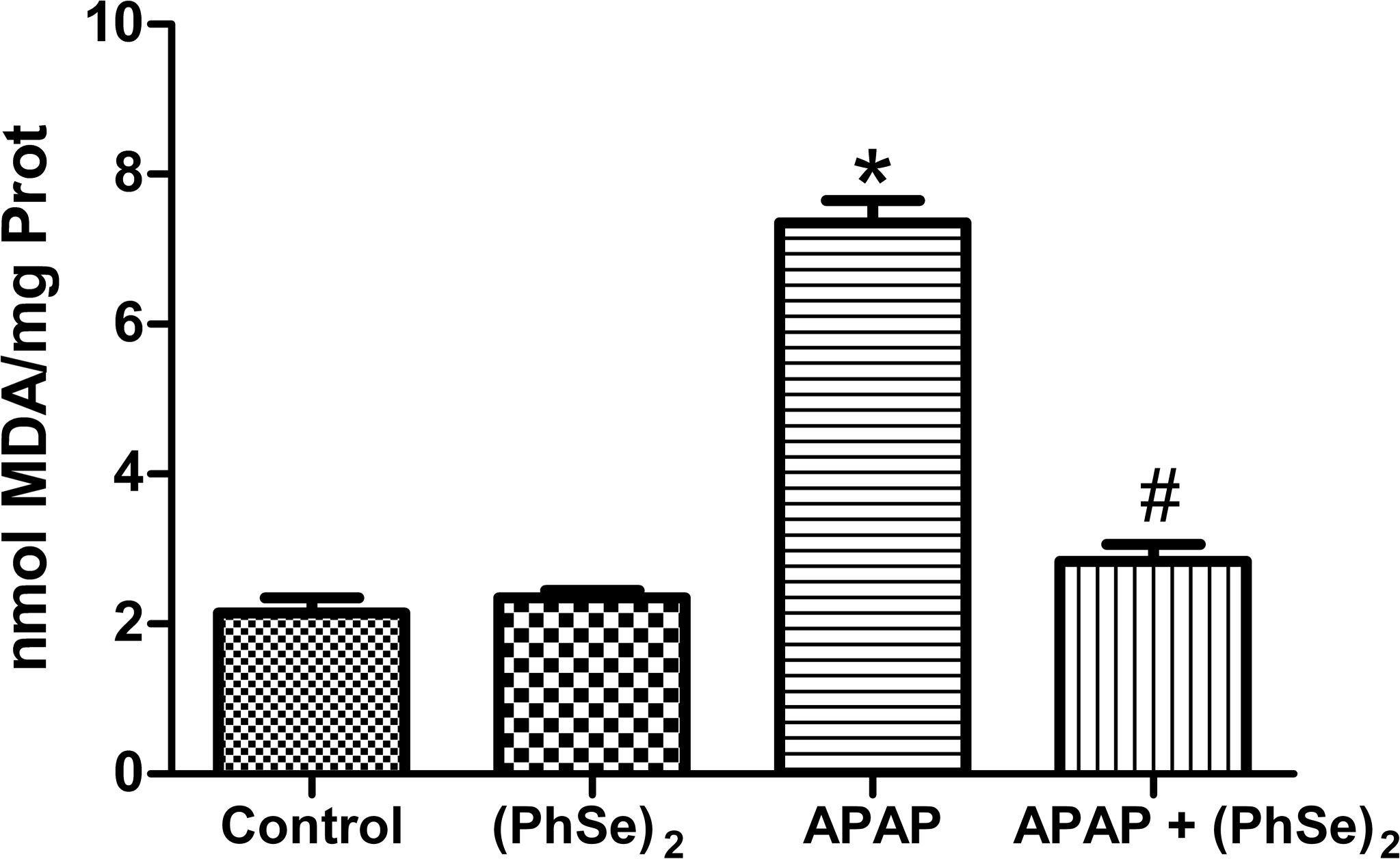
Effects of the administration of APAP and treatment with (PhSe)2 in the production of TBARS. Data are reported as mean ± SD of 6 animals per group. Significance was assessed by one-way analysis of variance (ANOVA), followed by Newman-Keuls’s test for post hoc comparison. *Indicates p < .05 as compared to the control group. #Indicates p < .05 as compared to the APAP group.
Analysis of Oxygen Reactive Species Production
Figure 4 shows that the intoxication with APAP caused an increase in the intracellular levels of reactive species, as evidenced by the increase in the production of DCF when compared to the control group (1.61-fold higher) (p < .0032). Treatment with (PhSe)2 effectively reduced the formation of intracellular reactive species when compared to the APAP group (p < .0032), keeping the formation of DFC at the basal level and reducing the damage caused by reactive oxygen species (ROS).
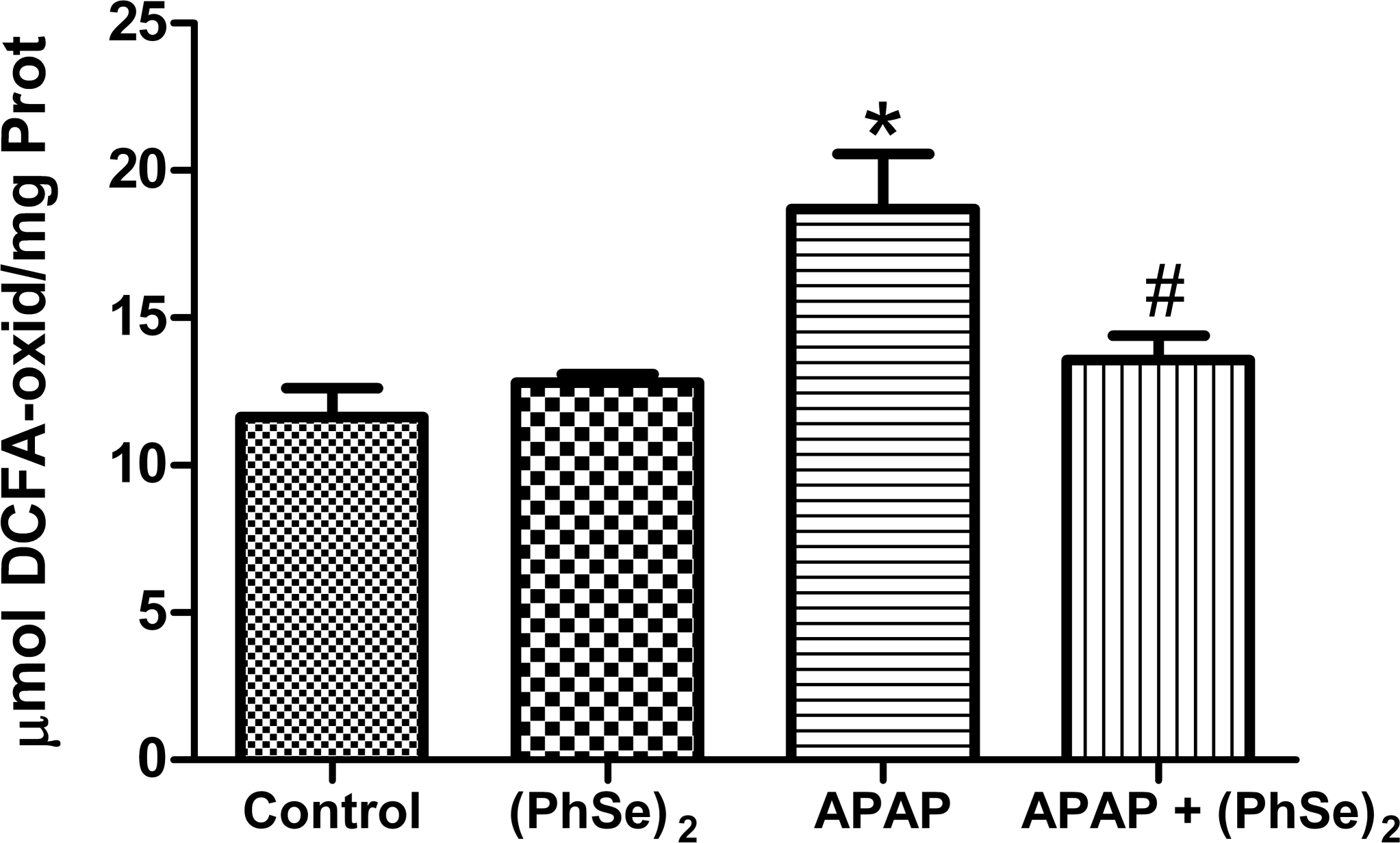
Effects of the administration of APAP and treatment with (PhSe)2 in the production of DCF-DA. Data are reported as mean ± SD of 6 animals per group. Significance was assessed by one-way analysis of variance (ANOVA), followed by Newman-Keuls’s test for post hoc comparison. *Indicates p < .05 as compared to the control group. #Indicates p < .05 as compared to the APAP group.
Myeloperoxidase Activity
Toxic doses of APAP initiated an inflammatory process, as evidenced by the marked increase in MPO activity when compared with the control group (18.7-fold higher) (p < .0001) (Figure 5). The group treated with (PhSe)2 showed a significant reduction in MPO activity when compared with the APAP group (p < .0001). No difference in MPO activity was found in treated group when compared with the control group (Figure 5), demonstrating a reduction in inflammatory processes.
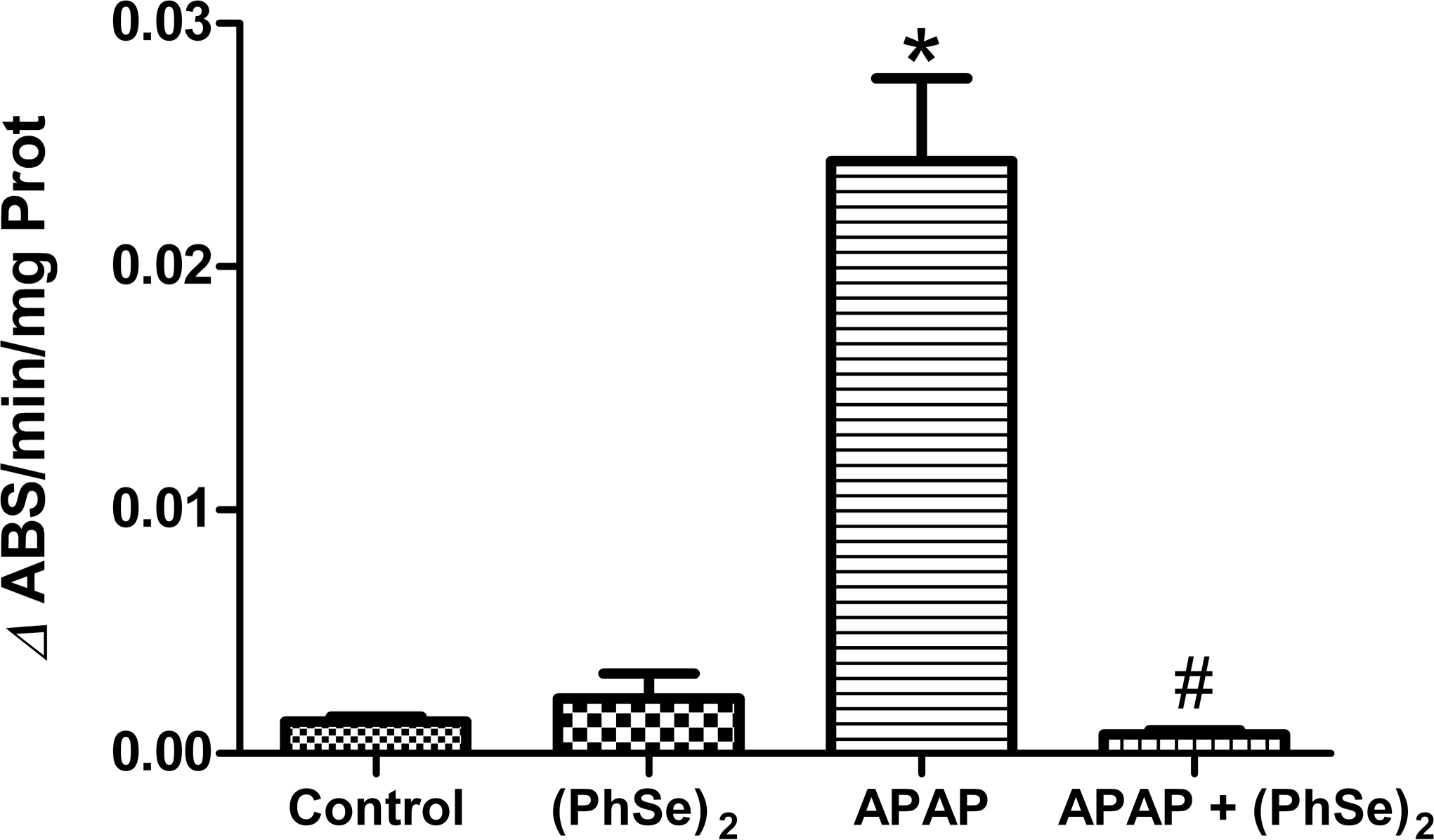
Effects of the administration of APAP and treatment with (PhSe)2 in the myeloperoxidase activity. Data are reported as mean ± SD of 6 animals per group. Significance was assessed by one-way analysis of variance (ANOVA), followed by Newman-Keuls’s test for post hoc comparison. *Indicates p < .05 as compared to the control group. #Indicates p < .05 as compared to the APAP group.
Catalase Activity
No significant difference in catalase activity was seen between treatment groups (data not shown).
Glutathione and Glutathione Disulfide Levels
An important factor associated with APAP-related toxicity is the decrease in GSH levels resulting from the generation of the APAP metabolite NAPQI. Figure 6 illustrates that the administration of APAP markedly decreased the GSH level relative to that in the control group (2.68-fold less) (p < .0001) and also decreased the GSSG levels (4.47-fold less than the control group) (p < .0005). Treatment with (PhSe)2 prevented the APAP-induced depletion of GSH observed in the APAP group (p < .0001) and maintained both the GSH and GSSG levels at baseline when compared with the control group, indicating an improvement in the antioxidant defense system. The GSH/GSSG ratio did not differ between groups.
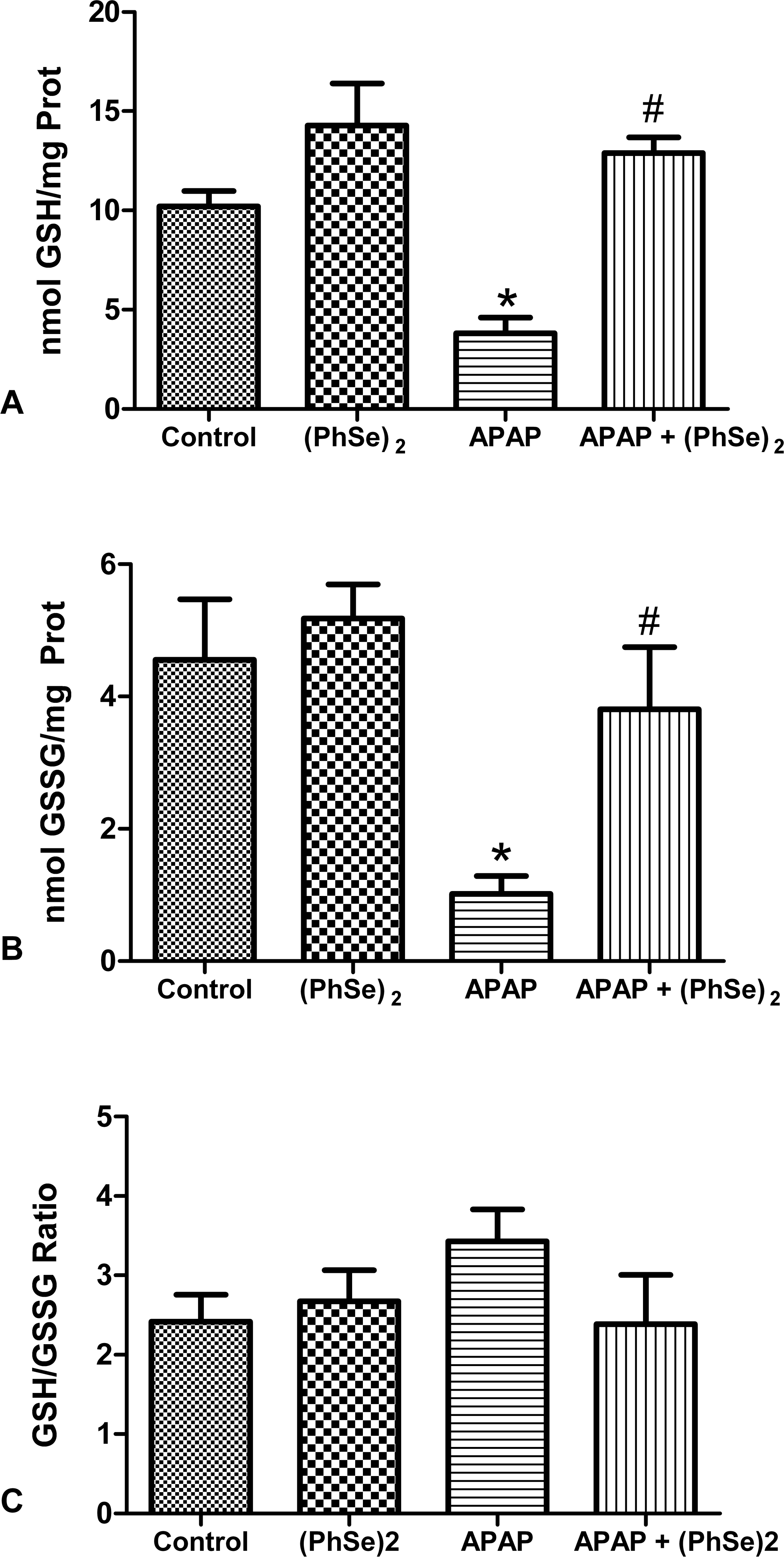
Effects of the administration of APAP and treatment with (PhSe)2 in the levels of GSH (A), GSSG (B), and ratio GSH/GSSG (C). Data are reported as mean ± SD of six animals per group. Significance was assessed by one-way analysis of variance (ANOVA), followed by Newman-Keuls’s test for post hoc comparison. *Indicates p < .05 as compared to the control group. #Indicates p < .05 as compared to the APAP group.
Histopathology
A histopathological assessment of the liver was performed for all groups. The livers of mice in the control (Figure 7A) and (PhSe)2 (Figure 7B) groups showed normal, well-defined histological structures with the absence of hepatocellular injury, fibrosis, and necrosis. The histopathological analysis of the livers from mice in the APAP group (Figure 7C) revealed signs of toxicity, with severe morphological changes and the presence of sinusoidal congestion (SC), vacuolar degeneration (VD) and nuclear pyknosis (NP), and the disorganization of the hepatic laminae. In contrast, the animals treated with (PhSe)2 (Figure 7D) exhibited a reduction in the APAP-induced changes, with less sinusoidal congestion, vacuolar degeneration, and nuclear pyknosis and the preservation of the hepatic laminae.
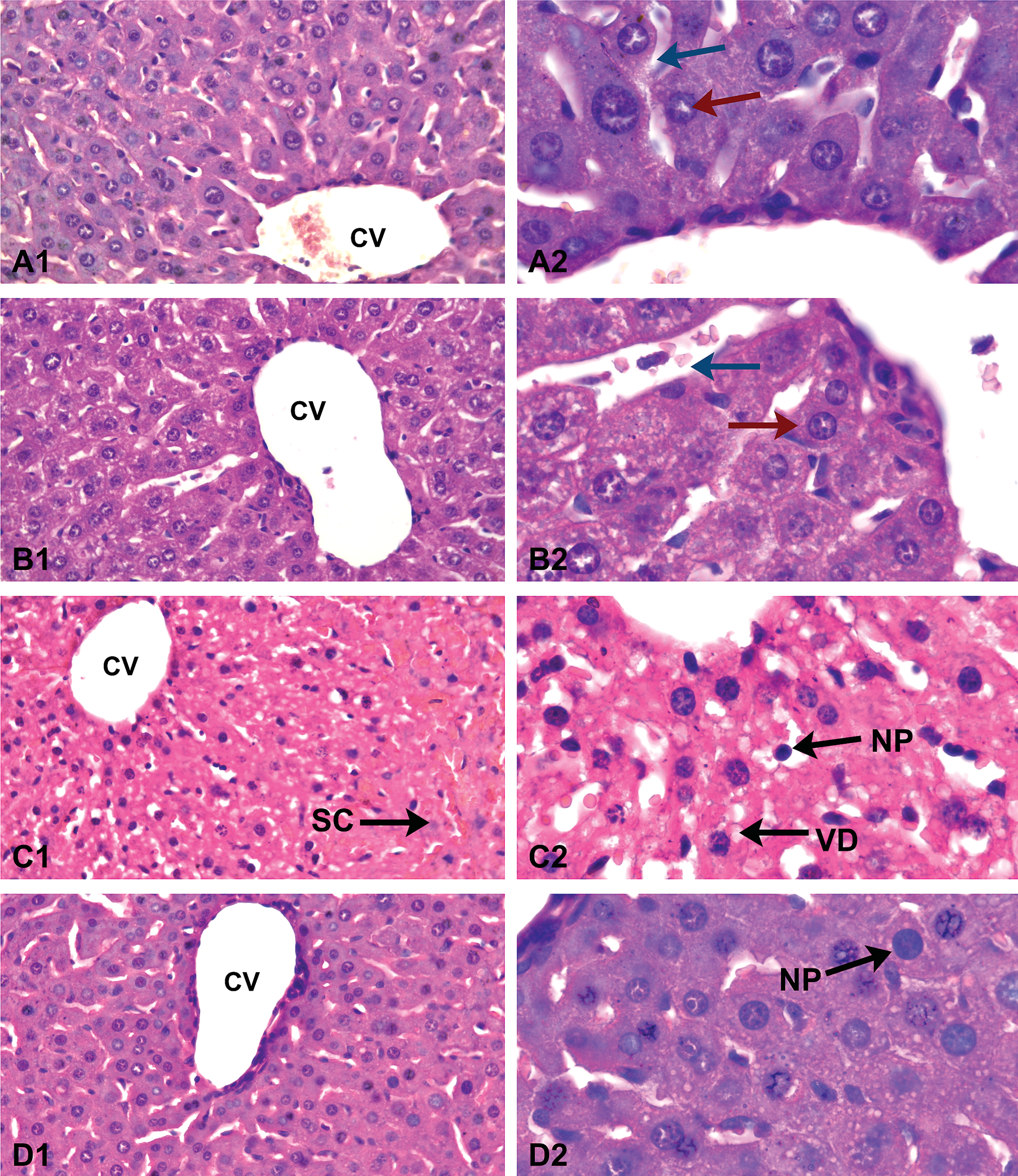
Liver from a control (A), with no changes and hepatocytes around the central vein (CV) arranged in cords separated by sinusoids. The sections from liver (PhSe)2 (B) showed normal histological appearance of the liver with hepatocytes adjacent to centrolobular vein (CV) with preserved architecture. APAP group (C) with histopathologic changes in liver showed nuclear pyknosis (NP), vacuolar degeneration (VD), sinusoidal congestion (SC), and disorganization of hepatic laminae. The sections from liver treated group (D) showed nuclear pyknosis (NP) and less vacuolar degeneration. Arrow red—hepatocytes, arrow blue—sinusoids, Figures A1–D1 (H&E stain 10×) and Figures A2–D2 (H&E stain 40×).
Discussion
In this study, we evaluated the ability of (PhSe)2 to protect against APAP-induced acute hepatic failure. Several studies have indicated that oxidative stress is an important factor in the development of APAP-associated hepatotoxicity (Bajt et al. 2004; Ghosh and Sil 2007), and GSH plays a key role in detoxification and the prevention of APAP toxicity in the liver (Nelson 1990; Richie, Lang, and Chen 1992). Both oxidative stress and LPO are early events associated with the generation of free radicals during the metabolism of APAP (Knight et al. 2001; Manov, Hirsh, and Iancu 2002). Moreover, toxicity from APAP can initiate an inflammatory process and increase tissue damage (Laskin and Laskin 2001; Luster et al. 2001). In our study, the administration of high doses of APAP resulted in the development of liver damage, oxidative stress, and inflammation, which were evidenced by increased LPO, increased levels of ROS, hepatic necrosis, and elevated levels of MPO activity. Treatment with (PhSe)2 in our study was able to reduce, in part or totally, the damage induced by an overdose of APAP.
To verify the damage caused by APAP administration, we analyzed the activity of serum aminotransferases. The increases in the serum ALT and AST activities were a result of cell damage or changes in membrane permeability, indicating that the liver damage was severe (Molander, Wroblewski, and Ladue 1955). In the group given APAP, we observed an increase in the serum AST and ALT levels. We also observed a reduction in hepatic cell viability based on the MTT assay, indicating severe tissue damage. The results obtained for the animals treated with (PhSe)2 show that (PhSe)2 was able to effectively decrease the severity of the APAP-induced tissue insult, as evidenced by the low levels of ALT and AST and the recovery of the cell viability, which indicated less hepatic damage.
(PhSe)2 is a compound with GPx-like activity and various other pharmacological properties (Borges et al. 2006; Nogueira, Quinhones, et al. 2003; Nogueira, Zeni, and Rocha 2004). (PhSe)2 can prevent LPO induced by various pro-oxidants (Borges et al. 2006; Brandão, de Oliveira, and Nogueira 2010; Rossato et al. 2002). In this study, animals intoxicated with APAP had high levels of TBARS, indicating increased LPO. (PhSe)2 was able to significantly reduce the LPO induced by APAP (based on the TBARS assay), which contributed to the maintenance of the integrity of cell membranes and tissue homeostasis. The estimate of ROS production using DCF showed that high doses of APAP promoted an increase in the generation of ROS. Treatment with (PhSe)2 resulted in a significant reduction in the generation of ROS and consequently reduced the damaging effects of ROS in mouse livers. These data reinforce the relationship between the toxicity of APAP and oxidative stress and also demonstrate the ability of (PhSe)2 to reduce the damage from oxidative stress induced by an overdose of APAP.
In addition to the well-characterized mechanism of direct APAP-induced hepatotoxicity, liver damage may also result from the disruption of the hepatic vasculature. APAP may generate perturbations in the endothelium, gap formation, and the coalescence of fenestrae. Subsequently, the sinusoid may collapse or disintegrate, reducing blood flow (Ito et al. 2003; McCuskey 2008; McCuskey et al. 2005). In our study, the histopathological analysis showed that an APAP overdose can cause severe damage in the liver. In the APAP group, we observed a disorganization of hepatic laminae with changes in the hepatic sinusoids and cords. The presence of sinusoidal, microvascular, portal, and centrilobular congestion (data not shown) is also indicative of a circulation backflow. Treatment with (PhSe)2 was effective in attenuating the damage caused by APAP in hepatic tissue, showing an improvement in the preservation of tissue structures with respect to the APAP group. An APAP overdose can also lead to an inflammatory response (James, Mayeux, and Hinson 2003). The enzyme MPO, which is present in neutrophils, is involved in the formation of ROS and in inflammatory processes. Myeloperoxidase can react with hydrogen peroxide and chloride anions to form free radicals and oxidizing agents (Lau and Baldus 2006; Podrez, Abu-Soud, and Hazen 2000). We found that animals that received an overdose of APAP showed a marked elevation in MPO activity, indicating the progression of an inflammatory process in liver tissue. The animals treated with (PhSe)2 showed a significant reduction in MPO activity, demonstrating reduced inflammation induced by APAP. These animals also exhibited reduced formation of reactive species, contributing to the decrease in tissue damage. Previous studies have suggested that the anti-inflammatory action of (PhSe)2 involves molecular mechanisms, such as the inhibition of protein kinase C (PKC); nitric oxide cell signaling; and inflammatory mediators, such as prostaglandins, bradykinin, and excitatory amino acids (Nogueira, Quinhones, et al. 2003; Savegnago et al. 2007). The findings of this study confirm that (PhSe)2 may be an effective anti-inflammatory agent for the treatment of acute hepatic failure induced by APAP.
The primary toxic effect of APAP is a result of its metabolite, NAPQI, which conjugates with GSH prior to excretion. The elevation of the level of NAPQI leads to depletion of hepatic GSH and to subsequent tissue damage (Jaeschke, Knight, and Bajt 2003; James, Mayeux, and Hinson 2003). GSH plays an important role in cellular redox homeostasis and is central to the antioxidant defense system (Circu and Aw 2010). In this work, APAP caused a marked reduction in the GSH level due to depletion by NAPQI, consequently reducing the GSSG level. The mechanisms involved in the antioxidant activity of (PhSe)2 are not entirely clear. Research has indicated that the antioxidant activity of (PhSe)2 is due in part to its GPx-mimetic action, which implies a concomitant consumption of GSH (Brandão, de Oliveira, and Nogueira 2010; Ogunmoyole et al. 2009). Our study showed that (PhSe)2 has a satisfactory antioxidant action and is able to reduce the amount of damage caused by APAP-induced oxidative stress; however, there was no difference in the level of GSH between the treated animals and those that received (PhSe)2 alone. Rosa and colleagues (2007) showed that low doses of (PhSe)2, as used in this study, did not alter the levels of GSH in liver and other tissues in mice. The effects obtained from treatment with (PhSe)2 could be result of a chemical reaction between (PhSe)2 and NAPQI, reducing the accumulation of this metabolite in liver tissue and consequently reducing the depletion of GSH levels and other toxic effects; these effects could also be due to an antioxidant action of (PhSe)2 (Li et al. 1994). Wilhelm et al. (2009) showed that pretreatment with (PhSe)2 was able to prevent APAP-induced hepatic damage, inducing a reduction in the level of oxidative damage associated with this process. In the present study, we evaluated the use of (PhSe)2 as a therapeutic treatment for intoxication with APAP. We observed that the administration of (PhSe)2 one hour after the animals had been given an overdose of APAP resulted in significant improvements in both biochemical and histopathological parameters. In addition to the reduction in the level of oxidative damage, we observed a restoration of the GSH level and a reduction in the level of inflammatory process.
Finally, we conclude that (PhSe)2 significantly reduced the acute hepatic failure induced by APAP, decreasing the damage caused by oxidative stress and inflammation and reducing the extent of the histopathological changes. These results suggest that this compound is a promising potential therapeutic agent for acute hepatic failure. However, more studies are needed to better understand the mechanisms by which (PhSe)2 reverses the effects of APAP.
Footnotes
This work was supported by the FINEP research grant “Rede Instituto Brasileiro de Neurociência (IBN-Net)” #01.06.0842-00 and INCT for Excitotoxicity and Neuroprotection—MCT/CNPq. M.H.S and N.J.R receive a fellowship by CAPES. J.C.B was recipient of PIBIC/CNPQ/UFSM fellowship. J.B.T.R and F.A.A.S. receive a fellowship by CNPq. S.C.P. is granted by the program “Contratación de Personal Investigador de Reciente Titulación Universitaria” from Consejería de Educación (Junta de Castilla y León, Spain) and European Social Fund (FEDER). CIBERehd is funded by Instituto de Salud Carlos III (ISCIII).