
Abstract
Acetaminophen (APAP) is the most commonly used analgesic and antipyretic drug in the world. However, hepatotoxicity caused by APAP overdose is the most frequent cause of acute liver failure worldwide and oxidative stress involved in the pathogenesis of APAP hepatotoxicity. Celastrol is a natural triterpenoid derived from Tripterygium wilfordii Hook F. that exhibits antioxidant, anti-inflammatory, and antitumor activities. In this study, we aimed to investigate the potential ameliorative effects of celastrol against APAP-induced cytotoxicity and oxidative stress. Human hepatocellular carcinoma cells (HepG2) were incubated with 20 mM of APAP for 24 h and posttreated with 50 nM, 100 nM, or 200 nM of celastrol for a further 24 h. The methylthiazolyldiphenyl-tetrazolium bromide, lactate dehydrogenase, and neutral red uptake assays showed celastrol posttreatments recovered cell viability and cell membrane integrity in a concentration-dependent manner. Celastrol posttreatments exerted a significant increase in the glutathione content and a decrease in the malondialdehyde and protein carbonylation levels. Also, celastrol posttreatments attenuated the APAP-induced oxidative stress by raising glutathione peroxidase, glutathione reductase, and catalase activities. However, superoxide dismutase activity did not change. In conclusion, celastrol treatment may improve cell viability and increase cellular antioxidant defense in HepG2 cells. These results suggest that celastrol may have the potential to ameliorate the APAP-induced oxidative stress and cytotoxicity.
Introduction
Acetaminophen (APAP) is a clinically safe and commonly used drug for its analgesic and antipyretic effects. However, overdose of APAP can lead to hepatotoxicity. 1 After ingestion at therapeutic doses, APAP is mainly metabolized by glucuronyl transferases and sulfotransferases in the liver which produce nontoxic water-soluble metabolites that are subsequently excreted in the urine. Approximately 10% of APAP is metabolized to its toxic metabolite N-acetyl-p-benzoquinone imine (NAPQI) by the liver enzyme CYP2E1. Normally, NAPQI is rapidly turned into a nontoxic form via covalent binding with glutathione (GSH). However, overdose consumption of APAP causes saturation in the glucuronidation and sulfation pathways. Excessive NAPQI formation results in GSH depletion and covalent binding to sulfhydryl groups of intracellular proteins. Increased NAPQI formation triggers cellular oxidative stress mechanism and causes reactive oxygen species (ROS) release in the hepatocytes, morphological and functional alterations in mitochondria, the mitochondrial deoxyribonucleic acid (DNA) damage, and interruption of adenosine triphosphate (ATP) production. The APAP-induced hepatocyte death results in severe liver damage and acute liver failure. 2 –4
The only clinically used antidote for APAP intoxication is N-acetyl cysteine (NAC). Early administration of NAC is essential in order to provide hepatoprotection. 5 Side effects such as nausea, vomiting, and anaphylactic reaction can be the limiting factors for NAC administration. 6,7 Thus, evaluation of novel alternative therapeutics is necessary for APAP-induced toxicity.
Celastrol is a quinone methide triterpene and is isolated from the root bark of the traditional Chinese medicinal plant, Tripterygium wilfordii Hook F. (thunder god vine) which belongs to Celastraceae family and grows in a wide area of south China. For hundreds of years, it has been traditionally used to treat autoimmune and inflammatory diseases and cancer. 8,9 The International Union of Pure and Applied Chemistry (IUPAC) name of celastrol is (2R,4aS,6aR,6aS,14aS,14bR)-10-hydroxy-2,4a,6a,6a,9,14a-hexamethyl-11-oxo-1,3,4,5,6,13,14,14b-octahydropicene-2-carboxylic acid and its chemical structure is provided in Figure 1. 10 Celastrol started receiving attention due to its potential therapeutic value evidenced from several research both in vitro and in vivo. Numerous studies have investigated the cellular and molecular targets of celastrol and the results from these studies showed that celastrol has antioxidant activity, reduces ROS generation, inhibits lipid peroxidation, suppresses nitric oxide (NO) production, and improves the cellular GSH cycle by increasing the intracellular GSH levels and the GSH/glutathione disulfide (GSSG) ratio. 8,11 –14

The chemical structure of celastrol.
In light of this evidence of celastrol’s antioxidant effects, we investigated the possible ameliorative effects of celastrol posttreatment on APAP-induced cytotoxicity and oxidative stress in HepG2 cells.
Materials and methods
Chemicals
Minimum essential media (MEM), fetal bovine serum (FBS), phosphate-buffered saline (PBS), penicillin–streptomycin solution, and trypsin-EDTA solution were purchased from Gibco Invitrogen Corp. (UK). Celastrol was of HPLC grade (purity ≥98%) and purchased from Sigma-Aldrich (USA). All other chemicals were of analytical grade and purchased from Sigma-Aldrich (MO, USA) unless otherwise stated.
Dose selection
For cell treatments, the appropriate concentrations of celastrol were determined on the basis of previously reported antioxidant properties of this compound in vitro and for this purpose, several doses of celastrol were tested with methylthiazolyldiphenyl-tetrazolium bromide (MTT) (data not shown) 15,16 in our pilot prestudies. The dose selection for APAP treatment was based on published studies with the same cell line. 17,18
Cell culture and treatments
HepG2 was purchased from the American Type Culture Collection (ATCC, VA, USA), and all the experiments were performed within 20 passages. The cells were grown with MEM containing 10% of FBS, 100 U of penicillin/mL, and 100 µg of streptomycin/mL in a humidified incubator supplied with 5% of CO2 at 37°C. The medium was changed once every 2 to 3 days. Prior to conducting the treatments, the cells were cultured for 24 h to ensure attachment.
Experimental procedure
For all of the assays used in this study, the cell treatment procedures were as follows. APAP and celastrol were dissolved in 100% of dimethyl sulfoxide (DMSO) and diluted with medium to the desired concentrations. Vehicle control cells received equal volume of DMSO (0.5%) as the treatments. The HepG2 cells were incubated with a medium containing 20 mM of APAP for 24 h to create APAP-induced cytotoxicity. After 24 h, the medium was renewed and the cells were posttreated with 50 nM, 100 nM, and 200 nM of celastrol for a further 24 h.
Cytotoxicity assays
MTT assay
To investigate the viability of HepG2 cells through the mitochondrial function, the MTT assay was used as previously described. 19 Briefly, the HepG2 cells were seeded in 96-well plates at 1 × 104 cells/well and grown for 24 h. After cell treatments as mentioned above, the medium was supplemented with 5 mg/mL of MTT in a 100 µL medium and the cells were incubated for 3 h at 37°C in the dark. After 3 h, the medium was removed and the wells were washed with PBS. Following the washing step, 100 µL of DMSO was added to dissolve the purple formazan crystals. The plate was gently shaken for 10 min to achieve complete dissolution and absorbance was measured at 570 nm using a microplate reader (Biotek, Epoc, VT, USA).
Neutral red uptake assay
The neutral red uptake (NRU) assay was performed following the protocol as described by Borenfreund and Puerner, 20 which is based on the measurement of the accumulation of neutral red dye in the lysosomes of viable cells. In brief, 1 × 104 cells/well were seeded in 96-well plates and grown for 24 h. Following cell treatments, the medium in each well was removed and replaced with a 150 μL medium containing 50 μg/mL of neutral red dye. In order for the live cells to uptake the dye, the cells were incubated for 3 h at 37°C. Following PBS wash, 150 µL of glacial acetic acid–ethanol–water (1:50:49) solution was added to the wells to solubilize the neutral red dye and the plate was incubated for a further 20 min with a gentle shake at 37°C. The optical density was read at 540 nm using a microplate reader.
For the cytotoxicity assays, the cell viability levels were expressed as a percentage of 0.5% DMSO-treated control cells.
Membrane integrity assay
Lactate dehydrogenase (LDH) is a cytosolic enzyme and the measurement of its leakage to the extracellular matrix due to cell membrane damage is an indicator of cell membrane integrity loss. For this purpose, the HepG2 cells were plated in 96-well plates at 1 × 104 cells/well and grown for 24 h. Following cell treatments, the LDH release in supernatants due to membrane damage was quantified using an LDH-cytotoxicity assay kit (RayBiotech, Inc. GA, USA) in accordance with the manufacturer’s protocol. DMSO of 0.5% was used as a negative control, while 1% Triton X-100 was used as a positive control. The optical density was measured at 495 nm using a microplate reader. The percentage of LDH release was determined based on the following formula:

Lipid peroxidation assay
Lipid peroxidation is an indicator of oxidative damage to lipids, and malondialdehyde (MDA) is the final product of the lipid peroxidation process. The assay is based on the amount of color change caused by the reaction of MDA with thiobarbituric acid (TBA). 21 For this purpose, the HepG2 cells were cultured in 25 cm2 flasks. Following cell treatments, 5 × 106 cells were harvested in 200 μL of PBS and sonicated for homogenization. The lipid peroxidation level was determined using QuantiChrom™ TBARS Assay kit (BioAssay Systems, CA, USA) according to the manufacturer’s protocol. Briefly, 100 μL of each sample was mixed with 200 μL of ice-cold TCA (10%) and incubated for 5 min on ice. This was followed by centrifugation at 14,000 rpm/min for 5 min. Then 200 μL of supernatants were collected in new tubes and were mixed with 200 μL of TBA, and the mixture was incubated at 100°C for 60 min. After cooling the mixture to room temperature, 100 μL of each sample was transferred in duplicate to 96-well plates and the absorbance was measured at 535 nm using a microplate reader. Results were expressed as fold of control.
Protein carbonylation
The measurement of protein carbonylation level can be used as an oxidative stress biomarker. 22 For this purpose, the HepG2 cells were cultured in 25 cm2 flasks. Following cell treatments, 1 × 106 cells were harvested in 1 mL of PBS and homogenized by sonication. The carbonylated protein level was determined using the protein carbonyl enzyme-linked immunosorbent assay (ELISA) kit (Bioassay Technology Laboratory, China) following the manufacturer’s instructions. The optical density was read at 450 nm using a microplate reader.
GSH level
GSH plays an important role in the cellular antioxidant system and the GSH level decreases under oxidative stress conditions. 23 Therefore, to determine the changes in the GSH level, the HepG2 cells were cultured in 25 cm2 flasks. Following cell treatments, 1 × 106 cells were harvested in 1 mL of PBS and were homogenized by sonication, and the GSH content of HepG2 cells was determined using the ELISA kit (Bioassay Technology Laboratory, China) to assay human GSH following the manufacturer’s procedure. The optical density was read at 450 nm using a microplate reader.
Antioxidant enzyme activity assays
To assay the activity of antioxidant enzymes, the HepG2 cells were cultured in 75 cm2 flasks. Following cell treatments, the cells were suspended in ice-cold PBS and homogenized by sonication. After centrifugation (15,000g for 10 min at 4°C), the obtained supernatant was used for antioxidant enzyme activities.
Glutathione peroxidase (GPx) is a protective enzyme against oxidative stress via controlling the formation of free radicals. The GPx activity was evaluated by measuring the nicotinamide adenine dinucleotide phosphate (NADPH) consumption spectrophotometrically at 340 nm for 4 min, which is proportional to the GPx activity in the sample. 24 For this purpose, the EnzyChrom™ Glutathione Peroxidase Assay kit (BioAssay systems, USA) was used according to the manufacturer’s protocols.
Glutathione reductase (GR) has an important role to maintain the cellular GSH pool against oxidative stress. The GR activity assay is based on the measurement of absorbance change caused by the reduction in DTNB [5,5′-dithiobis(2-nitrobenzoic acid)] at 412 nm. The rate of optical density change is directly proportional to the GR activity in the sample. 25 In this study, to evaluate the GR activity, the EnzyChrom™ Glutathione Reductase Assay kit (BioAssay systems, USA) was used according to the manufacturer’s protocols.
The antioxidant enzyme superoxide dismutase (SOD) catalyzes the dismutation of superoxide radicals to protect the cellular functions against free radicals. The SOD activity assay is based on the spectrometric measurement of the reduction in tetrazolium salt, WST-1 (2-(4-iodophenyl)-3-(4-nitrophenyl)-5-(2,4-disulfophenyl)-2H-tetrazolium, monosodium salt), by the superoxide anion at 440 nm. SOD scavenges the superoxide anion, thus the SOD activity can be quantified by measuring the decrease in the color development. 26 In this study, the rate of SOD activity was determined using the commercially available 19160-SOD determination kit (Sigma-Aldrich, USA), according to the manufacturer’s procedure.
Catalase is an important peroxisomal enzyme that degrades hydrogen peroxide (H2O2) by catalyzing its decomposition into water and oxygen. Catalase activity was determined by measuring the decomposition rate of substrate H2O2 spectrophotometrically for 5 min, which is proportional to the catalase activity in the sample. 27 For this purpose, the CAT100 Catalase assay kit (Sigma-Aldrich, USA) was used, according to the manufacturer’s protocol. Absorbance was measured at 520 nm using the Shimadzu UV 1800 spectrophotometer (Shimadzu, Japan).
Statistical analysis
All the experiments were performed as three replicates and the results were presented as the mean ± standard deviation. The statistical comparisons were evaluated using the one-way analysis of variance followed by the Tukey’s test for post hoc analysis, and the statistical significance was set at p < 0.05 (SPSS, version 21.0, USA).
Results
Evaluation of cytotoxicity and potential ameliorative effects of celastrol against APAP-induced toxicity in HepG2 cells
The APAP-induced cytotoxicity and the effects of celastrol in HepG2 cells were evaluated with the MTT and NRU assays, and the results demonstrated that APAP effected the cell viability by 54.07 ± 2.76% and 53.10 ± 2.81%, respectively (p < 0.05). The MTT assay results showed a significant recovery in cell viability with celastrol posttreatments to 67.49 ± 5.59% with 100 nM and 71.34 ± 2.74% with 200 nM celastrol concentrations compared to only APAP-treated cells (p < 0.05). However, the NRU assay results showed an insignificant recovery with celastrol posttreatments compared to only APAP-treated cells (p > 0.05) (Figure 2(a)).

Effects of APAP treatment and celastrol posttreatments on HepG2 cells viability (a) and cell membrane integrity (b). HepG2 cells were treated with 20 mM APAP for 24 h, after 24 h the medium was renewed and cells were posttreated with celastrol (50 nM, 100 nM, and 200 nM) for 24 h. Bar graphs were generated with Microsoft Excel 2016 (USA) and data are shown as the means ± SD (n:3); *p < 0.05 versus control cells; # p < 0.05 versus only APAP-treated cells. APAP: acetaminophen; MTT: 3-(4,5-dimethylthiazole-2-yl)-2,5-diphenyl tetrazolium bromide; LDH: lactate dehydrogenase; NRU: neutral red uptake; SD: standard deviation.
Effects of APAP and celastrol on membrane integrity in HepG2 cells
The LDH assay results showed that APAP treatment caused a significant increase in damage to the cell membrane integrity, and the LDH release increased from 21.73 ± 5.94% to 46.86 ± 2.81%. Celastrol posttreatments showed a significant decrease in damage to the cell membrane integrity compared to only APAP-treated cells in a dose-dependent manner. The LDH release decreased to 43.73 ± 4.23%, 43.66 ± 5.58%, and 27.80 ± 4.87%, and this decrease was significant with 200 nM celastrol concentration (Figure 2(b)).
Effects of APAP and celastrol on protein carbonylation, GSH levels, and lipid peroxidation in HepG2 cells
The amount of carbonyl formation in protein significantly increased from 194.78 ± 11.54 ng/mg protein to 321.00 ± 21.5 ng/mg protein with the APAP treatment and celastrol posttreatments caused a significant decrease in protein carbonylation with 100 nM and 200 nM concentrations to 246.53 ± 0.85 ng/mg protein and 220.64 ± 9.52 ng/mg protein, respectively (Figure 3(a)). The GSH level significantly decreased from 26.74 ± 1.22 ng/mg protein to 15.62 ± 0.33 ng/mg protein with 20 mM APAP treatment. As demonstrated in Figure 3(b), 50 nM, 100 nM, and 200 nM celastrol posttreatments attenuated the GSH decrease to 15.70 ± 0.57 ng/mg protein, 22.61 ± 0.45 ng/mg protein, and 23.05 ± 1.73 ng/mg protein, respectively, compared to only APAP-treated cells, and this attenuation was statistically significant at 100 nM and 200 nM concentrations (p < 0.05). The incubation of HepG2 cells with 20 mM of APAP caused a significant increase (4.95 fold) in lipid peroxidation, while 50 nM, 100 nM, and 200 nM of celastrol posttreatments decreased the lipid peroxidation significantly to 3.12-fold, 3.62-fold, and 2.36-fold, respectively, compared to only APAP-treated cells (Figure 3(c)).

Effects of APAP treatment and celastrol posttreatments on protein carbonylation (a), glutathione level (b), and lipid peroxidation (c) in HepG2 cells. HepG2 cells were treated with 20 mM APAP for 24 h, after 24 h the medium was renewed and cells were posttreated with celastrol (50 nM, 100 nM, and 200 nM) for 24 h. Control cultures received 0.5% DMSO. Bar graphs were generated with Microsoft Excel 2016 (USA) and data are shown as the means ± SD (n:3); * p < 0.05 versus control cells; # p < 0.05 versus only APAP-treated cells. APAP: acetaminophen; GSH: glutathione; MDA: malondialdehyde.
Effects of APAP and celastrol on antioxidant enzyme activities in HepG2 cells
The GR activity decreased significantly from 20.88 ± 0.73 U/mg protein to 10.85 ± 0.49 U/mg protein with 20 mM APAP treatment. After celastrol posttreatments, the GR activity increased to 13.93 ± 0.60 U/mg protein, 19.80 ± 0.56 U/mg protein, and 17.53 ± 0.13 U/mg protein with 50 nM, 100 nM, and 200 nM of celastrol concentrations, respectively, and this increase was significant with 100 nM and 200 nM compared to only APAP-treated cells (p < 0.05) (Figure 4(a)). The GPx activity decreased significantly with 20 mM APAP treatment from 138.41 ± 12.45 U/mg protein to 29.02 ± 7.91 U/mg protein. After celastrol posttreatments, the GPx activity increased to 58.56 ± 10.93 U/mg protein, 125.43 ± 16.08 U/mg protein, and 120.17 U/mg protein with 50 nM, 100 nM, and 200 nM of celastrol concentrations, respectively, and this increase was significant with 100 nM and 200 nM compared to only APAP-treated cells (p < 0.05) (Figure 4(b)). The SOD activity is presented in Figure 4(c) and the results did not show a significant inhibition with 20 mM APAP treatment, and celastrol posttreatments did not cause any change in the SOD inhibition rate (p > 0.05). Following 20 mM APAP treatment, the catalase activity reduced from 24.90 ± 1.57 µmol/min/mg protein to 9.88 ± 0.76 µmol/min/mg protein. After celastrol posttreatments, the catalase activity increased significantly to 24.34 ± 5.53 µmol/min/mg protein and 23.09 ± 2.00 µmol/min/mg protein with 100 nM and 200 nM concentrations, respectively (p < 0.05) (Figure 4(d)).
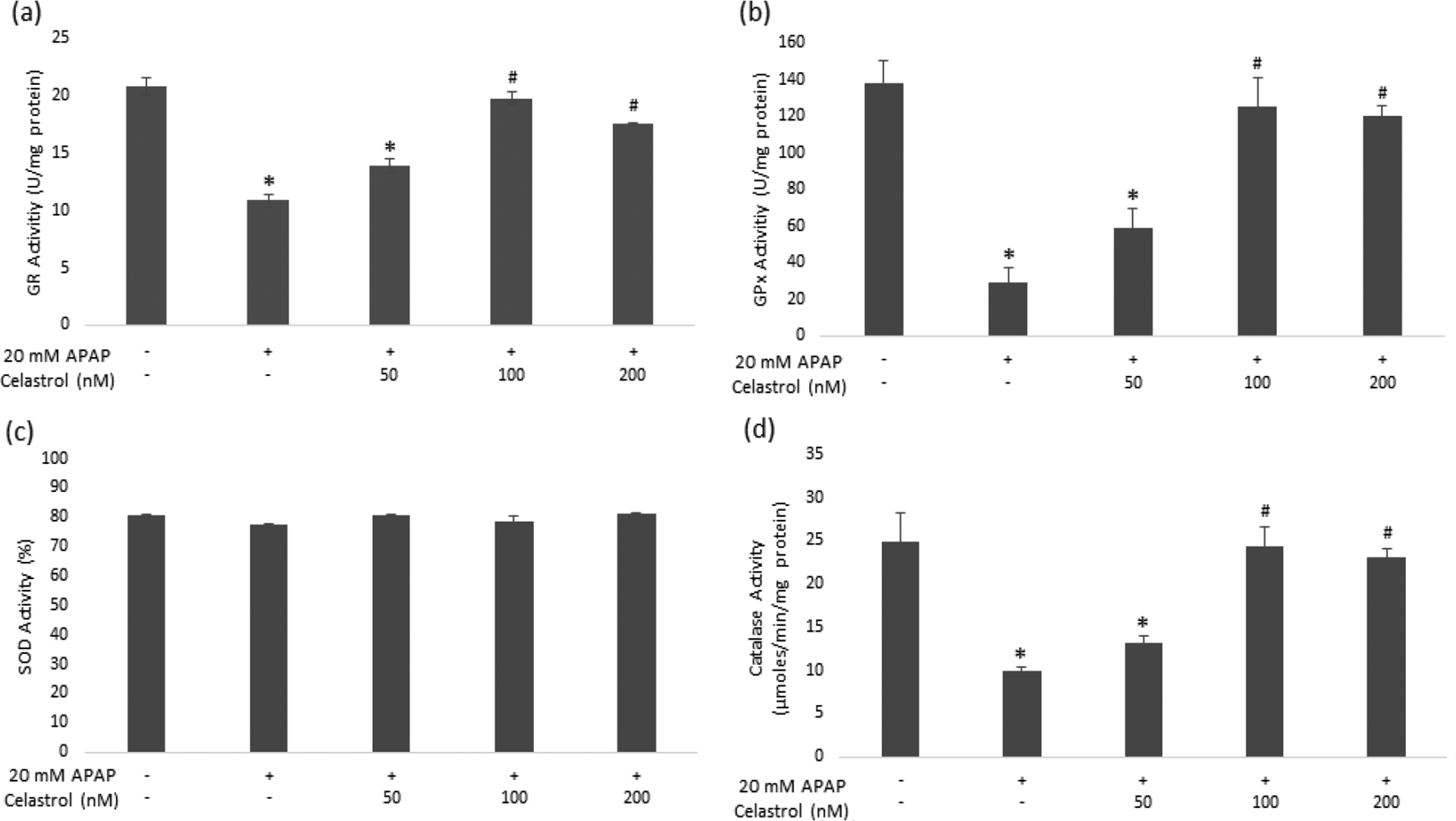
Effects of APAP treatment and celastrol posttreatments on different antioxidant enzyme activities GR (a), GPx (b), SOD (c), and catalase (d) in HepG2 cells. HepG2 cells were treated with 20 mM APAP for 24 h, after 24 h the medium was renewed and cells were posttreated with celastrol (50 nM, 100 nM, and 200 nM) for 24 h. Control cultures received 0.5% DMSO. Bar graphs were generated with Microsoft Excel 2016 (USA) and data are shown as the means ± SD (n:3); * p < 0.05 versus control cells; # p < 0.05 versus only APAP-treated cells. APAP: acetaminophen; GR: glutathione reductase; GPx: glutathione peroxidase; SOD: superoxide dismutase.
Discussion
APAP is a widely used drug and it is safe for use because of its analgesic and antipyretic effects in therapeutic doses. However, due to the widespread use, overdoses of APAP represent common problems and result in acute liver failure. The main mechanisms that are related with APAP toxicity are depletion of GSH by reactive metabolite NAPQI, increase in reactive oxygen and nitrogen species, and oxidative stress-mediated ATP depletion by loss of mitochondrial membrane potential in hepatocytes. It was shown that improving the GSH and antioxidant status may prevent the APAP-induced hepatotoxicity. However, there is a need for further studies to understand the approach of antioxidant therapy for APAP-induced hepatotoxicity. 28,29
Celastrol is a quinone methide triterpene isolated from T wilfordii Hook F. and has been reported to exhibit cytoprotection and antioxidant activity. 30 –32 The present study showed that celastrol posttreatment was effective against APAP-induced cytotoxicity. Celastrol recovered decreased cell viability and membrane integrity of APAP-treated HepG2 cells.
Protein carbonylation is a type of protein oxidation and the levels of carbonylated proteins accurately indicate oxidative damage. 33 It has been reported that APAP can cause elevation in protein carbonlyation. 28 Moreover, the carbonylated protein level in the cells significantly increased after APAP treatment. Previous reports demonstrate the protective and ameliorative effects of celastrol on protein carbonylation in cell culture conditions. 34,35 Also, our results showed that after APAP administration, celastrol posttreatment decreased the protein carbonyl content in HepG2 cells. GSH is one of the most important intracellular antioxidants and its depletion by APAP may cause oxidative stress. There are studies showing that celastrol can increase the GSH content in the cells. 11,36,37 . Our GSH assay results revealed that celastrol posttreatment increased the GSH content in HepG2 cells, which had been decreased by APAP treatment. MDA, the end product of lipid peroxidation, is widely used as an oxidative stress marker. Many studies reported that natural compounds with antioxidant properties can reduce APAP-induced lipid peroxidation in vitro and in vivo. 18,38 –40 We found that APAP-induced increase in the MDA level decreased with all the tested concentrations of celastrol.
Cells have cellular antioxidant enzyme systems for protection against oxidative stress. The main antioxidant enzymes are SOD, catalase, GR, and GPx. SOD converts superoxide radicals to H2O2. Catalase and GPx dissociate the end product of the dismutation reaction, H2O2, into oxygen and water. GR regenerates oxidized GSH to the reduced form. 41 Therefore, the changes in cellular antioxidant enzyme systems can be considered as the important biomarkers of antioxidant response.
Previous reports indicated that oxidative stress is one of the main causes responsible for APAP-induced hepatotoxicity. 28,42,43 Celastrol exhibits antioxidant properties by increasing the activities of GPx, GR, SOD, and catalase. 12,14,37,44 Our results showed that APAP treatment leads to decreased activities of GR, GPx, and catalase in the HepG2 cells and that celastrol posttreatments lead to an increased activities of GR, GPx, and catalase. However, the SOD activity did not show any significant change with APAP treatment and celastrol posttreatments. Similarly, Chen et al. 45 found that celastrol had no significant effect on SOD expression levels in tumor cells. Some reports claim that celastrol increases the production of ROS. Many of these studies use higher doses than the studies showing the antioxidant properties of celastrol, thus, can produce different effects. 45 –47 However, similar to current literature, we demonstrated that celastrol exhibits antioxidant effects in low concentrations.11, 45 Our study showed for the first time that celastrol had positive effects on APAP-induced cytotoxicity and oxidative stress parameters in HepG2 cells. Nevertheless, considering that our experiment design did not include a known antidote for APAP-induced toxicity such as NAC, it was not possible to compare the ameliorative effect of celastrol. Including a positive control would improve the quality of the results obtained.
In conclusion, we identified that APAP-induced cytotoxicity and membrane integrity loss were significantly recovered by celastrol posttreatments in HepG2 cells. Celastrol posttreatments decreased carbonylation of proteins, improved the GSH content in HepG2 cells, and ameliorated lipid peroxidation level. Also, celastrol posttreatments improved cellular antioxidant defense in HepG2 cells. These results indicated that celastrol can improve antioxidant status after APAP-induced cytotoxicity. This ameliorative effect of celastrol may be related to its ability to increase the antioxidant capacity. Our findings support that celastrol may have a therapeutic value to prevent or ameliorate the effects of APAP-induced cytotoxicity. Further studies are needed to evaluate the role of celastrol as a possible antidote for APAP toxicity. Different models such as other cell lines, in vivo animal models, and eventually human trials should be designed.
Footnotes
Acknowledgement
The authors would like to thank the Research Fund of Istanbul University for the financial support towards this study.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: Research Fund of Istanbul University (Grant numbers 49622, 51660 and BEK-2016-20061).