
Abstract
Weak peroxisome proliferator–activated receptor (PPAR) α agonists (fibrates) are used to treat dyslipidemia. This study compared the effects of the potent and selective PPARα agonist CP-778875 on peroxisomal β-oxidation and cardiac and/or skeletal muscle injury with those of the weak PPARα agonist fenofibrate. We hypothesized that these muscle effects are mediated through the PPARα receptor, leading to increased β-oxidation and consequent oxidative stress. CP-778875 (5 or 500 mg/kg) and fenofibrate (600 or 2,000→1,200 mg/kg, dose lowered because of intolerance) were administered to rats for six weeks. Standard end points, serum troponin I, heart and skeletal muscle β-oxidation of palmitoyl-CoA, and acyl co-oxidase (AOX) mRNA were assessed. Both compounds dose-dependently increased the incidence and/or severity of cardiomyocyte degeneration and necrosis, heart weight, troponin I, and skeletal muscle degeneration. Mean heart β-oxidation (3.4- to 5.1-fold control) and AOX mRNA (2.4- to 3.2-fold control) were increased with CP-778875 500 mg/kg and both doses of fenofibrate. β-Oxidation of skeletal muscle was not affected by either compound; however, a significant increase in AOX mRNA (1.6- to 2.1-fold control) was observed with CP-778875 500 mg/kg and both doses of fenofibrate. Taken together, these findings were consistent with PPARα agonism and support the link between increased cardiac and skeletal muscle β-oxidation and resultant muscle injury in the rat.
Introduction
Peroxisome proliferator–activated receptor (PPAR) agonists are used clinically or are being developed for the treatment of dyslipidemia (PPARα), type 2 diabetes (PPARγ), and obesity (PPARβ or PPARδ). Although PPARα agonists have been widely used in humans for decades, the agents currently in use are fairly weak agonists with a variable safety and efficacy profile. In order to improve efficacy and maintain or improve safety profiles for drugs targeting these receptors, companies are searching for PPAR agonists with higher potency and specificity.
PPARα agonists such as fenofibrate or chlofibrate are currently used clinically to treat dyslipidemia with a very good safety record and none of the cardiovascular risks experienced with PPARγ agonists (Graham et al. 2010; Nissen and Wolski 2010). However, in an apparent contradiction, the administration to rats of potent and highly selective PPARα agonists such as CP-778875 (Figure 1; binding selectivity > 180× against PPARβ and > 1500× against PPARγ, functional activity selectivity > 23× against PPARβ and > 400× against PPARγ) results in an increased incidence and/or severity of murine progressive cardiomyopathy, and skeletal muscle degeneration and necrosis (Hodel 2002; Pruimboom-Brees et al. 2006). Interestingly, no effects on cardiac and skeletal muscle were noted in a six-week monkey toxicity study at very high doses of this compound (unpublished data), resulting in speculation that such effects were specific to rodents, and that monkeys—and by extension, humans—were somehow resistant to these effects.
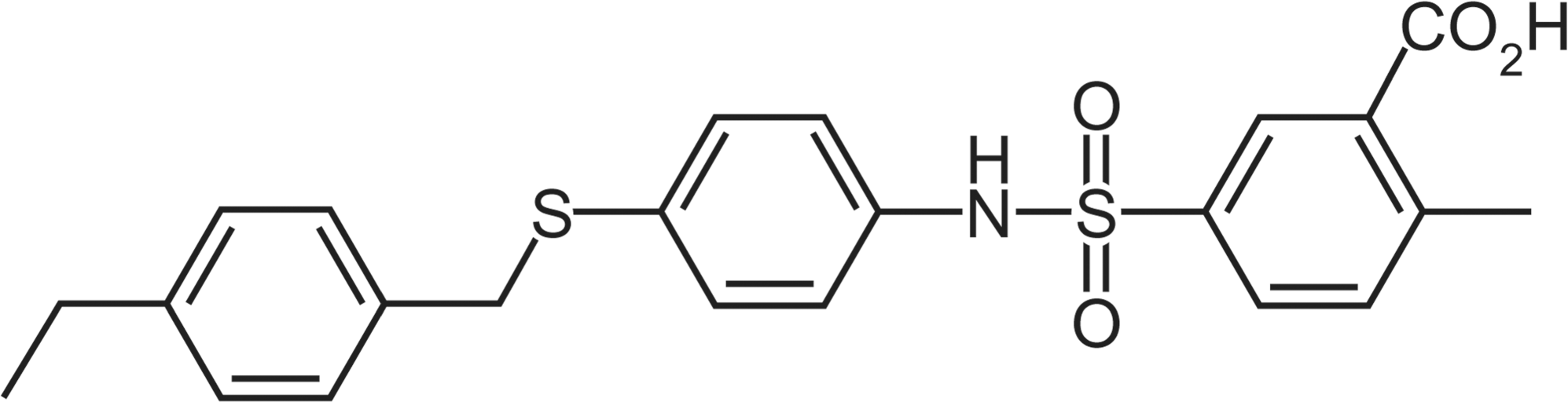
Chemical structure of CP-778875.
We hypothesized and proved that this rat-specific effect was caused by a drug-related increase in peroxisomal β-oxidation and consequent oxidative stress injury (Pruimboom-Brees et al. 2006), which was not produced in monkeys or humans. This result is important since PPARα agonists are being developed for the treatment of atherosclerosis, with a consequent low tolerance for cardiovascular safety issues.
A troubling gap in this logic is that there is currently little record in the literature of cardiac or skeletal muscle effects observed in rats with weak PPARα agonists. If our hypothesis is correct, similar degrees of cardiac and skeletal muscle degeneration should result with both weak and potent PPARα agonists (like CP-778875) at doses that produce the similar levels of peroxisomal β-oxidation. However, despite an extensive literature on PPAR agonists, there are no such studies comparing potent and weak PPARα agonists for effects on cardiac and skeletal muscle pathology at appropriate doses based on functional effects in these target tissues.
Therefore, the current study was designed to compare the cardiac and skeletal muscle effects in rats of CP-778875 with the weaker PPARα agonist fenofibrate (PPARα transactivation IC50 for fenofibrate = 30 mM, CP-778875 = 79 nM) at doses that produce comparable levels of peroxisomal β-oxidation. Although there have been no preclinical literature reports of cardiac myocyte necrosis with fenofibrate, it was chosen as a model compound to test this hypothesis since it has been safely used in humans for many years and is a relatively selective PPARα agonist (10× PPARα to PPARγ activity; Wilson et al. 2000).
Material and Methods
Animals and Husbandry
Ten-week-old Sprague-Dawley rats (Crl: CD[SD] IGS BR) were obtained from Charles River Breeding Laboratories (Kingston, NY). Animals were acclimated to our laboratory for three weeks prior to dosing. Rats were randomly assigned to treatment groups according to body weight using a computer program. Each rat was identified by a unique number tattooed on the tail. Rats were housed individually in hanging stainless-steel cages equipped with an automatic watering valve. The animal room environment was controlled (70°F ± 5°F, humidity 50% ± 10%, twelve-hour light/dark cycle). Certified laboratory diet (PMI Feed, Inc., 5002) and drinking water were provided ad libitum. Animals were fasted overnight prior to blood sampling for clinical pathology and prior to necropsy. The protocol and any amendment(s) or procedures involving the care and use of animals in this study were reviewed and approved by the Pfizer Global Research and Development Institutional Animal Care and Use Committee prior to the study. The animal care and experimental procedures of this study were conducted in compliance with the U.S. Animal Welfare Act and the ILAR Guide (1996).
Test Substance, Experimental Design, Observations, and Measurements
Fenofibrate (99% pure) was supplied by Sigma Aldrich (St. Louis, MO), and CP-778875 (98.9% pure) was synthesized and supplied by Pfizer Global Research and Development. Fenofibrate was prepared daily as a suspension in 0.5% methylcellulose, and CP-778875 was prepared weekly as a suspension in 0.5% methylcellulose/0.05% sodium lauryl sulfate vehicle. Control animals received 0.5% methylcellulose/0.05% sodium lauryl sulfate vehicle. The dose volume was 10 mL/kg.
Vehicle, CP-778875, and fenofibrate were administered orally by gavage once daily to groups of ten male and ten female (main study) rats. Control animals received vehicle (0.5% methylcellulose/5% sodium lauryl sulfate) at a dose volume of 10 mL/kg. CP-778875-treated rats received 5 or 500 mg/kg, whereas fenofibrate treated rats received 600 or 2,000 → 1,200 mg/kg (2,000 mg/kg for twenty-one days and 1,200 mg/kg for nineteen days). Doses of CP-778875 were selected based on previous studies (unpublished) in which 25 mg/kg was a threshold dose for cardiac myonecrosis and/or myofibrosis and skeletal muscle degeneration and regeneration. Fenofibrate doses were chosen based on an equivalence of receptor transactivation, AOX mRNA, and systemic exposure to that achieved for CP-778875. For CP-778875, the 5 mg/kg dose was an anticipated no-observable-adverse-effect level (NOAEL), and 500 mg/kg was an anticipated maximum tolerated dose that was expected to result in mild cardiomyocyte necrosis and increased serum troponin I. For fenofibrate, the 600 and 2,000 mg/kg doses were expected to result in peroxisomal proliferation, mild cardiomyocyte necrosis, and increased serum troponin I. Fenofibrate 2,000 mg/kg was not tolerated, and the dose was lowered to 1,200 mg/kg on Day 22 (main-study and toxicokinetic [TK] females) or Day 23 (TK males). For TK and biochemical assessments, additional groups of six males and six females were administered the same doses.
Animals were observed for signs of toxicity and for any changes in appearance or behavior (predose, immediately after dosing, one hour after dosing, and several hours after dosing). Body weights and food consumption were recorded weekly.
Clinical Pathology Assessments
Blood samples for hematology and serum chemistry determinations were collected by jugular venipuncture from main study animals on Day 41, prior to necropsy. Rats were bled in a fasted state following CO2: O2 anesthesia. Standard hematology parameters were assessed using an Advia 120 automated analyzer (Siemens Diagonistics, Tarrytown, NY). Blood samples taken from the vena cava at necropsy were used to determine prothrombin and activated partial thromboplastin times, which were measured using the STA Compact Automated Coagulation System (Stago) analyzer (Diagnostica Stago, Parsippany, NJ). Standard serum clinical chemistry assessments were measured on the Hitachi chemistry analyzer (Roche Diagnostics, Indianapolis, IN), and serum troponin I was measured with the Bayer Advia Centaur analyzer (Siemens Diagnostics, Tarrytown, NY).
Toxicokinetics
For CP-778875–treated rats, serum was obtained at 0.5, one, four, eight, and twenty-four hours after dosing (three rats/sex/dose/time point). CP-778875 was assayed using a LC-MS/MS method. For fenofibrate-treated rats, serum was obtained at one, two, four, six, and twenty-four hours after dosing (three rats/sex/dose/time point). Fenofibrate was assayed using HPLC/MS. Toxicokinetic calculations were performed in WinNonlin (version 3.2, released June 11, 2001, Pharsight Corporation, St. Louis, MO), and mean data were used in all TK analyses. The area under the mean serum concentration-time curve (AUC0-24 h ) was estimated using the linear trapezoid approximation. Cmax was defined as the maximum mean serum concentration observed, and Tmax was defined as the time at which mean Cmax was first observed.
Peroxisomal β-Oxidation and AOX mRNA Levels
Measurements were made as previously described (Pruimboom-Brees et al. 2006). Peroxisomal β-oxidation was determined in tissue homogenates (Lazarow 1981). Briefly, frozen tissue samples were gradually thawed in Dulbecco’s phosphate-buffered saline (PBS). Samples were then minced and homogenized in nine volumes (liver) or five volumes (heart and skeletal muscle) of cold 0.25 M sucrose. The homogenate was then centrifuged at 600 × g for ten minutes. The supernatant was frozen in 1-mL aliquots.
On the day of assay, samples were thawed and assayed first for protein content using the bicinchoninic acid method. Following the addition of Triton X-100, each sample was mixed, and then aliquots were transferred to sample cups. Reaction mixture (20 mM NAD, 0.33 M DTT, 10 mM Coenzyme A, 1 mM FAD, 1 mg/mL bovine serum albumin, and 100 mM KCN in 0.05 M HEPES) and 0.5 mM palmitoyl A were separately placed in the appropriate reagent cups. A sixty-second incubation at 37°C was followed by thirty absorbance readings at 340 nm, taken every ten seconds in order to determine the change in absorbance (▵A) per minute. For each sample in the Cobas-Fara COAV program, the resulting ▵A/min is multiplied by a calculation factor F (F = 1/∊ × 103 mL/L × 0.25 cm2/sample volume in mL where ∊ = 0.622 cm−1 mM−1, the millimolar extinction coefficient for NADH) to yield µmol × L−1 × minute−1, the numerical value of which is printed out along with the corresponding sample number by the Cobas-Fara. This value is then multiplied by the homogenate dilution factor (e.g., 9 or 5, depending on the tissue) and divided by 1,000 g/L to yield the final sample palmitoyl CoA activity as µmoles NAD reduced/g liver × minute. Because of a 1:1 stoichiometry between NADH and acetyl CoA, the rates of NAD reduction are equivalent to µmoles of acetyl CoA formed per minute.
The activity of the cyanide-insensitive peroxisomal acyl CoA oxidizing system was determined by measuring palmitoyl-CoA–dependent reduction of NAD at 340 nm. AOX mRNA levels were determined by Quantigene. Flash-frozen samples were homogenized in tissue preparation buffer (TPB) containing 50 mM HEPES, 2% lithium lauryl sulfate, 0.5% Micro-O-Protect (Roche Diagnostic, Indianapolis, IN), 8 mM EDTA, 500 mM lithium chloride, and 3.8 mg/mL proteinase K (EM Science, Merck KGaA, Darmstadt, Germany). A volume of 6–24 µL of tissue homogenate was applied to 100 µL of Quantigene lysis buffer (Genospectra, Inc., Fremont, CA) containing the probe sets for mouse AOX in a COSTAR ninety-six-well plate. Subsequently, 90 µL was applied to each well of the capture plate containing 100 µL of the TPB. After an overnight incubation at 53°C, the plate was developed according to the manufacturer’s protocol. Briefly, plates were allowed to sit at room temperature for fifteen minutes; they were then washed twice with Quantigene wash buffer, and 100 µL of Amplifier reagent was added to each well.
Following an hour of incubation at 53°C, plates were allowed to sit at room temperature for fifteen minutes and were washed twice with wash buffer, and 100 µL of Label Probe reagent was applied to each well. After one hour of incubation at 53°C, plates were washed six times with wash buffer, and 100 µL of substrate was added. The plates were incubated for thirty minutes at 37°C and immediately read on a Dynex luminometer. The capture plates and all reagents were purchased from Genospectra, Inc. or prepared as described in the Quantigene manufacturer’s protocol.
Pathology
On study Day 41, main study rats were anesthetized by CO2: O2 inhalation and euthanized by exsanguination. Following a gross necropsy examination, the weights of liver and heart were recorded, and liver, skeletal muscle, and heart were collected and placed in fixative. Following fixation, tissues were trimmed, dehydrated, embedded in paraffin, sectioned, mounted on glass slides, and stained for histopathologic evaluation. Toxicokinetic rats were euthanized in a similar manner. Samples of the liver (right medial lobe), skeletal muscle (gastrocnemius block from the right leg, which included the soleus), and approximately half the heart were collected, frozen at −60°C, and stored at −80°C until evaluation for peroxisomal proliferation by β-oxidation analysis. Samples of the liver (left medial lobe), skeletal muscle (gastrocnemius including soleus from the left leg), and approximately half the heart were collected, frozen at −60°C, and stored at −80°C until evaluation for AOX mRNA.
Statistical Analysis
Treated group means were compared with the control group mean. Dunnett’s multiple comparison procedure (Dunnett 1955; Dunnett 1964) was used if a preliminary Bartlett’s test (Sokal and Rohlf 1969) or F test for variance of homogeneity was not significant at the α = .05 level. If there was significant variance heterogeneity, the Cochran-Cox modified t test (Cochran and Cox 1957) was used for comparison between treated and control group means. Statistical significance of the comparisons was indicated at both the α = .05 and .01 levels. Tests were two-tailed.
Results
In-life Observations
CP-778875 was generally well tolerated at 5 and 500 mg/kg. At 500 mg/kg, one of six TK males died on Day 31 from an undetermined cause. Clinical signs in 500 mg/kg CP-778875–treated rats were limited to loose stools (6/20) and salivation (12/20). Statistically significant decreases in absolute body weight (87–92% of control on Days 15, 22, 29, and 36) were noted in males at 500 mg/kg (Figure 2A). In the 500 mg/kg males, cumulative body weight gain on Day 36 was also statistically significantly decreased (46% of control). At 500 mg/kg CP-778875, weekly mean food consumption was statistically significantly decreased in males (Figure 2C, 78–88% of control on Days 8 and 15) for females (Figure 2D, 90% of control on Day 8). Taken together, these data indicate that 500 mg/kg exceeded the maximum tolerated dose.
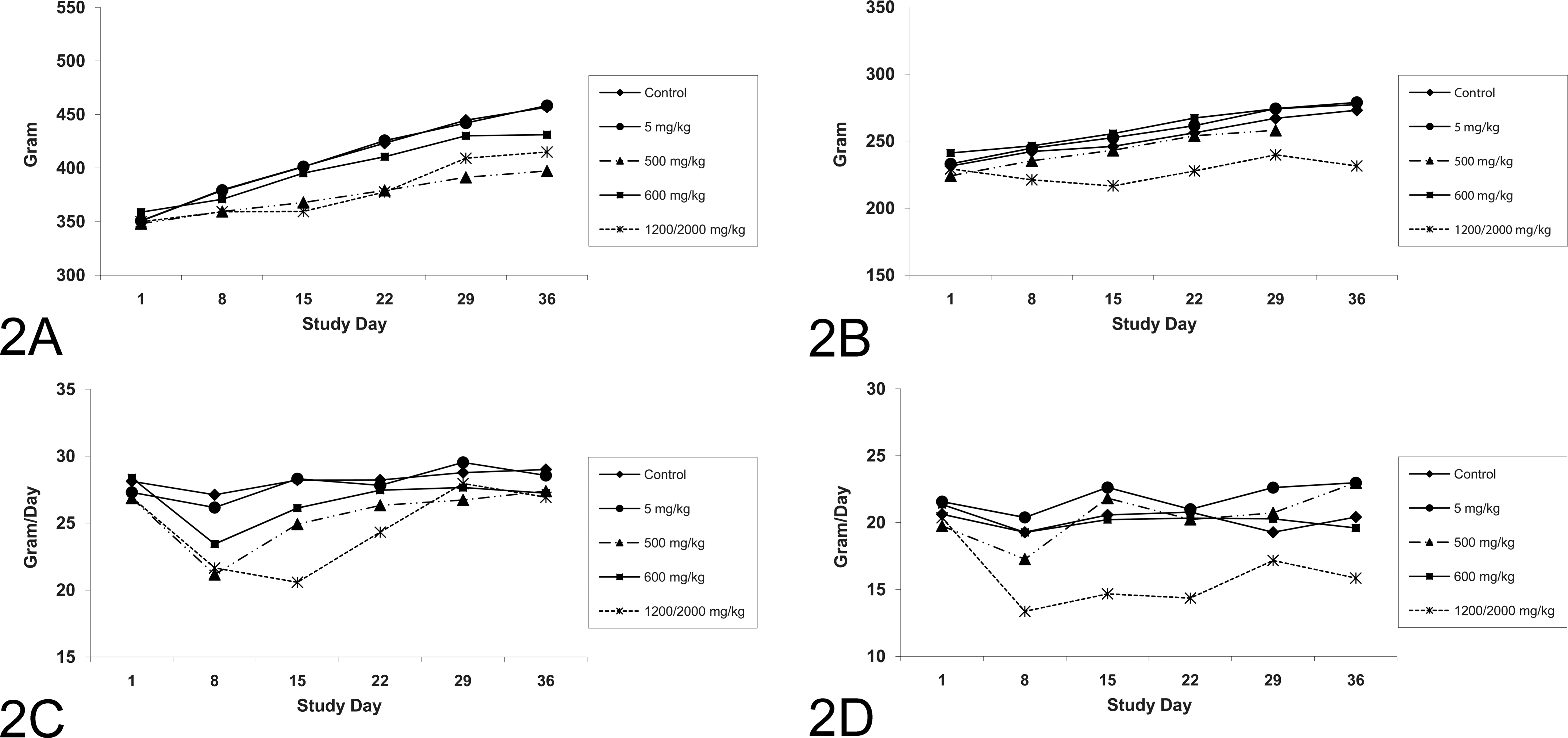
Time course for body weight (mean, N = 10 rats/group) for (A) male and (B) female rats; and food consumption (mean, N = 10 rats/group) for (C) male and (D) female rats.
Fenofibrate was well tolerated at 600 mg/kg but was poorly tolerated at 2,000 mg/kg, which necessitated a lowering of the dose to 1,200 mg/kg on Day 22 or 23. In the main study animals that received 2,000→1,200 mg/kg, there was an unscheduled death from an undetermined cause on Day 24. Another 2,000→1,200 mg/kg main study animal died on Day 4 as a result of a multicentric lymphoma that was considered unrelated to treatment with fenofibrate. Animals administered 2,000→1,200 mg/kg had clinical signs related to deteriorating health status, including hunched posture (8/20), stained fur or urine staining (13/20), rough fur (17/20), decreased activity (1/20), and pale skin (2/20). Consistent with the lack of tolerance of the 2,000→1,200 mg/kg dose of fenofibrate, mean body weights were decreased in males (Figure 2A; Days 15, 22, 29, and 36, 89–91% of control) and females (Figure 2B; Days 8, 15, 22, 29, and 36, 85–91% of control). In the 2,000→1,200 mg/kg males and females, cumulative body weight gain on Day 36 was also statistically significantly decreased (59% of control for males, 5% of control for females). Statistically significant decreases in mean food consumption were also noted in 2,000→1,200 mg/kg fenofibrate males (Figure 2C; Days 8, 15, and 36, 0.73–0.93% of control) and females (Figure 2D; Days 8, 15, 22, and 36, 0.69–0.78% of control). These data indicate that the 600 mg/kg dose of fenofibrate was well tolerated, but the 2,000→1,200 mg/kg dose exceeded the maximum tolerated dose.
Cardiac Findings
Serum troponin I is a noninvasive, sensitive, specific, and widely used clinical and preclinical biomarker for cardiac muscle damage. Consistent with our hypothesis that potent and weak PPARα agonists could both produce cardiac muscle damage at biologically comparable doses, there was an increase in individual and group mean troponin I values (Table 1, Figure 3) in both sexes at 500 mg/kg CP-778875 and at 600 and 2,000→1,200 mg/kg fenofibrate. At 500 mg/kg CP-778875, five of ten male rats had increased troponin I values (two- to sixteen-fold control group mean), whereas two of ten females had increased values (three- to five-fold control group mean). Two of ten males dosed with 600 mg/kg fenofibrate had increased troponin I values (seven- to twelve-fold control group mean), and four of eight dosed with 2,000→1,200 mg/kg fenofibrate had increased troponin I values (four- to five-fold control group mean). Corresponding increases in females administered 600 or 2,000→1,200 mg/kg fenofibrate occurred in two of ten (three- to five-fold) and eight of ten (three- to sixteen-fold) rats.
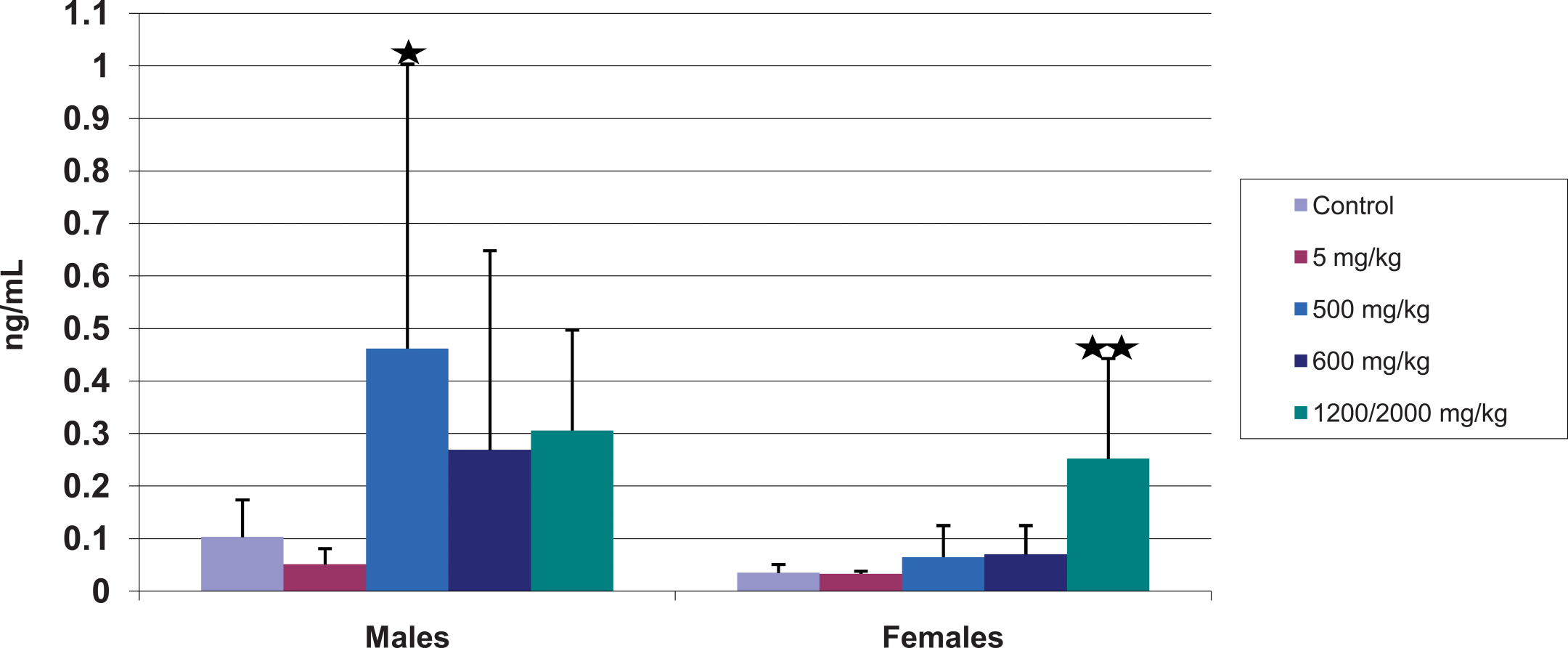
Serum troponin I (mean ± standard deviation, n = ten rats/group) on Day 41 for control, CP-778875 5 mg/kg, CP-778875 500 mg/kg, fenofibrate 600 mg/kg, and fenofibrate 2,000→1,200 mg/kg–treated rats. Significantly different from control, *p < .05, **p < .01.
Selected serum chemistry findings (mean ± SD).
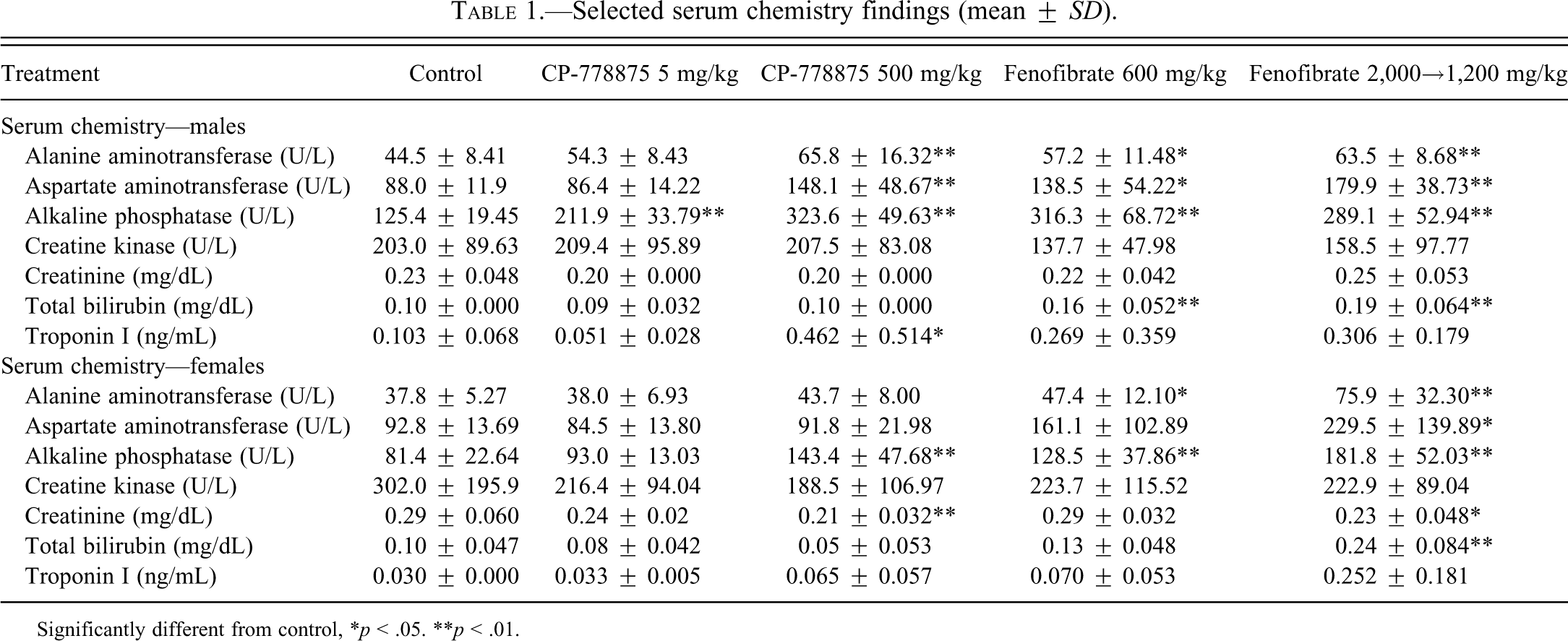
Significantly different from control, *p < .05. **p < .01.
Although mean troponin I values were statistically significant only for the 500 mg/kg CP-778875 males because of inter-animal variability, individual animal values were increased above the current control values for the 600 or 2,000→1,200 mg/kg fenofibrate–treated males and females. The increases in mean troponin I values correlated microscopically with an increase in the incidence and/or severity of myonecrosis/fibrosis and mononuclear cell infiltration in the heart (as described below). In general, the troponin I values were higher in control, CP-778875, and fenofibrate-treated male than in corresponing female rats, which is consistent with the microscopic findings of myonecrosis/fibrosis being higher in male rats and is consistent with a recent literature report regarding baseline serum cardiac troponin I concentrations in rats (Herman et al. 2011). Individual-animal and mean serum creatine kinase concentrations were not increased by treatment with CP-778875 or fenofibrate. These findings demonstrate the utility of troponin I as a biomarker for detecting PPARα-induced cardiac muscle damage.
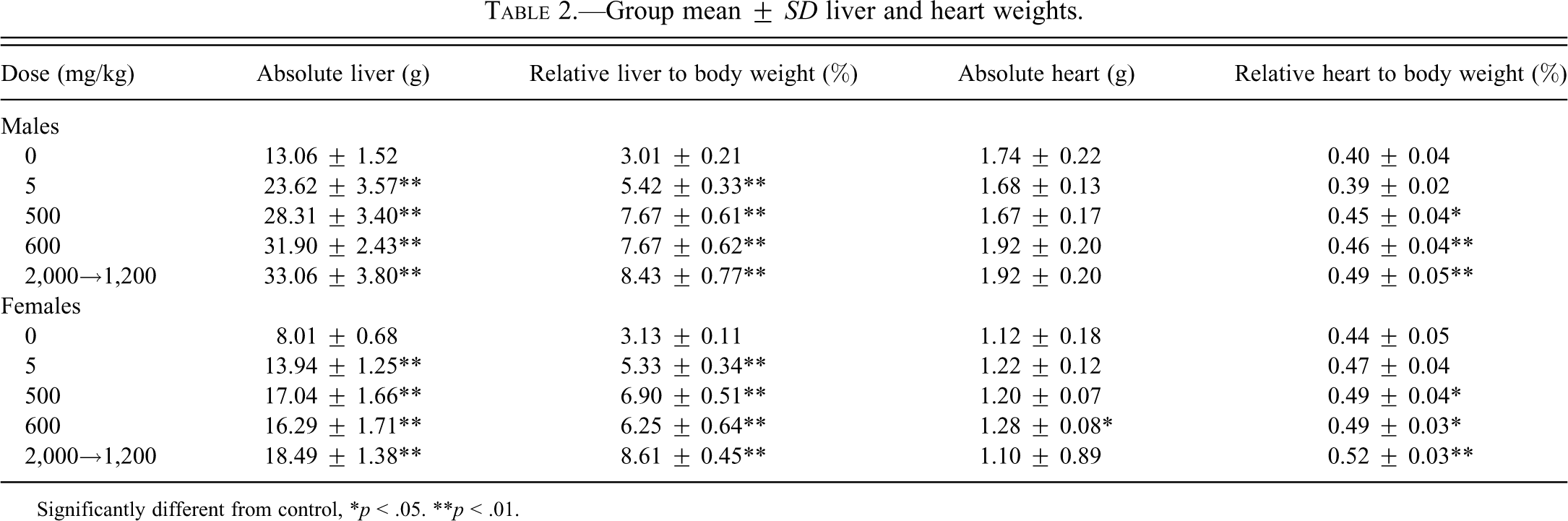
Compound-related increases in absolute and/or relative (to body and/or brain weight) heart weights (1.1- to 1.2-fold control) were observed in both sexes at 500 mg/kg CP-778875 and both doses of fenofibrate (Table 2). However, although there was no absolute histological correlate for this observation on an individual-animal basis, it is unlikely to be related to PPARα agonism, since the literature suggests that chronic PPARα agonism is associated with increased myosin light chain-2 transcription and cardiac hypertrophy (Hamano et al. 2001; Finck et al. 2002). In the heart (Table 3), there was a dose-dependent increase in the incidence and/or severity of myonecrosis/fibrosis in both sexes at 500 mg/kg CP-778875 and both doses of fenofibrate that correlated with elevated serum troponin I values. Myonecrosis/fibrosis associated with mononuclear cell infiltration is a common background finding in Sprague-Dawley rats that is known to increase in incidence and severity with age (Mohr et al. 1992) and that is more frequent in males. In the present study, myonecrosis/fibrosis (Figures 4A–4C) was graded as minimal, mild, or moderate when fewer than twelve myofibers, fewer than fifty myofibers, or more than fifty myofibers were affected, respectively. In males, there was a compound-related increase in the incidence of mild myonecrosis/myofibrosis at 500 mg/kg CP-778875 and both doses of fenofibrate and a compound-related increase in the severity of myonecrosis/myofibrosis, and a grade moderate was observed only at 500 mg/kg CP-778875 (2/10 males). In CP-778875–treated males at 500 mg/kg, the myocardial finding was subacute with more frequent and larger mononuclear cell infiltrates, whereas in fenofibrate-treated males at 1,200→2,000 mg/kg, the myocardial finding was more chronic, with myofibrosis being particularly prominent in a few individuals. In females, with the exception of 1/10 rats diagnosed with mild myonecrosis/myofibrosis and mononuclear cell infiltration at 2,000→1,200 mg/kg fenofibrate, the major compound-related effect was an increased incidence of minimal myonecrosis/myofibrosis and mononuclear cell infiltration at 500 mg/kg CP-778875 and both doses of fenofibrate.
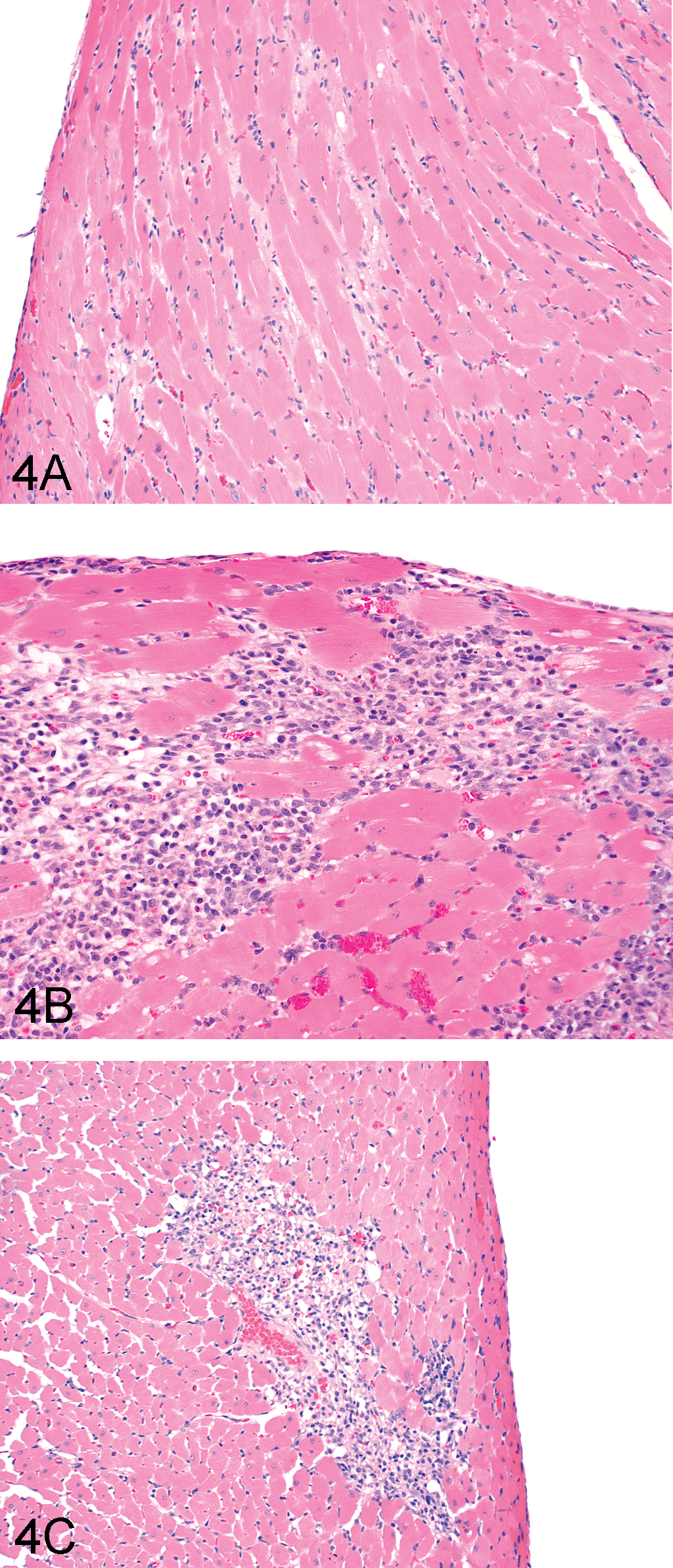
Representative photomicrographs of heart tissue from fenofibrate 600 mg/kg (A, B) and CP-778875 500 mg/kg (C) treated rats. Tissues were stained with hemotoxylin and eosin. Original magnification was 100× for (A) and (C), and 40× for (B). Findings in (A) were mild myocardial fibrosis with a few monocytes, whereas findings in (B) were mild myocardial necrosis with many monocytes and little fibrosis. In (C), findings included mild myocardial necrosis with many monocytes and little fibrosis.
Group mean ± SD liver and heart weights.
Significantly different from control, *p < .05. **p < .01.
Microscopic findings.
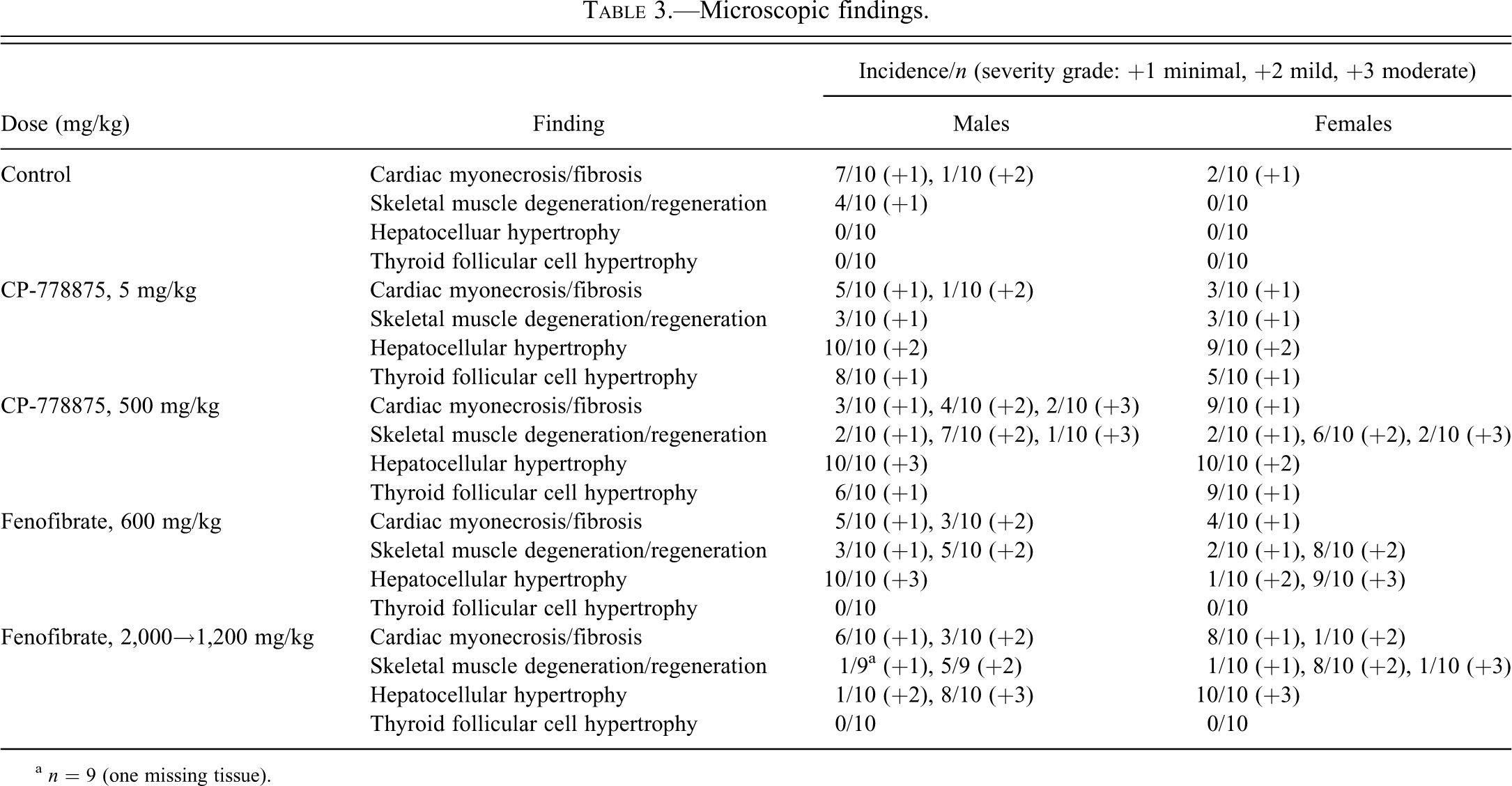
a n = 9 (one missing tissue).
Skeletal Muscle Findings
In female rats, significant decreases in serum creatinine levels (Table 1; 72–79% control) were observed at 500 mg/kg CP-778875 and at 2,000→1,200 mg/kg fenofibrate and correlated microscopically with myofiber degeneration/regeneration in skeletal muscles (Table 3). In the gastrocnemius/soleus (Figure 5A, 5B), minimal myofiber degeneration (with occasional mononuclear cell infiltrates) and regeneration (myofiber basophilia) is of a type commonly seen in control Sprague-Dawley rats maintained at Pfizer Drug Safety Research and Development. Myofiber degeneration/regeneration was considered minimal when one to three foci (with one to three myofibers per foci) were involved per section. Mild to moderate myofiber degeneration/regeneration was considered pharmacological (Pruimboom-Brees et al. 2006) and was observed in both sexes at 500 mg/kg CP-778875 and both doses of fenofibrate.
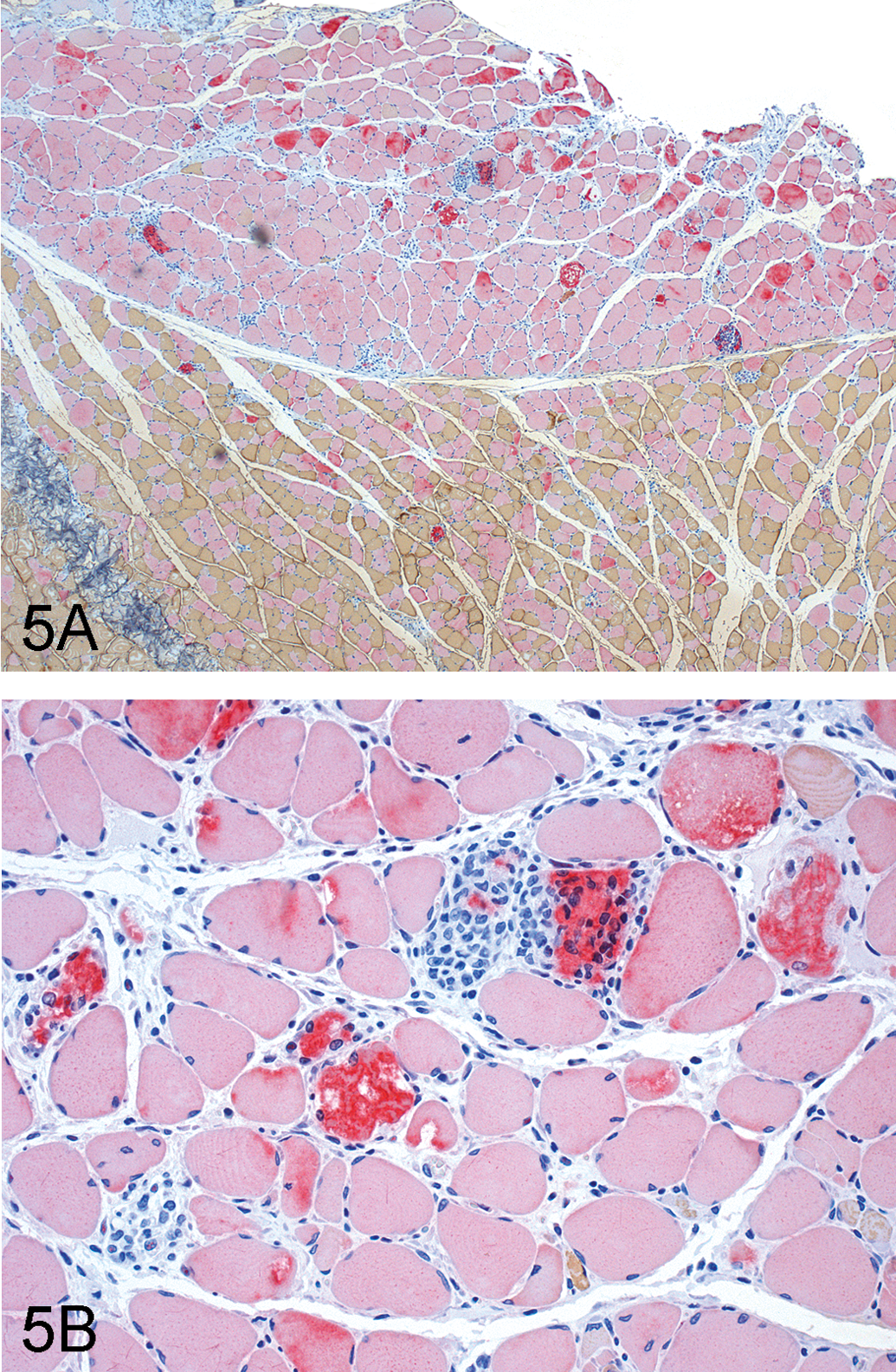
Representative photomicrographs of skeletal muscle tissue from fenofibrate 2,000→1,200 mg/kg–treated rats. (A) Moderate skeletal muscle degeneration/regeneration (section stained by double immunohistochemistry and counterstained with hemotoxylin, original magnification 40×), myosin heavy chain fibers Type 1 (slow fibers, red) and Type 2 (fast fibers, brown). (B) Moderate skeletal muscle degeneration/regeneration (hemotoxylin and eosin, original magnification was 200×).
Liver Findings
Dose-related increases in serum liver enzymes (1.3- to 2.6-fold control) included alkaline phosphatase in males at 5 mg/kg CP-778875 and in both sexes at 500 mg/kg CP-778875 and at 600 and 2,000→1,200 mg/kg fenofibrate, ALT and AST in males at 500 mg/kg CP-778875 and in both sexes at 600 and 2,000→1,200 mg/kg fenofibrate, and total bilirubin in males at 600 mg/kg fenofibrate and both sexes at 2,000→1,200 mg/kg fenofibrate (Table 1). These findings are consistent with hepatic peroxisomal proliferation and microsomal enzyme induction associated with PPARα agonism. A compound-related increase in bile acids was observed in females at 2,000→1,200 mg/kg fenofibrate (5.9-fold control), and a few animals presented very high values (3/10 females, 127.8, 174.8, 178.2 µmol/L). This finding is likely consistent with “regurgitation” of bile acids from the hepatocytes into the blood secondary to hepatocellular hypertrophy and compression of bile canaliculi. With both compounds, significant increases in mean absolute and relative (to body and/or brain weight) liver weights (1.7- to 2.8-fold control) were observed in both sexes at all doses (Table 2). This finding is consistent with PPARα-mediated hepatic peroxisome proliferation and microsomal induction and correlated microscopically with mild to moderate centrilobular hypertrophy, which was observed at all doses with both compounds. Hepatocellular hypertrophy often resulted in subcapsular necrosis and inflammation as a result of the increased fragility of the enlarged liver and in retention in bile within the bile canaliculi, which resulted from compression of bile canaliculi by hypertrophied hepatocytes (Hall’s stain–positive intracellular/extracellular pigments). This microscopic finding also correlated with serum chemistry markers and is consistent with hepatic peroxisomal proliferation and microsomal induction associated with PPARα agonism.
Other Findings
Also secondary to PPARα-mediated hepatic microsomal induction, minimal hypertrophy of follicular cells (Table 3) was observed in the thyroid at 5 and 500 mg/kg CP-778875 in both sexes, and minimal hypertrophy of basophilic cells was observed in the pituitary of 4/10 males at 500 mg/kg CP-778875 (Greaves 1990).
Peroxisomal Proliferation and AOX mRNA Analyses
Peroxisomal β-oxidation was assessed in the liver, heart, and skeletal muscle by palmitoyl-CoA β-oxidation (Figure 6A–6C) and AOX mRNA (Figure 7A–7C). AOX is the rate-limiting enzyme in peroxisomal β-oxidation. Increases in palmitoyl Co-A substrate oxidation indicate post-transcriptional increases in AOX activity.
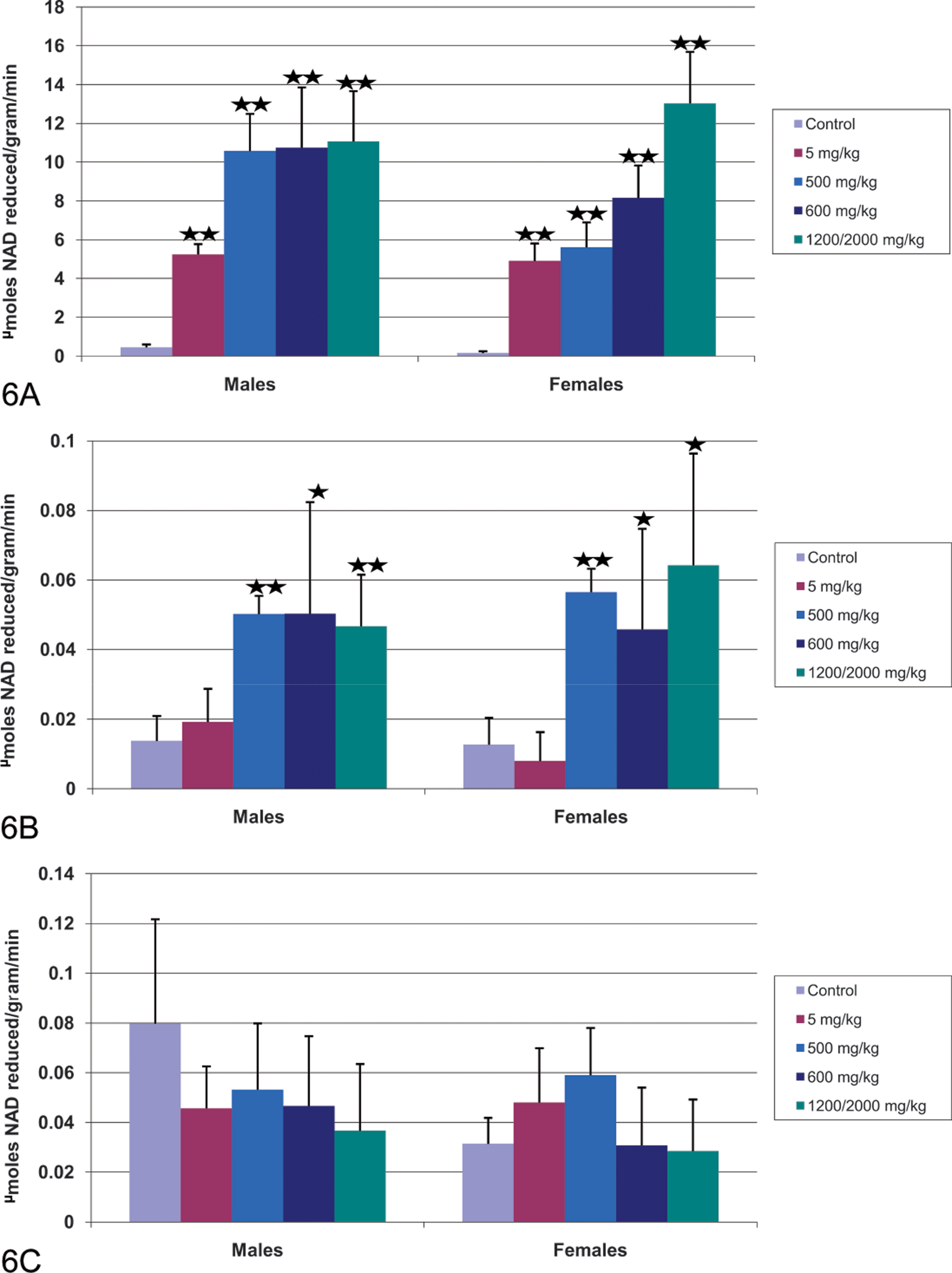
Effect of CP-778875 and fenofibrate on peroxisomal β-oxidation of palmitoyl CoA in (A) liver, (B) heart, and (C) skeletal muscle. Bars represent the mean ± standard deviation (n = five or six rats/group). Significantly different from control, *p < .05. **p < .01.
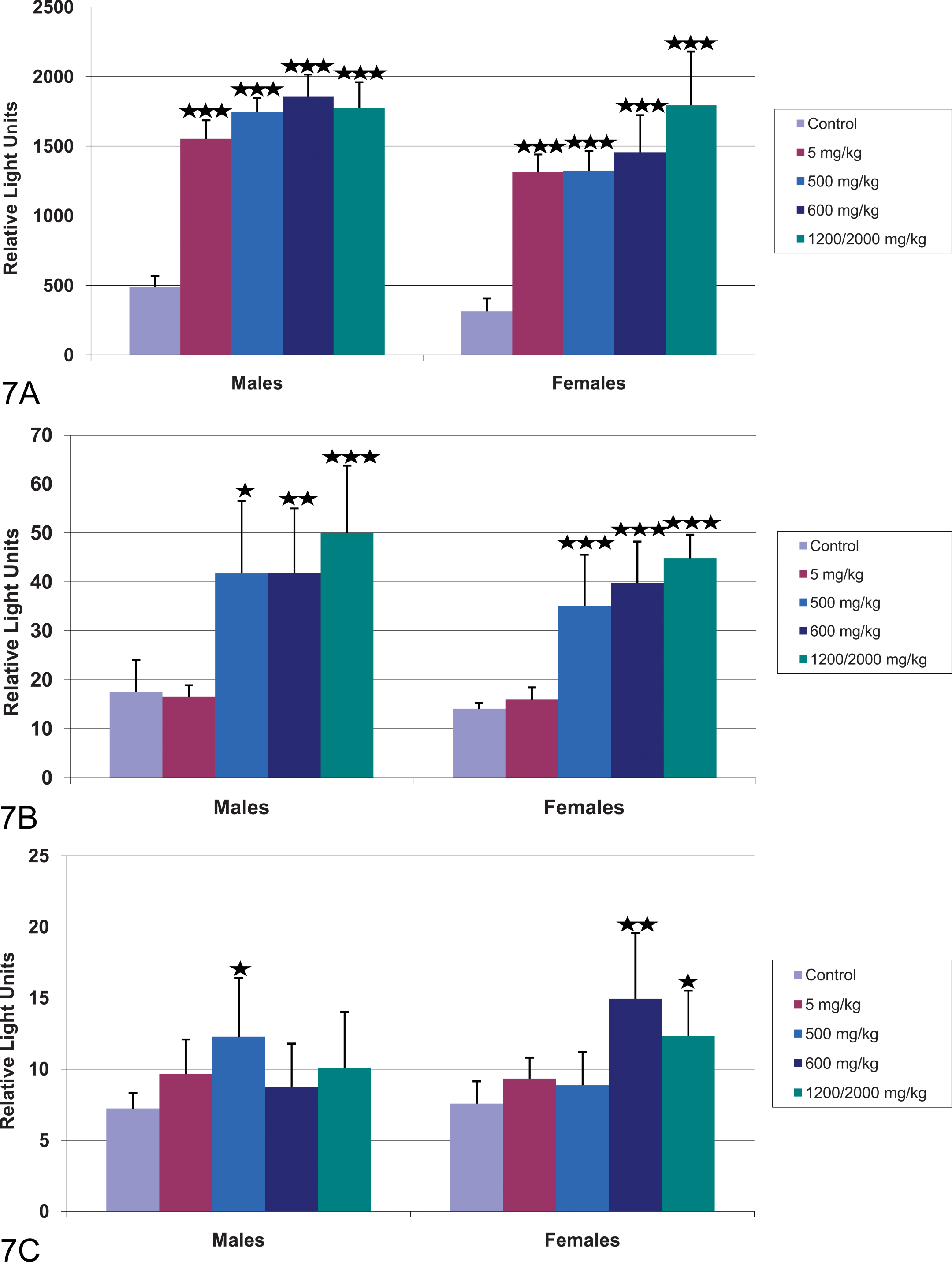
Effect of CP-778875 and fenofibrate on AOX mRNA levels in (A) liver, (B) heart, and (C) skeletal muscle. Bars represent the mean ± standard deviation (n = five or six rats/group). Significantly different from control, *p < .05. **p < .01. ***p < .001.
Significant dose-dependent increases in mean liver palmitoyl-CoA β-oxidation (11.5- to 34.8-fold control) and AOX mRNA levels (3.2- to 4.2-fold control) were observed in rats administered 5 or 500 mg/kg CP-778875 (Figures 6A and 7A). Mean heart β-oxidation values (3.6- to 4.4-fold control) and AOX mRNA levels (2.4- to 2.5-fold control) were significantly increased at 500 mg/kg (Figures 6B and 7B). CP-778875 did not affect β-oxidation in skeletal muscle within the limit of detection, however, a significant increase in mean skeletal muscle AOX mRNA was noted in males (1.9-fold control) administered 500 mg/kg (Figure 7C).
Peroxisomal β-oxidation also occurred in fenofibrate-treated rats. At 600 and 2,000→1,200 mg/kg fenofibrate, mean liver (23.5- to 80.7-fold control) and heart (3.4- to 5.1-fold control) palmitoyl-CoA β-oxidation values were significantly increased (Figures 6A and 6B). Mean liver (3.6- to 5.7-fold control) and heart (2.4- to 3.2-fold control) AOX mRNA values were also significantly increased in male and female rats administered fenofibrate 600 and 2,000→1,200 mg/kg (Figures 7A and 7B). In skeletal muscle, fenofibrate did not affect β-oxidation, however, a significant increase in mean skeletal muscle AOX mRNA was observed in female rats (1.6- to 2.1-fold control; Figure 7C).
Toxicokinetics
For CP-778875, systemic exposure (assessed by AUC0–24 h and Cmax ) increased with increasing dose (Table 4). AUC0–24 h on Day 36 was 3,020 and 826,000 ng·h/mL for the respective 5 and 500 mg/kg doses, whereas Cmax on Day 36 was 1,060 and 55,800 ng/mL for the respective 5 and 500 mg/kg doses. This result provides AUC0–24 h multiples of 8× (5 mg/kg) and 2,096× (500 mg/kg) as compared with the upper estimate of the human clinical efficacious systemic exposure AUC value of 394 ng·h/mL at a dose of 6 mg (Terra et al. 2008). On Day 1, the mean Cmax and AUC0–24 h increased by 68- and 354-fold, respectively, with a 100-fold increase in dose (from 5–500 mg/kg). On Day 36, a 100-fold increase in dose resulted in a 53- and 374-fold increase in mean Cmax and AUC0–24 h, respectively. Systemic exposure on Day 36 was similar to the exposure on Day 1 for the 5 and 500 mg/kg dose groups. Mean Cmax values on Day 14 in the 5 and 500 mg/kg dose groups were 0.90- and 0.69-fold of Day 1 values, respectively. The corresponding mean AUC0–24h were 0.95- and 0.73-fold of Day 1 values, respectively.
Toxicokinetics.
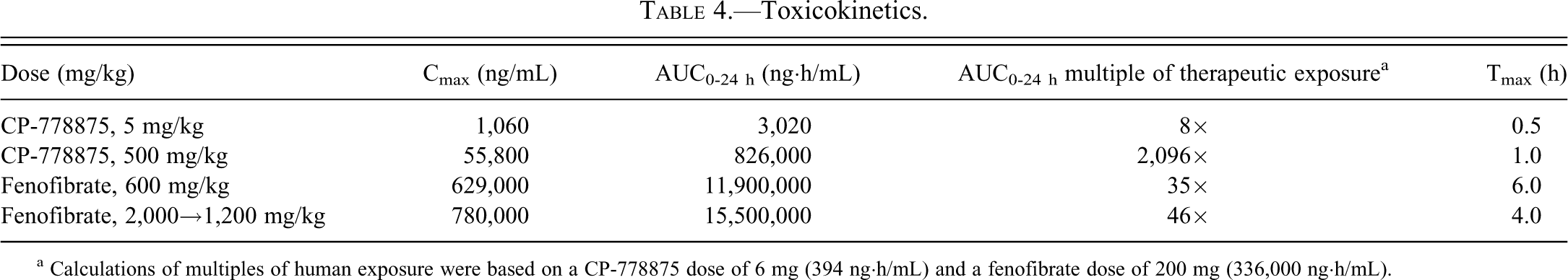
aCalculations of multiples of human exposure were based on a CP-778875 dose of 6 mg (394 ng·h/mL) and a fenofibrate dose of 200 mg (336,000 ng·h/mL).
For fenofibrate, systemic exposure increased slightly with increasing dose (Table 4). AUC0–24 h on Day 36 was 11,900,000 and 15,500,000 ng·h/mL for the respective 600 and 1,200 mg/kg doses, whereas Cmax on Day 36 was 629,000 and 780,000 ng/mL for the respective 600 and 1,200 mg/kg doses. This finding provides AUC0–24 h multiples of 35× (600 mg/kg) and 46× (1,200 mg/kg) as compared with a reported human clinical systemic steady-state exposure AUC value of 336,000 ng·h/mL at a dose of 200 mg (Miller and Spence 1988). On Day 1, the mean Cmax and AUC0–24 h increased by 1.5- and 1.6-fold, respectively, with an approximate 3.3-fold increase in dose (from 600 to 2,000 mg/kg). Similar comparison was not made on Day 36 because the 2,000 mg/kg dose was lowered to 1,200 mg/kg after twenty-one days of dosing at 2,000 mg/kg. Systemic exposure on Day 36 was slightly higher than the exposure on Day 1 for the 600 mg/kg dose group. Both mean Cmax and AUC0–24 h values of Day 36 in the 600 mg/kg dose group were 1.7-fold of Day 1 values.
Discussion
To safely conduct human clinical trials with a potent PPARα agonist, we leveraged the extensive clinical experience with a safe, relatively selective, but weak PPARα agonist (fenofibrate). Therefore, the primary objective of this study was to compare the effects of the highly potent and selective PPARα agonist CP-778875 on peroxisomal β-oxidation and cardiac muscle injury with those induced by a weak PPARα agonist (fenofibrate), which has an extensive safety record. We hypothesized that the cardiac effects of CP-778875 and fenofibrate are mediated through the PPARα receptor, leading to increased cardiac β-oxidation and consequently, oxidative stress (Gilde et al. 2003; Pruimboom-Brees et al. 2006). To test this hypothesis and because of the significant differences between those two compounds in regard to the potency toward the PPARα receptor, the doses of fenofibrate were selected to achieve equivalence of receptor transactivation and subsequent AOX mRNA up-regulation. Based on a pilot study, this dose was estimated to be greater than 3,000 mg/kg, which was a poorly tolerated dose. Therefore, the fenofibrate doses chosen for the present study were a well-tolerated dose (600 mg/kg) and a maximum tolerated dose (2,000→1,200 mg/kg). The doses of CP-778875 were an anticipated NOAEL (5 mg/kg) that was in excess (8×) of the estimated clinically efficacious concentration (Terra et al. 2008) and a maximum tolerated dose (500 mg/kg) that was well in excess of the clinically efficacious concentration (2,096×).
Similar to the results for 500 mg/kg CP-778875, there were dose-related increases in the incidence and/or severity of cardiac myonecrosis/fibrosis (accompanied by elevated serum troponin levels and heart weight) in rats administered 600 or 2,000→1,200 mg/kg fenofibrate. Consistent with a link between these cardiac effects and increased peroxisomal β-oxidation caused by PPARα agonism, the present study also demonstrated an increase in palmitoyl-CoA β-oxidation and AOX mRNA in the heart of rats administered 500 mg/kg CP-778875 and 600 or 2,000→1,200 mg/kg fenofibrate. Taken together, these data strongly suggest that the ligand-dependent activation of PPARα leads to sustained increase in cardiac peroxisomal β-oxidation. Subsequently, cellular oxidative stress (glutathione depletion, and up-regulation of the glutathione system and enzymes that detoxify reactive oxygen species such as glutathione reductase, glucose-6-phosphate dehydrogenase) is likely to result in cellular degeneration and necrosis (Pruimboom-Brees et al. 2006). Additional oxidative stress may also have resulted from mitochondrial dysfunction (fenofibrate inhibits Complex I; Nadanaciva et al. 2007). Thus, two structurally unrelated PPARα agonists caused, at equipotent doses, similar cardiac histopathology, clinical chemistry, and biochemical changes, thereby supporting the hypothesis. It is noteworthy that the large clinical safety database that exists for fenofibrate suggests this finding observed in rats at very high toxicological doses does not occur in humans at clinical doses.
Interestingly, 100 mg/kg fenofibrate has been shown to reduce cardiac hypertrophy, inflammation, and fibrosis in wild-type mice in a chronic pressure overload model (Duhaney et al. 2007). However, in PPARα-deficient mice treated with fenofibrate, there was an increased mortality, adverse changes on ventricular wall dimensions, increased myocardial hypertrophy, and cardiac fibrosis (Duhaney et al. 2007). Thus, in some models fenofibrate exerts a beneficial effect on cardiac remodeling via PPARα activation, whereas it also induces myocardial injury independent of PPARα in another model.
Although CP-778875 500 mg/kg and both doses of fenofibrate resulted in an increase in skeletal muscle degeneration/regeneration, there was no correlative increase in skeletal muscle palmitoyl-CoA β-oxidation, despite an increase in AOX mRNA. The apparent lack of concordance is probably a result of the much greater sensitivity of the mRNA measurements compared with the biochemical assay. This finding may also be compounded by the presence of numerous Type II fibers (energy source from glycolysis) among the Type I fibers (energy source from β-oxidation) in the gastrocnemius muscle used to measure β-oxidation in this study, which would tend to dilute an effect occurring only in the Type I fiber type (i.e., within the soleus). Histologically, we have shown that necrosis only occurs in the Type I fibers, which is consistent with the hypothesis of Pruimboom-Brees et al. (2006), linking increased β-oxidation with muscle injury in both skeletal and cardiac muscle.
In rodent models, PPARα agonists cause skeletal (De Souza et al. 2006; Faiola et al. 2008) and cardiac muscle toxicity. These muscle toxicities are best detected by microscopic examination of tissues; however, assessment of biomarkers such as troponin I (cardiac toxicity) and creatinine kinase (skeletal muscle toxicity) are also important, as they are commonly used for detecting muscle injury in humans. A limitation in the use of troponin in human clinical trials is that elevated troponin does not predict active, irreversible myocardial cell injury (Wallace et al. 2004). Thus elevations of troponin I indicate that lethal cardiac cell injury has already occurred. A goal of phase 1 human clinical trials is to safely identify the maximum tolerated dose. With potential cardiotoxic drug candidates, this is a difficult goal to achieve, as it is unacceptable to dose-escalate until cardiac toxicity is observed.
Translation of PPARα agonist cardiac and skeletal muscle effects noted in rats to human risk assessment is not straight forward. Hepatic effects of PPARα treatment have been extensively studied; however, responses in skeletal (De Souza et al. 2006) and cardiac muscle are less understood. In humans, fibrates have been implicated in the occurrence of myalgias (Bannwarth 2002; Hodel 2002; Roca et al. 2002). In rodents, administration of PPARα agonists induces hepatic peroxisome proliferation, but primates are relatively refractory to these effects (Cariello et al. 2005; Klaunig et al. 2003). Human patients taking fibrates at clinically relevant doses for eight weeks did not have elevated hepatic AOX mRNA (Roglans et al. 2002). Rodent PPARα expression has been estimated to be ten-fold greater than that in humans on both the mRNA and protein levels (Palmer et al. 1998; Tugwood et al. 1998). The percentage of functional human PPARα is expected to be lower than that in the rat because of the expression of inactive human PPARα splice variants (Palmer et al. 1998). In addition, in vitro studies using gain-of-function PPARα and small-molecule PPARα agonist demonstrate a significant, species-specific regulation of the AOX promoter by PPARα. The cynomolgus and human AOX promoter, which contrast radically from the rat AOX promoter, are not acutely induced by PPARα (Kane et al. 2006), which casts doubt on the predictive ability and physiological relevance of rodents for the assessment of PPAR-related safety monitoring.
Footnotes
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article. The authors received no financial support for the research, authorship, and/or publication of this article.