
Abstract
A number of therapeutic immunomodulatory biologics, including antibodies, fusion proteins, and recombinant proteins, have been causally linked with serious adverse effects in humans. In nearly all cases, these serious adverse effects have been directly associated with the immunomodulatory biologic’s intended pharmacologic activity or exaggerated pharmacology. Examples of immunomodulatory biologics known to cause serious adverse effects in the clinic ranging from immunostimulation and cytokine release syndrome (e.g., TGN1412) to immunosuppression with increased risk of opportunistic infections (e.g., TNF-α antagonists, anti-integrins) are presented. Specific examples of the nonclinical testing strategy used for the clinical risk assessment of these immunomodulatory biologics are discussed, with an emphasis on the clinical relevance and predictivity of the models. Infectious challenge animal models, in particular, were critically evaluated for their utility in evaluating clinical risk assessment versus understanding mechanism of action. The nonclinical safety testing strategy for an immunomodulatory biologic should be custom tailored to interrogate the biology of the immunologic target in order to best assess potential clinical risk. This nonclinical strategy should include mechanistic and efficacy models of pharmacologic activity and immunologic signaling pathways, in vitro immunologic assays such as cytokine release, and immunophenotypic assessment by flow cytometry, immunohistochemistry, and/or immunofluorescence, as appropriate.
Introduction
A number of therapeutic immunomodulatory biologics, including antibodies, fusion proteins, and recombinant proteins, have been causally linked with serious adverse effects in humans. In nearly all cases, these serious adverse effects have been directly associated with the immunomodulatory biologic’s intended pharmacologic activity or exaggerated pharmacology (Table 1). Immunomodulatory biologics can bind to a variety of immunologic targets, including immune cells, such as T and B lymphocytes or antigen-presenting cells, and soluble immune mediators such as cytokines or chemokines. Their modes of action (MOAs) encompass cell depletion, suppression of immune function, prevention of leukocyte homing or trafficking, neutralization of cytokines and chemokines, and immune stimulation. Immunomodulatory biologics have been approved for a wide range of disease indications, including rheumatoid and psoriatic arthritis, inflammatory bowel disease (IBD, both ulcerative colitis and Crohn’s disease), multiple sclerosis, psoriasis, ankylosing spondylitis, and transplant rejection. Because immunomodulatory biologics have a wide range of clinical indications, immunologic targets, and MOAs, the nonclinical safety evaluation for each biologic should be tailored to interrogate its specific MOA and the interaction of the biologic with its target, in addition to consideration of the more routine immunotoxicology evaluations, such as immunophenotyping of peripheral blood lymphocytes and evaluation of the T lymphocyte–dependent antibody response (Brennan et al. 2010; Descotes 2009; Weir 2008). Some of these specific approaches for nonclinical assessment of clinical risk for immunomodulatory biologics will be discussed in greater detail here.
Adverse clinical events associated with immunomodulatory biologics.

b Orthoclone (muromonab-CD3) package insert, 2010.
eTysabri (natalizumab) Web site.
fGenentech, Inc. Press release, April 8, 2009.
This review will provide a historical overview of the serious adverse effects related to immunomodulatory biologics, from the identification of the clinical risk of opportunistic infections with TNF-α inhibitors to the cytokine release syndrome elicited by the CD28 agonist TGN1412 and the emergence of progressive multifocal leukoencephalopathy (PML) in patients treated with the anti-α4 integrin, natalizumab. Specific examples of the nonclinical testing strategy used for the clinical risk assessment of several immunomodulatory biologics are discussed in detail. The nonclinical models and assay systems used to demonstrate mechanism of action, efficacy, and toxicity are presented for each biologic, with an emphasis on the clinical relevance and predictivity of the models. The nonclinical approaches taken are evaluated for several specific biologic examples, and different options and/or approaches are discussed. Infectious challenge animal models, in particular, were critically evaluated and found to demonstrate poor correlation between the degree of compromise demonstrated in the model and the incidence or severity of adverse events seen in the clinic. The poor predictivity of these infectious challenge models for immunomodulatory biologics highlights the challenges of clinical risk assessment for these molecules and emphasizes the need for a wide range of nonclinical studies, including mechanistic studies using specialized techniques, to fully evaluate the effect on immune function and inform potential clinical risk.
Therefore, it is recommended that the nonclinical testing strategy for an immunomodulatory biologic should be custom tailored to the immunologic target biology and the mechanism of action of the biologic. This approach characterizes the liabilities and benefits—the yin and yang—of target modulation, and thus, best assesses the potential clinical risk. This nonclinical strategy should consider both in vitro and in vivo models and use mechanistic and efficacy models of pharmacologic activity and immunologic signaling pathways in addition to traditional immunotoxicology evaluations.
TNF-α Inhibitors and Increased Risk of Opportunistic Infections
TNF-α inhibitors represent one of the first groups of biologic immunomodulators to target a specific immune effector molecule. This group includes the TNFR2-Fc Enbrel (etanercept) and the anti-TNF-α monoclonal antibodies (MAbs) Humira (adalimumab), Remicade (infliximab), Simponi (golimumab), and Cimzia (certolizumab pegol). As evidenced by the relatively large number of approved TNF-α inhibitors, this class has proven to be clinically effective, and targeting TNF-α is a very popular immunomodulatory strategy. Despite their popularity and clinical effectiveness across a number of indications, including rheumatoid and psoriatic arthritis, plaque psoriasis, and IBD, the entire class of TNF-α inhibitors carries an increased clinical risk of tuberculosis (TB) that has been recognized for over a decade (package inserts for Enbrel [etanercept], Humira [adalimumab], Remicade [infliximab], Simponi [golimumab], and Cimzia [certolizumab pegol]). A 2004 review cites the incidence of Mycobacterium tuberculosis infection at 143.8/100,000 in United States patients for infliximab, and 34.5/100,000 for etanercept versus a background infection incidence of 5.6/100,000 (Wallis et al. 2004). Similarly, treatment with both infliximab and etanercept has also been associated with an increased risk of granulomatous infection with a number of other organisms, including Histoplasma capsulatum, Candida spp., and Listeria spp. As a result of the increased risk of opportunistic granulomatous infections, all TNF-α inhibitors carry label statements regarding the risk of developing TB as well as the risk of developing opportunistic fungal infections (package inserts for Enbrel [etanercept], Humira [adalimumab], Remicade [infliximab], Simponi [golimumab], and Cimzia [certolizumab pegol]. There is also an increased risk of demyelinating disease with TNF-α inhibitors that is unrelated to the risk of opportunitic infections; however, this risk is small (Bernatsky et al. 2010).
Both anti-mouse TNF-α and mouse TNFR2-Fc induced a dose-dependent decrease in survival of mice with acute and chronic M. tuberculosis infections in a mouse model of TB infection. However, both molecules had minimal or no effect on bacterial burden in the lung (Plessner et al. 2007). Although animal models of host resistance for TB have been useful for identifying the clinical hazard of increased TB infection with TNF-α inhibitors, the lack of clinically relevant positive controls, particularly immunomodulatory biologic controls, by which the degree of compromise in these models can be gauged, has thus far made infectious challenge models poorly predictive of the clinical risk of opportunistic infection for other biologics.
TGN1412 and Cytokine Release Syndrome
TGN1412 is an anti-CD28 IgG4 agonistic antibody developed by TeGenero AG of Würzburg, Germany, that directly activates human T lymphocytes by binding to the CD28 stimulatory co-receptor without cross-linking the T lymphocyte receptor (Bhogal and Combes 2006). A phase 1 clinical trial for TGN1412 was initiated in March 2006 in London using normal healthy male volunteers aged twenty-one to thirty-five. Six volunteers received a single IV infusion of 0.1 mg/kg TGN1412, and two received placebo. All infusions were given ten minutes apart. Within ninety minutes after dosing was initiated, all six subjects receiving TGN1412 became severely ill and had to be admitted to the ICU. The subjects all developed profound angioedema and multiple organ system failure, with several subjects requiring dialysis. All volunteers survived, but one had to have his fingers and toes amputated. Subsequently, this acute illness following TGN1412 dosing was identified as cytokine release syndrome, as increased levels of TNF-α, IFN-γ, and IL-2, -6, and -10 were detected in treated volunteers (Suntharalingam et al. 2006).
The original in vitro studies conducted by TeGenero using human peripheral blood mononuclear cells (PBMC) co-cultured with TGN1412 in an aqueous solution assay did demonstrate polyclonal T cell expansion accompanied by the secretion of “anti-inflammatory cytokines, most notably of IL-10” (TGN1412 Investigator’s Brochure). The specific in vitro cytokine release assay data were redacted from the Investigator’s Brochure, but the conclusion was that cytokine release was not identified as a serious clinical risk (TGN1412 Investigator’s Brochure).
The cynomolgus monkey was chosen as the animal model for toxicity evaluation because TGN1412 binds with high affinity to a six-amino-acid epitope on an extracellular loop of the CD28 molecule that is 100% homologous between cynomolgus monkeys and humans. In addition, the expression levels of CD28 molecules and T cell subset ratios were felt to be comparable between macaque and human T cells (Bhogal and Combes 2007; TGN1412 Investigator’s Brochure). In the FIH-enabling toxicity study, weekly doses of 0, 5, or 50 mg/kg were administered IV to cynomolgus monkeys for four weeks. Notable findings included an expanded T lymphocyte population; increased size of lymph nodes; and transient, moderate (three- to eighteen-fold), dose-dependent elevations in IL-2, IL-5, and IL-6, consistent with expected pharmacology of TGN1412. Overall, the cynomolgus monkeys tolerated TGN1412 very well, and the top toxicity study dose of 50 mg/kg was the no observed adverse effects level (NOAEL) (TGN1412 Investigator’s Brochure; Bhogal and Combes 2007).
So why then were the nonclinical studies, including the toxicity study in cynomolgus monkeys, not predictive of the cytokine release syndrome seen in the Phase 1 clinical trial?
It is now recognized that nonhuman primates are very poor predictors of lymphocyte stimulation, proliferation, and cytokine release in humans. Although cultured cynomolgus PBMCs show binding and a limited response to immobilized TGN1412, these cells do not show the strong activation that human T cells demonstrated following TGN1412 binding, supporting the hypothesis that there are key differences in the response of human and nonhuman primate lymphocytes to TGN1412 via CD28 signaling (Stebbings et al. 2007; Waibler et al. 2008). Early hypotheses included altered signal transduction in nonhuman primate CD28 compared to human CD28, possibly caused by slight differences in the amino acid sequence in the CD28 transmembrane region (Bhogal and Combes 2007; Waibler et al. 2008), and hyperresponsiveness of human T cells to polyclonal activation caused by the lack of the CD33-related siglec inhibitory signaling molecule that is present in nonhuman primates (Nguyen et al. 2006). The most plausible explanation to date, however, is that CD4+ effector memory T lymphocytes, which have been identified as the primary effector cells responsible for the cytokine release syndrome seen following TGN1412 administration, lack CD28 expression in macaques such as cynomolgus monkeys, and thus would have been unable to respond to TGN1412 (Eastwood et al. 2010).
In addition to the poor predictivity of the toxicity study in the cynomolgus monkey model for TGN1412’s cytokine release syndrome, the in vitro cytokine release assay used by TeGenero may not have been sensitive enough to accurately predict in vivo cytokine release (Stebbings et al. 2007). Currently, it is recommended that multiple in vitro assay formats should be evaluated for cytokine release assays, including immobilized MAb versus MAb in solution, PBMCs versus whole blood, inclusion of endothelial cells, and so on (Bugelski et al. 2009; Findlay et al. 2010). Additionally, use of appropriate positive controls (e.g., CD28 agonists, anti-CD3 such as OKT3), PBMCs and/or whole blood from at least three individuals, and evaluation of the biologic at a minimum of three different concentrations are considered best practice. Ultimately, the MOA of the MAb/biologic and in vivo model system(s) should drive the choice of the appropriate cytokine release assay format(s) (Bugelski et al. 2009; Findlay et al. 2010).
Regulatory authorities, particularly the European Medicines Agency and the Medicines and Healthcare Products Regulatory Agency (UK) have now instituted the requirement to evaluate/consider using the minimally anticipated biological effect level (MABEL) in selecting the first in human (FIH) dose for potential high-risk medicinal products as a result of the TGN1412 incident. Data used to determine MABEL should consider all available in vitro and in vivo data such as receptor occupancy and pharmacologic/PD dose response. The use of MABEL to determine FIH dose should not be the default; the decision on whether to use MABEL, pharmacologically active dose, or NOAEL should be MOA and data-driven. Additionally, the totality of the nonclinical pharmacology and safety data should be used to determine whether or not a biologic is considered high risk. Since 2007, when the concept of MABEL was introduced, we have been successful in using NOAEL or pharmacologically active dose, rather than MABEL, in order to select FIH starting doses for immunomodulatory biologics that were not considered to be high-risk, so long as the decision has been data driven and could be scientifically justified.
Anti-Integrins and PML
Integrins are heterodimeric adhesion molecules composed of α and β subunits that mediate a wide variety of cell–cell and cell–matrix interactions that are involved in growth and development, wound healing, hemostasis, and vascular transmigration of leukocytes at sites of inflammation (Arnaout et al. 2005). Homing of leukocytes during immune surveillance is mediated by the interaction of specific α4 integrins on leukocytes with endothelial cells at sites of inflammation. The interaction of α4β1 with VCAM-1 mediates systemic/peripheral homing, which provides immunity to systemic antigens, whereas the interaction of α4β7 with MAdCAM-1 mediates mucosal homing, which is responsible for immunity to mucosal antigens (Cheroutre and Madakamutil 2005). Tysabri (natalizumab) is an IgG4 MAb that binds to the α4-subunit of α4β1 and α4β7 integrins, thereby blocking their binding to VCAM-1 and MAdCAM-1, respectively. Tysabri was approved in November 2004 for relapsing multiple sclerosis (MS); however, marketing was suspended and clinical trials halted in February 2005 because of the occurrence of PML in two patients receiving Tysabri in combination with Avonex (interferon β-1a). Progressive multifocal leukoencephalopathy is caused by reactivation of latent JC polyoma virus infection, although the exact mechanism of viral reactivation and spread is unknown. An additional case of PML was diagnosed retrospectively in a patient in an open-label Crohn’s disease clinical trial who died in 2003. In June 2006, the FDA approved an application for resumed marketing of Tysabri indicated as monotherapy for patients with relapsing forms of MS with a black box warning on PML; in January 2008, Tysabri was approved for patients with moderate to severe Crohn’s Disease (FDA:Post-market drug safety information on Natalizumab [marketed as Tysabri] Web site; Tysabri [natalizumab] Web site).
There have been 124 cases of PML, with twenty-three deaths worldwide, in patients treated with Tysabri as of May 2011. These data indicate a strong trend of increased incidence of PML with increasing number of Tysabri infusions. For patients treated with Tysabri between one and two years, the incidence of PML is 0.49 per 1,000, whereas in patients receiving Tysabri for two years or longer, the incidence of PML increases sharply to 2.76 per 1,000 (Multiple Sclerosis Resources Centre Tysabri Web site). Another anti-integrin, Raptiva (efalizumab), is an anti-integrin that was indicated for the treatment of moderate to severe plaque psoriasis. Although Raptiva targets a different integrin, αL/LFA-1/CD11a, than Tysabri does, the αL/LFA-1/CD11a integrin is broadly expressed by all leukocytes, as is the α4β1 integrin targeted by Tysabri. Raptiva was voluntarily withdrawn from the United States market in 2009 because of an increased risk of PML (Genentech, Inc. Press release, April 8, 2009). Although the exact pathogenesis of PML occurrence in patients treated with anti-integrins is unclear, one hypothesis is that inhibition of systemic lymphocyte homing allows reactivated JC virus to reach the central nervous system (CNS) and infect oligodendrocytes (Tyler 2008).
rhuMAb Beta7: An Anti-Integrin More Specifically Targeted to Mitigate Adverse Effects
rhuMAb Beta7 specifically binds to the β7 integrin and blocks the interaction of α4β7 integrin on circulating leukocytes with MAdCAM-1 on HEV in mucosal tissues, such as the gastrointestinal (GI) tract, and the interaction of αEβ7 integrin on intraepithelial lymphocytes with E-cadherin on epithelial cells, such as those in the GI tract epithelium (Cheroutre and Madakamutil 2004). It is currently being developed for treatment of IBD (ulcerative colitis and Crohn’s disease). Because of the occurrence of PML in patients treated with the anti-α4 integrin Tysabri, options that were considered for the risk assessment of PML with rhuMAb Beta7 included animal models of latent virus reactivation and mechanistic studies to differentiate the MOA of rhuMAb Beta7 from that of anti-integrins that block the α4 integrin. Several animal models of latent virus reactivation were considered. In the mouse papovavirus K (related to JC virus) model, papovavirus K demonstrates latency after eight months, with the kidney a major site of viral latency (similar to JC virus). The K virus is reactivated and can be detected in tissues following immunosuppression with potent immunosuppressive drugs such as cyclophosphamide, or by complete ablation of T lymphocytes with anti-CD3 or a combination of anti-CD4 and CD8 (Greenlee and Dodd 1984; Greenlee et al. 1991). As this model requires potent immunosuppression to reactivate the latent virus and lacks an appropriate immunomodulatory biologic as a positive control, this and other animal models of latent virus reactivation were rejected because of their lack of clinical predictivity.
An alternative strategy to assess the risk of rhuMAb Beta7, and to differentiate it from anti-α4 integrins, was to comprehensively interrogate rhuMAb Beta7’s MOA. These investigations convincingly demonstrated that rhuMAb Beta7 specifically affected mucosal homing, but not systemic homing, of leukocytes in several different experimental model systems. First, in the CD45RBhigh-reconstituted severe combined immunodeficiency (SCID) mouse model of IBD, rhuMAb Beta7 was able to block radiolabeled lymphocyte homing to the inflamed colon but had no effect on lymphocyte homing to the spleen, a peripheral lymphoid organ (Figure 1; Stefanich et al. 2011).
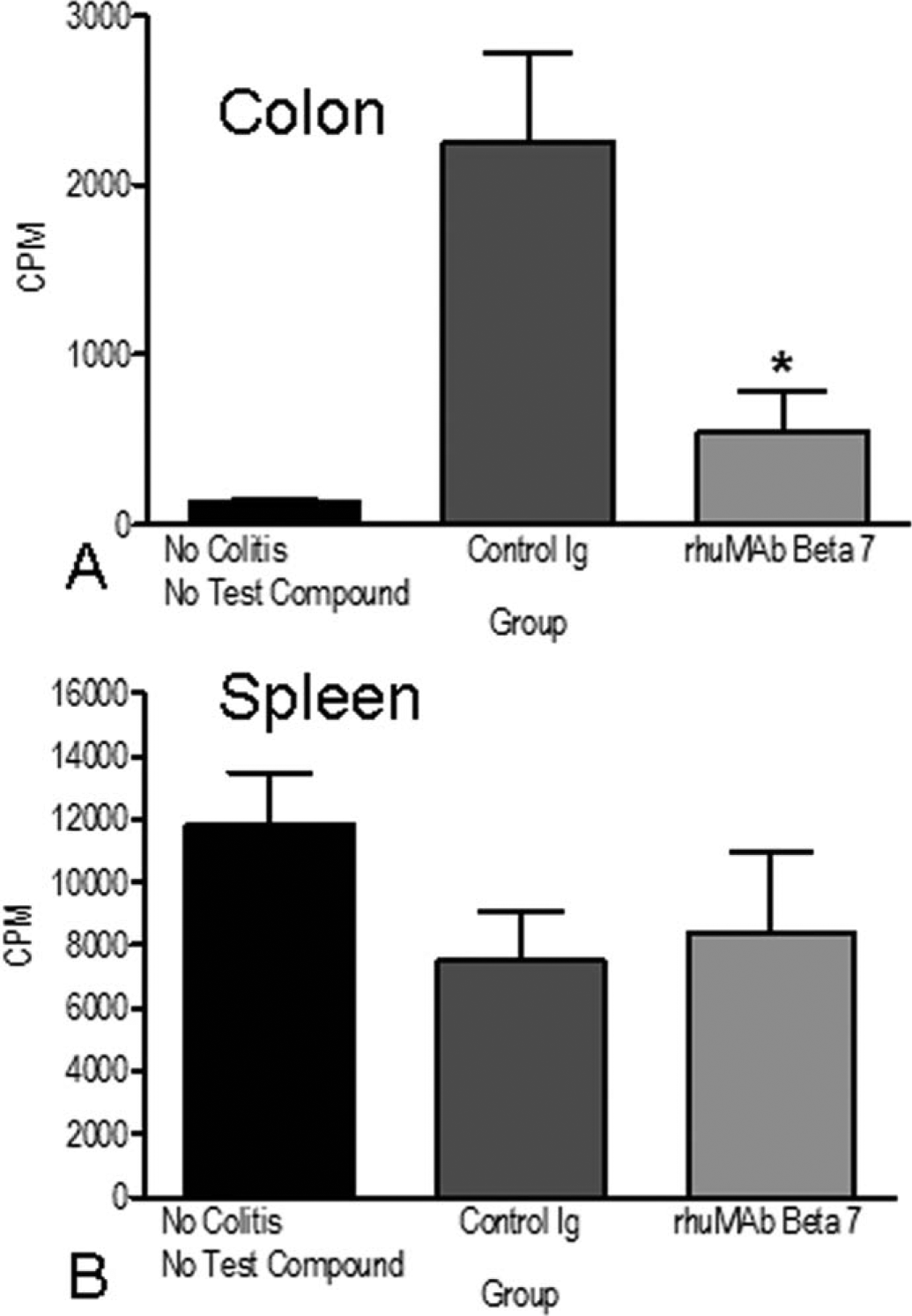
In vivo homing of lymphocytes to the inflamed colon of CD45RBhighreconstituted severe combined immunodeficiency mice. Severe combined immunodeficiency mice were reconstituted with Cr51-labeled CD45RBhigh CD4∔ T cells. In this model of colitis, rhuMAb Beta7 significantly blocked lymphocyte recruitment and homing to the inflamed colon (panel A; as measured by gamma count of harvested organs) when compared to a control Ig (control IgG vs rhuMAb Beta7, p <.001), but had no effect on lymphocyte homing to the spleen, a peripheral lymphoid organ (panel B). Untreated mice without colitis had essentially no homing to the colon; nearly all labeled lymphocytes trafficked to the spleen. Values shown are group means ± standard deviation counts per minute. Asterisk indicates p < .05. Reprinted from Br J Pharmacol (2011),
A second study in cynomolgus monkeys demonstrated that rhuMAb Beta7 specifically increased peripheral blood β7Hi CD45RA- “gut homing memory” CD4+ T lymphocytes but had no effect on β7Low CD45RA- “peripheral homing memory” CD4+ T lymphocytes or β7Low-Intermediate CD45RA+ naïve CD4+ T lymphocytes (Figure 2; Stefanich et al. 2011). The observed increase in β7Hi expressing CD45RA- “gut homing memory” CD4+ T lymphocytes in peripheral blood strongly suggested that rhuMAb Beta7 was blocking the homing of this lymphocyte subset to mucosal tissues but did not affect lymphocyte trafficking to peripheral lymphoid tissues. This hypothesis was confirmed by histologic analysis of GI tract tissues from twenty-six-week toxicity studies in both mice and cynomolgus monkeys. In these studies, rhuMAb Beta7 induced a dose-dependent reversible decrease in lymphocytes limited to the GI-associated lymphoid tissue (GALT) in cynomolgus monkeys and in the intestinal lamina propria in the mouse (Figure 3). Despite the decrease in intestinal lymphocytes evident in both species, there was no evidence of GI tract infection or of any of other adverse finding in either species, indicating that rhuMAb Beta7’s observed effect on the GI tract was pharmacologic but not associated with any toxicity.
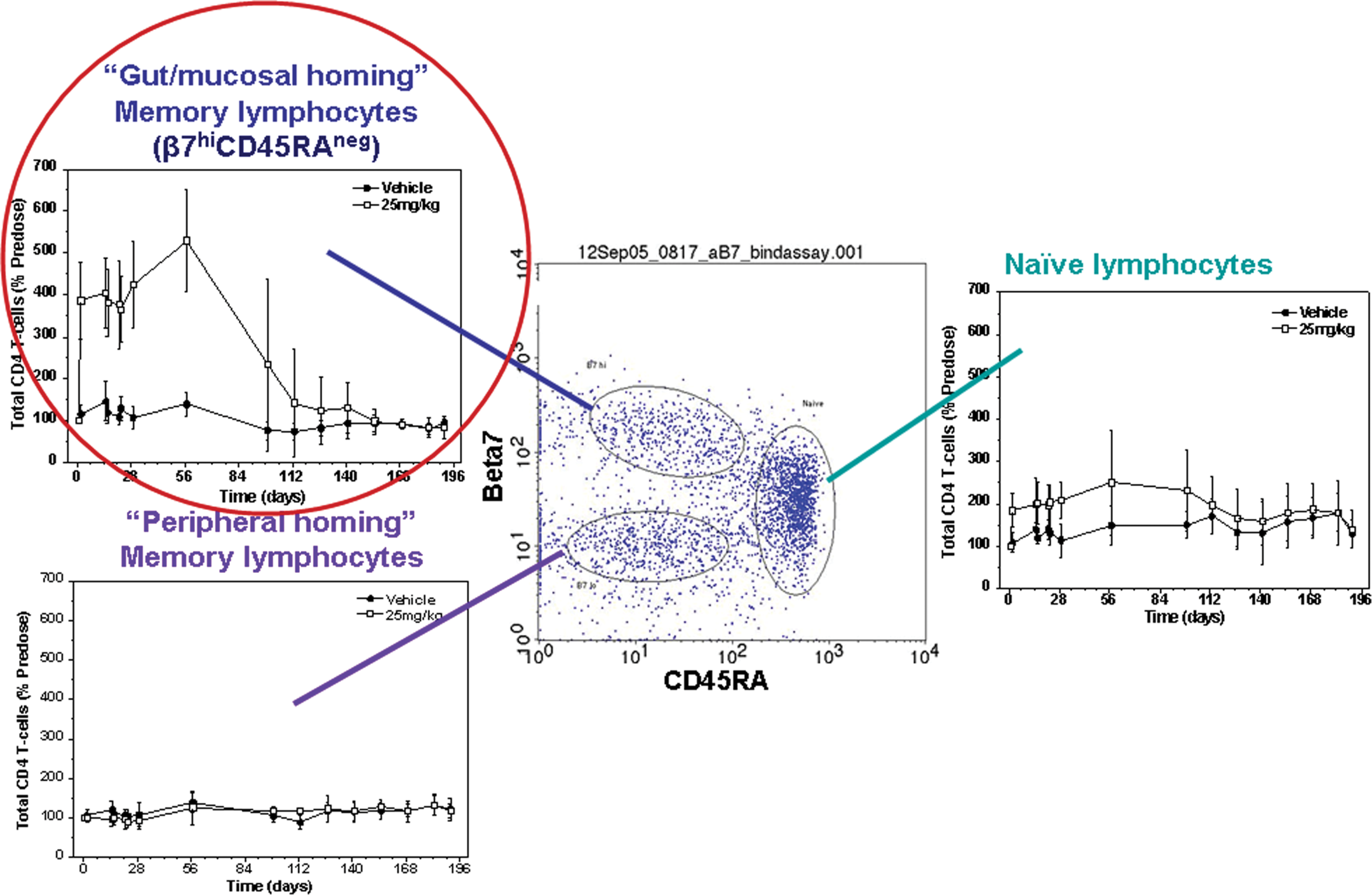
Pharmacodynamic biomarker fluorescence activated cell sorting (FACS) analysis of cynomolgus monkey peripheral blood following rhuMAb Beta7 treatment. The center FACS scatter dot plot illustrates three subsets of peripheral blood CD4+ lymphocytes, subdivided according to their homing properties, that were monitored by flow cytometry as rhuMAb Beta7 PD biomarkers: CD45RA−β7.
high(“gut/mucosal homing memory”), CD45RA−β7.
low(“peripheral homing memory”), and CD45RA+β7.
intermediate (“naïve”) CD4+ cells. This FACS scatter plot has CD45RA expression on the x axis and β7 expression on the y axis, and is gated on CD4+ cells. This scatter dot plot is representative of the staining pattern seen in untreated cynomolgus monkeys. The upper left plot (circled in red) illustrates a marked increase (four- to five-fold) in absolute peripheral blood CD45RA−β7.
highCD4+(“gut/mucosal homing memory”) T cell counts following administration of rhuMAb Beta7 versus vehicle to cynomolgus monkeys. In contrast, the upper right and lower left plots illustrate essentially no changes in absolute peripheral blood CD45RA+β7.
intermediateCD4+ (“naïve”) T cell counts and absolute CD45RA−β7.
lowCD4+ (“peripheral homing memory”) T cell counts, respectively, following administration of rhuMAb Beta7 versus vehicle to cynomolgus monkeys. This CD4+ T cell FACS analysis illustrates that rhuMAb Beta7 acts specifically on the CD45RA−β7.
highCD4+(“gut/mucosal homing memory”) T cell subset in peripheral blood by the presumed mechanism of blocking homing of this subset to gut/mucosal tissues. All absolute T cell subset counts are shown as a percentage of predose baseline. Reprinted from Br J Pharmacol (2011),
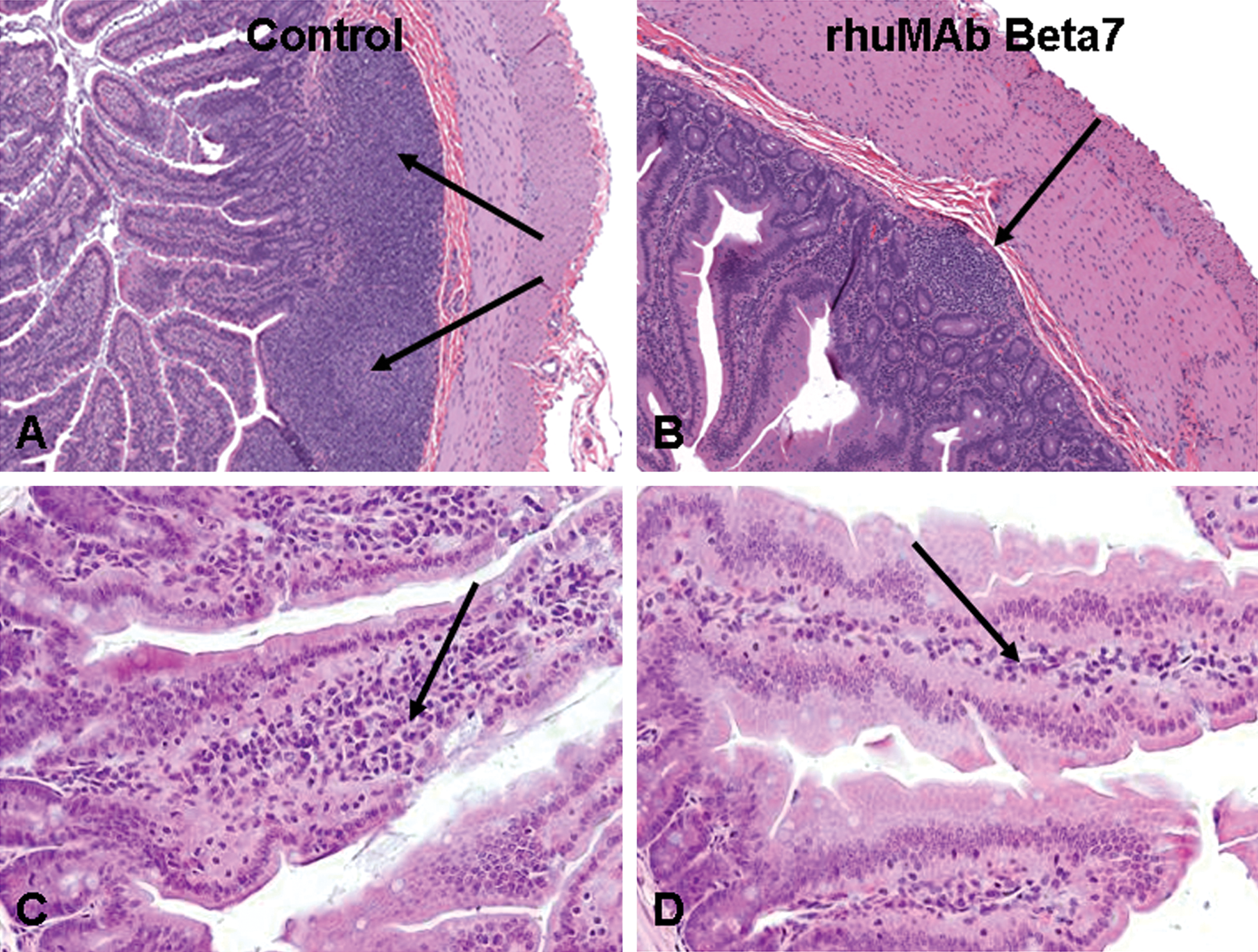
rhuMAb Beta7 decreases lymphocytes in the GI tract of cynomolgus monkeys and CD1 mice following twenty-six weeks of treatment. Cynomolgus monkeys treated with rhuMAb Beta7 weekly for twenty-six weeks exhibited a decrease in lymphocytes within their gastrointestinal lymphoid tissue (arrow in panel B) compared to control monkeys (arrows in panel A). Sections of ileum are illustrated. Similarly, CD1 mice treated with rhuMAb Beta7 twice weekly for twenty-six weeks exhibited a decrease in lymphocytes within their small intestinal lamina propria (arrow in panel D) compared to control monkeys (arrows in panel C). Sections of jejunum are illustrated. This decrease in lymphocytes was reversible following a twenty-six-week (cynomolgus monkey) or twenty-week (mouse) treatment-free recovery period, and was not accompanied by any other pathologic findings in the gastrointestinal tract or elsewhere.
Finally, in the myelin basic protein T-cell receptor transgenic mouse model of EAE, mouse anti-β7 had no effect on disease severity via several different assessment measures, including improvement of survival and leukocytic infiltration into the CNS by histologic evaluation. In contrast, mouse anti-α4 completely prevented EAE-induced mortality and significantly decreased disease severity, including a significant decrease in leukocytic infiltration into the CNS (Figure 4; Stefanich et al. 2011).
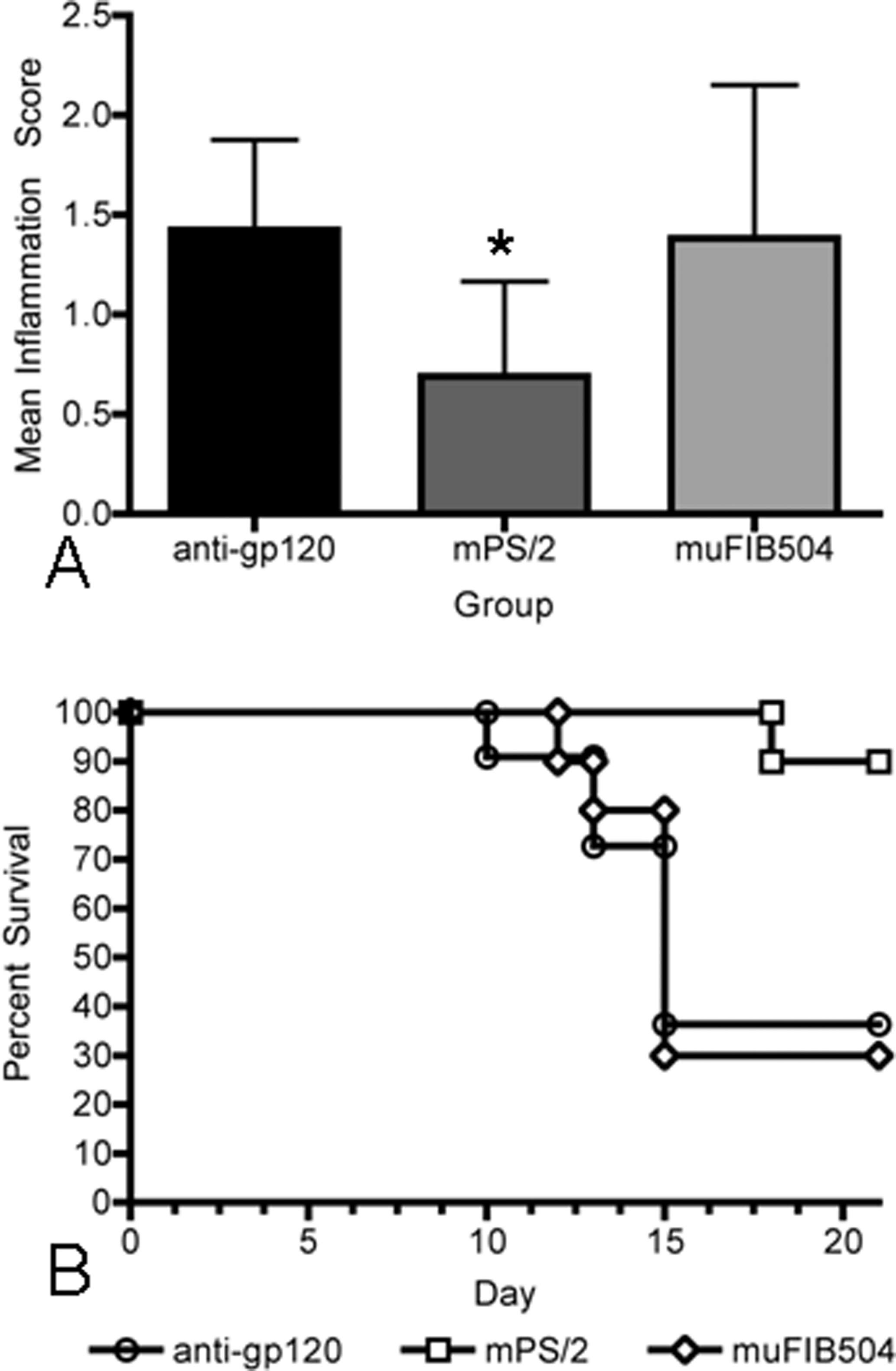
Anti-Beta7 in mouse myelin basic protein T-cell receptor (MBP-TCR) transgenic model of EAE. (A) Mean central nervous system (CNS) histologic inflammation scores ± standard deviation in brain and spinal cord of MBP-TCR mice with EAE. Anti-mouse α4 MAb (mPS/2) significantly reduced mean CNS inflammation, as assessed by histologic scores (asterisk indicates p = .009) compared to control anti-gp120, whereas anti-mouse β7 (muFIB504) had no effect on histologic inflammation in the CNS compared to control anti-gp120. (B) Percent survival of MBR-TCR transgenic mice with severe EAE. Anti-mouse α4 MAb (PS/2) significantly improved survival from EAE compared to control anti-gp120, whereas anti-mouse β7 (muFIB504) had no effect on EAE survival compared to control anti-gp120. The difference between these survival curves is significant (log rank test, p < .001). Anti-gp120 = control mouse IgG1 antibody (circles); mPS/2 = anti-mouse α4 (squares); muFIB504 = anti-mouse β7 (rat-mouse chimeric antibody, diamonds). n = 10 mice per group. Reprinted with modifications from Br J Pharmacol (2011),
These MOA studies and the study directly comparing the effects of anti-β7 to those of anti-α4 convincingly demonstrate that rhuMAb Beta7 has a very different effect on leukocyte homing than do anti-α4 integrins, namely, that rhuMAb Beta7 specifically targets mucosal homing of leukocytes but does not affect systemic homing, including trafficking to the CNS. The results from these studies suggest that rhuMAb Beta7’s risk of inducing PML should be lower than that of anti-integrins such as Tysabri and Raptiva, which target broad leukocyte populations involved in systemic homing and trafficking. Thus, studies designed to evaluate specific MOA may be more relevant than infectious challenge models in overall clinical risk assessment, particularly since there are currently no appropriate infectious challenge models that are predictive of clinical risk of PML for immunomodulatory agents such as rhuMAb Beta7.
Risk Assessment of BAFF Blockers
BAFF is a member of the TNF superfamily and binds to three different members of the TNFR family that are expressed on B lymphocytes: TACI, BR3, and BCMA. TACI and BCMA also bind to another TNF superfamily ligand, APRIL (Wu et al. 2000; Thompson et al 2001). TACI is an inhibitory receptor; TACI -/- mice develop B cell proliferation and autoimmunity (Seshasayee et al 2003). As a B-cell inhibitor, TACI-Ig has the theoretical risk of increasing opportunistic infections. TACI-Ig’s risk of opportunistic infection was evaluated in an acute mouse influenza model. In this model, TACI-Ig induced the expected decrease in B cells, but not T cells, in peripheral blood by immunophenotyping, as well as a dose-dependent decrease in spleen weight. In addition, TACI-Ig induced a dose-dependent decrease in influenza-specific IgG and IgM in both lung and serum; however, TACI-Ig did not affect viral clearance or survival in this influenza model (Roque et al. 2006).
There is no mechanism-based reason to suspect that blocking TACI, a B cell receptor, would have any effects in an influenza model, which is a T lymphocyte–dependent viral resistance model. Additionally, there was no clinically relevant biologic positive control for this model. Based on MOA and lack of adequate controls, the use of this host resistance model for TACI-Ig was unnecessary and resulted in data that were difficult to interpret. In contrast, risk assessment of a similar molecule, BR3-Fc, was approached differently, using detailed B lymphocyte subset immunophenotyping by flow cytometry, IHC, and IF, as well as functional assays.
Like TACI, BR3 is a member of the TNF superfamily, but unlike TACI, BR3 binds only to BAFF, and the interaction of BAFF with BR3 is a key factor for B-cell survival. BR3 -/- and BAFF -/- mice lack mature B cells, findings consistent with BAFF and BR3 being key B-cell survival factors (Gross et al 2001; Yan et al. 2001). In normal mice, BR3-Fc causes a marked decrease in splenic follicular B cells (Lin et al. 2007). In a NZB/W (F1) mouse model of spontaneous lupus nephritis, BR3-Fc ameliorated disease severity as assessed by improvement in survival, anti-dsDNA antibody titer, proteinuria, and glomerulonephritis as evaluated by renal histopathology (Kayagaki et al. 2002).
In order to investigate the MOA and evaluate target liability of BR3-Fc, a BR3-Fc pilot safety and proof of activity study in cynomolgus monkeys was designed to assess the effects of BR3-Fc on B-cell subsets and the functional recall response to both T lymphocyte–dependent (tetanus and KLH) and T lymphocyte–independent (Pneumovax 23) vaccinations. In this study, using CD21-CD27 dual-label FACS on CD20+ lymphocytes to differentiate memory and naïve B-cells (Vugmeyster et al. 2004), BR3-Fc induced a statistically significant decrease in both naïve and memory B lymphocytes in cynomolgus monkey lymphoid tissues following eighteen weeks of treatment (Vugmeyster et al. 2006). BR3 was shown to have greater expression on CD21Hi B cells (marginal zone B cells) in lymphoid tissues, and BR3-Fc induced the greatest decrease in this tissue subpopulation of B-cells (Figure 5; Vugmeyster et al. 2006).
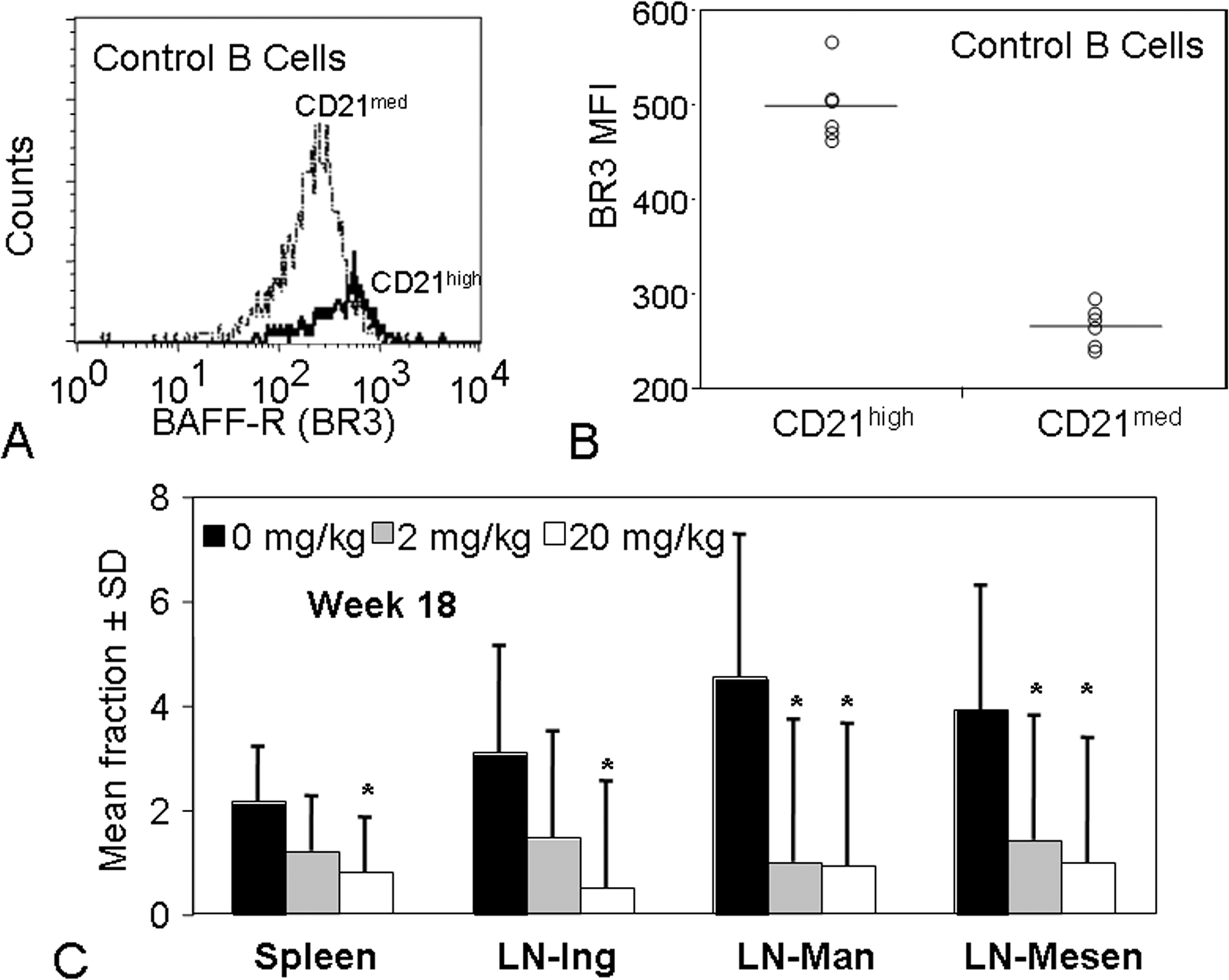
Higher BR3 expression in CD21high (marginal zone) B cells and BR3-Fc-mediated targeting of these CD21high B cells in the spleen and lymph nodes of cynomolgus monkeys. Cynomolgus monkeys were treated weekly with 0, 2, or 20 mg/kg of BR3-Fc for eighteen weeks. (A and B) Spleen samples from animals in the 0 mg/kg control group were stained with antibodies to CD20, CD21, and BR3 and assessed for expression of BR3. Panel (A) illustrates representative FACS expression of BR3 for B cells expressing CD21med (dotted line) or CD21high (bold, solid line). Panel (B) is a summary of BR3 mean fluorescence intensity (MFI) for the two different B cell subsets. Lines correspond to the average BR3 MFI for each subset (CD21med and CD21high) and demonstrate that the CD21high B cells have a higher level of BR3 expression than do CD21med B cells. (C) The CD21high (marginal zone) B cell fraction of lymphocytes was assessed by flow cytometry at eighteen weeks after the first dose, as described in the text. The mean CD21high B cell fraction ± SD is shown. LN-ing, LN-man, LN-mesen are inguinal, mandibular, and mesenteric lymph nodes, respectively. Asterisks denote decreases that are statistically different from control (p < .05). Reprinted from Am J Pathol (2006)
Dual-label IHC and IF staining can be used to differentiate B cell subsets in the normal cynomolgus monkey splenic lymphoid follicles. Dual-label CD20/SMA IHC delineates B lymphocytes in the OMZ (OMZ B cells reside outside the SMA+ band of myofibroblasts that encircle the lymphoid follicle), whereas dual-label IgD/F-Actin IF delineates the B lymphocytes in the mantle zone and IMZ (Figure 6; Vugmeyster et al. 2006). Dual-label CD20/SMA IHC staining and morphometric quantification demonstrated that BR3-Fc decreased the area of the entire splenic lymphoid follicle, and in particular, the OMZ (Figure 7), whereas dual-label IgD/F-Actin IF further demonstrated that BR3-Fc decreased the cynomolgus monkey splenic IgD+ area (mantle zone + IMZ; Figure 8). These results confirmed and further refined the effects of BR3-Fc in lymphoid tissues as demonstrated by flow cytometric analysis (Vugmeyster et al. 2006).
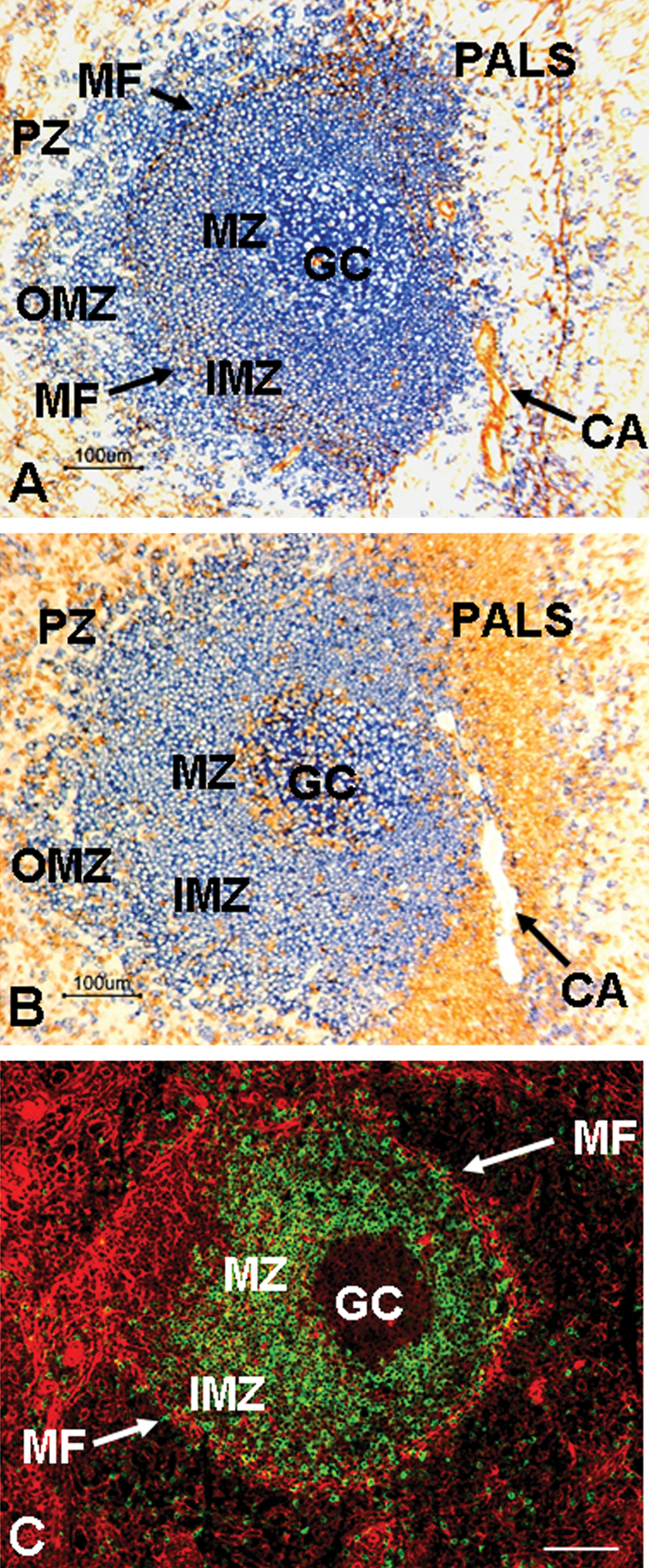
Histomorphology of a normal cynomolgus monkey splenic lymphoid follicle. Sections of cynomolgus splenic lymphoid follicle stained for the B cell marker CD20 (blue) and α-smooth muscle actin (SMA/brown; panel A), CD20 (blue) and the T lymphocyte marker CD3 (brown; panel B), and IgD (green) and F-actin (red; panel C). Panels A and B are serial paraffin-embedded sections; panel C is a frozen section. Panel A illustrates how the band of SMA+ myofibroblasts (MF) divides the inner marginal zone (IMZ) from the outer marginal zone (OMZ). Panel B illustrates the periarteriolar lymphoid sheath (PALS) of CD3+ T cells that surrounds the central arteriole (CA) adjacent to the
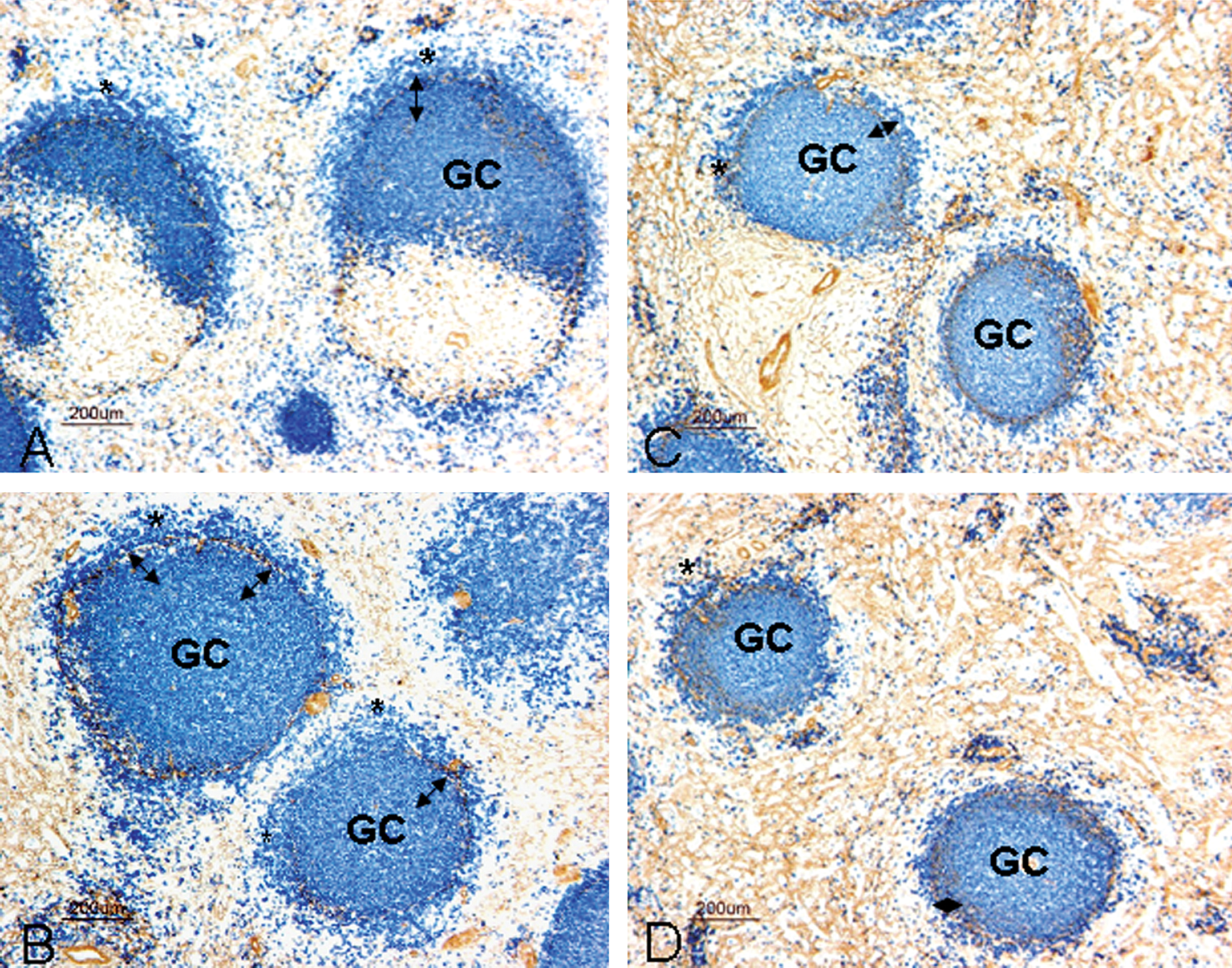
Dual-label IHC for CD20 (blue) and smooth muscle actin (brown) on spleens from BR3-Fc–treated cynomolgus monkeys illustrates a decrease in follicular CD20+ immunostaining as well as in the outer marginal zone of BR3-Fc–treated spleens. Representative paraffin-embedded sections of spleen dual labeled for CD20 (blue) and SMA (brown) from two vehicle control animals (A, B) and two animals given 20 mg/kg BR3-Fc (C, D). These panels illustrate that BR3-Fc induced a decrease in both total follicle size—particularly in the CD20+ follicular lymphocytes between the germinal center (GC) and the SMA+ band, indicated by double headed arrows—and in the outer marginal zone area (CD20+ lymphocytes outside the SMA+ band, identified by asterisks). Bars = 200 µm. Reprinted with modifications from Am J Pathol (2006).
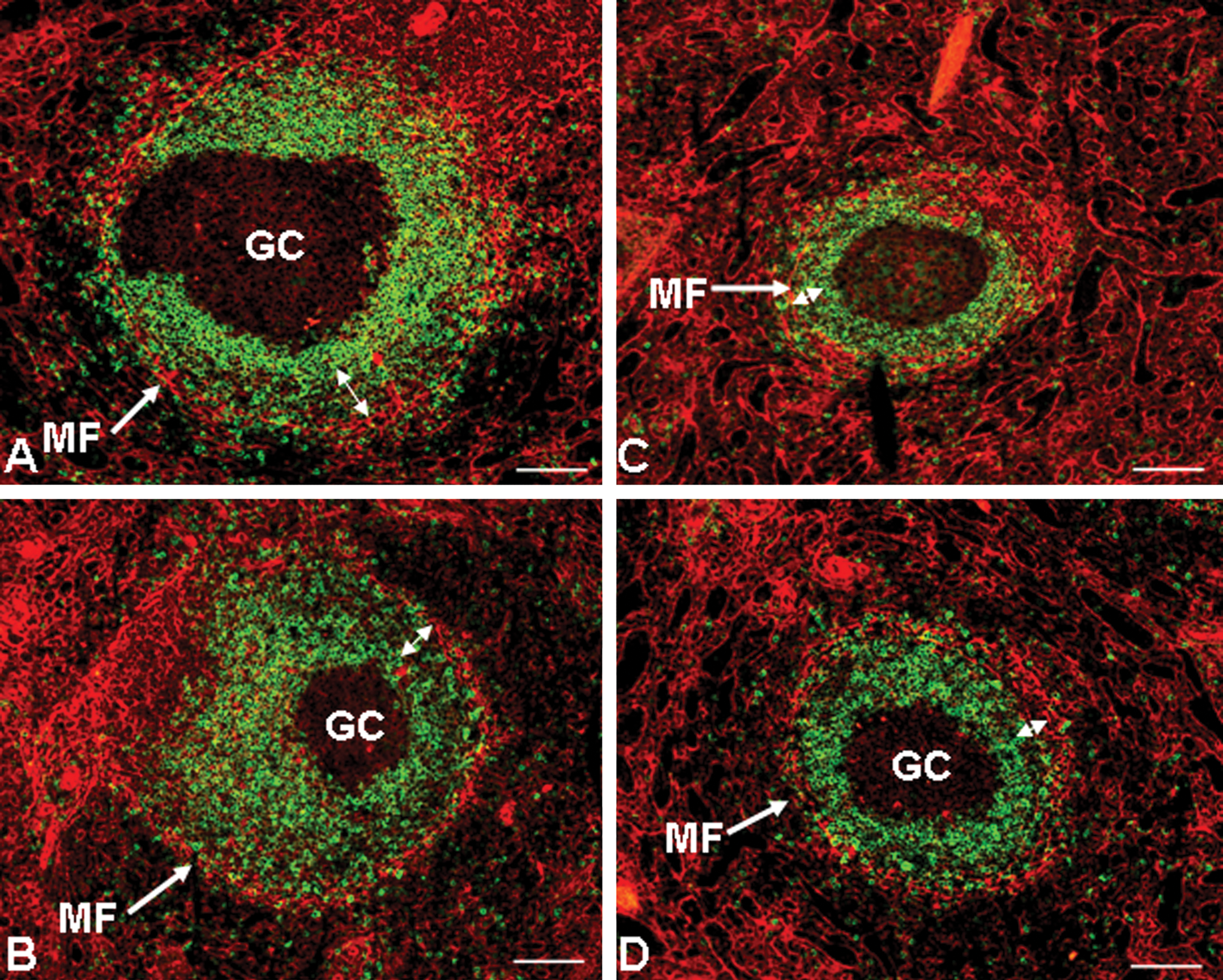
Dual-label immunofluorescent (IF) staining for IgD (green) and F-actin (red) on spleens from BR3-Fc–treated cynomolgus monkeys illustrates a decrease in IgD+ lymphocytes in BR3-Fc–treated spleens. Representative frozen sections of spleen dual labeled for IgD (green) and F-actin (red) from two vehicle control animals (A, B) and two animals given 20 mg/kg BR3-Fc (C, D). These panels illustrate that BR3-Fc induced a decrease in the follicular area expressing IgD, particularly in the lymphocytes that were strongly IgD+ (the mantle zone). In addition, BR3-Fc also induced a decrease in the IgD+ lymphocytes in the weakly to moderately strongly IgD+ lymphocytes in the IMZ (illustrated by double-headed arrows as the area between the strongly IgD+ mantle zone and the red band of myofibroblasts). Abbreviations: GC, germinal center, MF, myofibroblasts. Bars = 100 µm. Reprinted with modifications from Am J Pathol (2006).
Overall, the cynomolgus monkey pilot toxicity and proof of activity study demonstrated that BR3-Fc induced the anticipated B-cell reductions in both peripheral blood and in lymphoid tissues. Primarily naïve B cells were reduced in peripheral blood, but both naïve and memory B cells were decreased in lymphoid tissues as assessed by flow cytometry, with marginal zone B cells most affected. Dual-label IHC and IF staining confirmed and further refined the flow cytometric findings by demonstrating that marginal zone B cells (both IMZ and OMZ) were markedly reduced in lymphoid tissue. All B cell effects were reversible at the end of a twenty-three-week treatment-free recovery period. Normal antigenic immune responses to both T cell–independent antigens (Pneumovax 23) and to T cell–dependent antigens (tetanus and KLH) were observed (Vugmeyster et al. 2006). Therefore, BR3-Fc’s risk assessment based on this study was that the targeted effect on lymphoid marginal zone with no effect on either T cell–dependent or T cell–independent antigenic response suggested that the clinical risk of opportunistic infections in patients given BR3-Fc should be low.
IL-22 as a Specific Example of a Yin and Yang Immunomodulatory Biologic
IL-22 is a member of the IL-20 subfamily within the IL-10 family of cytokines. It induces epidermal hyperplasia and alterations in epidermal differentiation, including expression of the anti-microbial S100A7 (psoriasin), the hyperproliferative keratin CK16, and the transcription factor pStat3, in reconstituted human epidermis. These effects on cultured human epidermis are very similar to features found in psoriatic epidermis (Figure 9). Additionally, IL-22 induces β-defensins, VEGF, IL-20, and several chemokines in reconstituted human epidermis; again, these are features that mimic psoriasis (Sa et al. 2007).
IL-23, a cytokine known to be involved in the pathogenesis of psoriasis, was injected into the ears of IL-22 KO mice and their wild-type littermates to further elucidate the role of IL-22 in epidermal hyperplasia and inflammation,. In this study, IL-22 KO mice showed a marked reduction of IL-23–induced ear thickening, as assessed by quantification of gross ear thickness, epidermal thickness, and dermal inflammation (Figure 10; Zheng et al. 2007). Similar results were seen using an anti-IL-22 MAb (Zheng et al. 2007). Lending further support to the importance of IL-22 in the pathogenesis of epidermal hyperplasia, inflammation, and psoriasis, an anti-IL-22 MAb was found to greatly reduce disease severity in the CD45RBHi immune transfer mouse model of psoriasis (Ma et al. 2008).
Based on results from these studies, IL-22 plays a very important role in the pathogenesis of cutaneous inflammation, including cutaneous autoimmune diseases, such as psoriasis. IL-22 upregulates numerous molecules that are important in cutaneous innate immunity, including antimicrobial peptides such as the S100 proteins and defensins. As IL-22 plays an important role in cutaneous inflammation and innate immunity, we next asked whether IL-22 also played a role in host defense, using the GI tract as a model system.
Attaching and effacing (A/E) bacterial pathogens, such as enterohemorrhagic E. coli and enteropathogenic E. coli are a major cause of diarrhea, morbidity, and mortality worldwide.
Citrobacter rodentium is a naturally occurring A/E bacterial pathogen of mice. In immunocompetent mice, C. rodentium causes mild-moderate colonic mucosal hyperplasia and is a self-limiting disease with no mortality. In contrast, in immunocompromised mice, morbidity, mortality, and colonic lesions are much greater. Using C. rodentium infection in mice as a model of A/E bacterial disease, we investigated the role of IL-22 in host defense to this pathogen using IL-22 KO mice.We found that IL-22 KO mice were more susceptible to C. rodentium colitis than their WT counterparts, as assessed by increased mortality, increased weight loss, and histologic evidence of more severe colitis with ulceration, sometimes transmural, and deeper bacterial invasion into colonic crypts in IL-22 KO mice than in WT mice (Figure 11; Zheng et al. 2008).
This important role for IL-22 in GI mucosal host defense was further elucidated using treatment with an anti-IL-22 MAb. Treatment with anti-IL-22 at the time of bacterial inoculation also increased mortality and weight loss, and it worsened colitis severity histopathologically in the C. rodentium colitis model. In contrast, treatment with anti-IL-22 on Day 8 following bacterial inoculation had no effect on disease severity. In addition, IL-22 induced antibacterial peptides S100A8, S100A9, RegIIIγ, and RegIIIβ from treated mouse colons, but only RegIIIγ and RegIIIβ appear critical during C. rodentium infection, as both failed to be upregulated in IL-22 KO mice versus WT mice, and treatment with muRegIIIβ-Ig was able to partially protect IL-22 KO mice from C. rodentium–induced weight loss and mortality (Zheng et al. 2008).
As a result of these and other studies, it is now well established that IL-22 is a Th17 cytokine that plays an important role in bridging acquired and innate immunity in mucocutaneous inflammation and defense. IL-22 is produced by T cells polarized to a Th17 phenotype by IL-6, TGF-β, and IL-23, and it produces molecules involved in defense and repair/regeneration of mucocutaneous epithelial barriers in the skin, GI tract, and lung (Ouyang et al. 2009; Sonnenberg et al. 2011). Because IL-22 bridges acquired and innate immunity, but also plays a role in autoimmune diseases, such as psoriasis, it is a quintessential yin and yang immunomodulatory molecule, having both beneficial (tissue defense, repair, and regeneration) and deleterious (pro-inflammatory) effects based on the disease context. Thus, the development of IL-22–based therapeutics pose unique challenges, and the specific immunotoxicity testing program for IL-22 should take into account whether IL-22 is to be antagonized or agonized, the specific clinical disease indication, and the method of delivery.
Overall Conclusions and Recommendations
The risk assessment for immunomodulatory biologics should comprehensively characterize the MOA of the biologic. This characterization should include but not be limited to: in vitro cytokine release assays, flow cytometric immunophenotyping of lymphocyte cell subpopulations, IHC/IF staining and quantification, evaluation of in vivo animal models of efficacy, incorporation of appropriate pharmacologic/MOA and immunotoxicity assessments in toxicology studies, and other functional assays, as appropriate. The ultimate goal of the immunotoxicity program should be to understand as much about the immunomodulatory biologic’s MOA as possible to best assess its risk/benefit ratio for the intended patient population. The routine use of infectious challenge models is not recommended, as the predictivity of these models for clinical risk assessment of biologics has not been established and relevant immunomodulatory biologic positive controls for these models have not been validated. These models can be of use, however, for the identification of specific mechanism-based and model-dependent hazards.
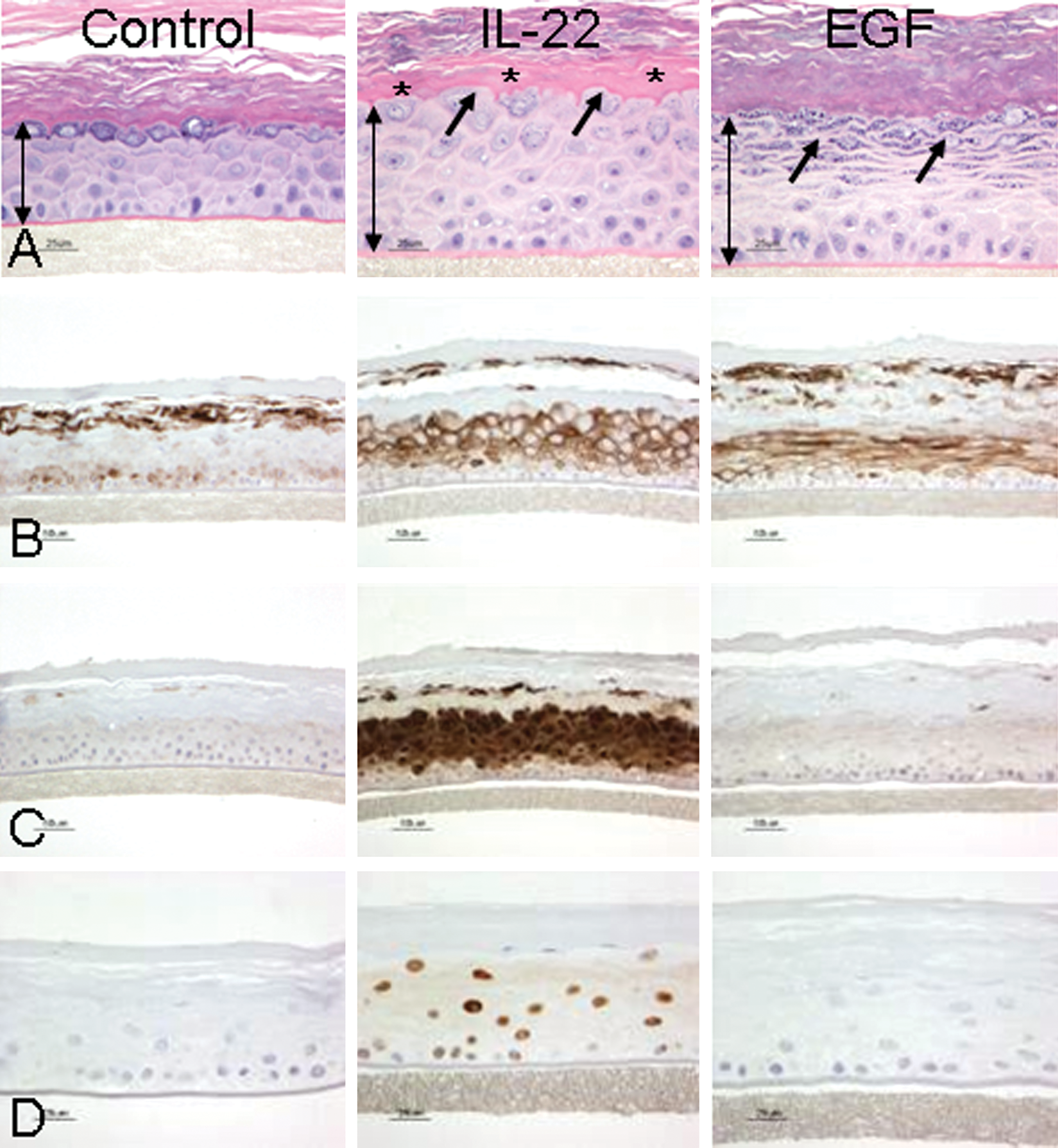
Effects of IL-22 on epidermal differentiation. Four-day cultures of reconstituted human epidermis (RHE). Images in (A) demonstrate that IL-22 induced epidermal hyperplasia, as denoted by the increased length of the double-headed arrows compared to the untreated control. In addition, IL-22 also uniquely altered normal epidermal differentiation as demonstrated by hypogranulosis, or a decrease in the granular cell layer (arrows), and hyalinization of the lower stratum corneum (asterisks*). In contrast, EGF also induced epidermal hyperplasia but did not induce the other changes in epidermal differentiation. Similarly, images in B, stained for cytokeratin 16; images in C, stained for psoriasin (S100A7); and images in D, stained for pSTAT3 all demonstrate that IL-22 alters epidermal differentiation and activates pSTAT3 as compared to EGF. These effects of IL-22 on reconstituted human epidermis are very similar to those seen in psoriatic epidermis. Modified from the original figure published in Sa et al. (2007). J Immunol
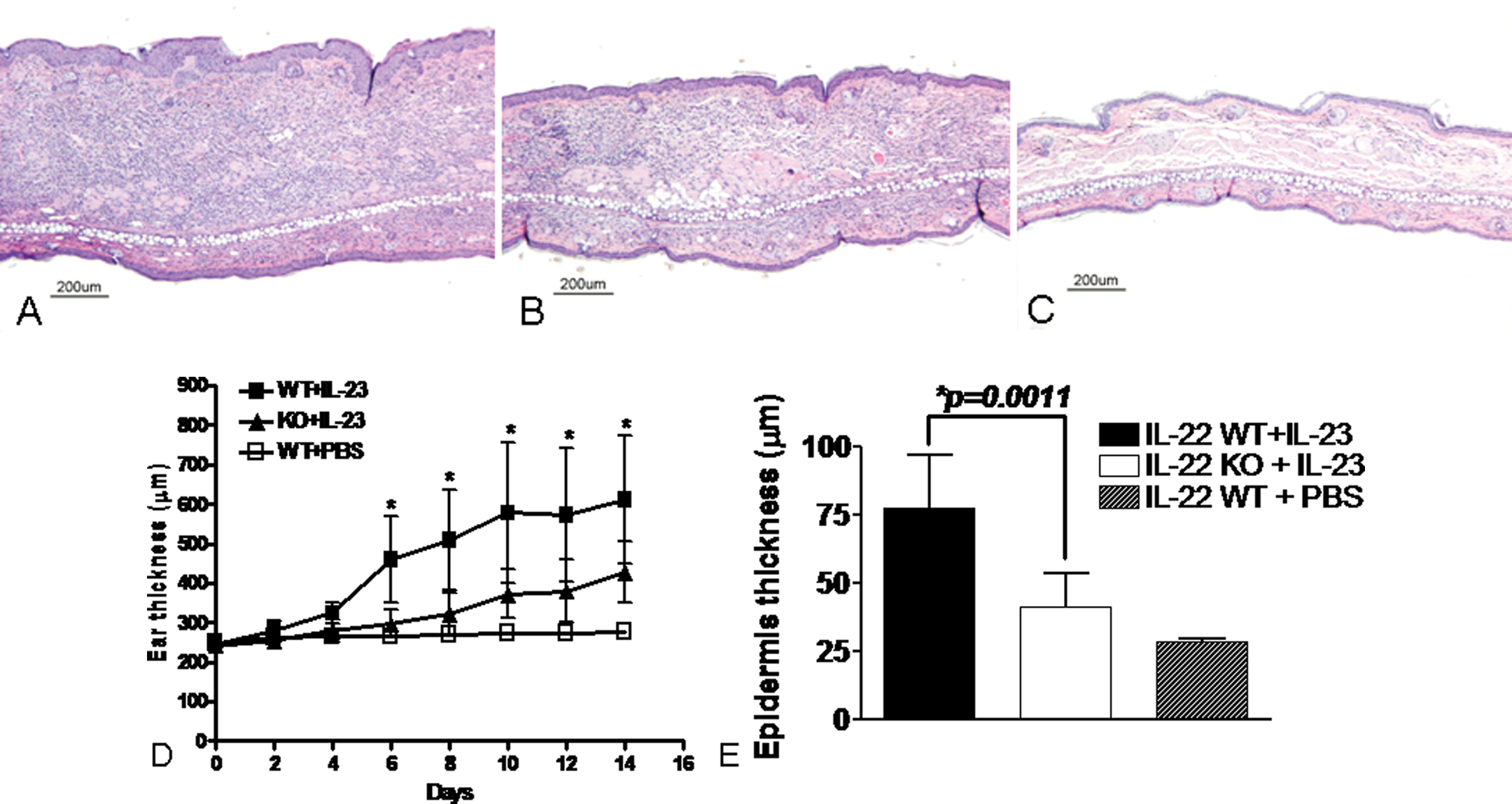
IL-22 deficiency reduces IL-23–induced ear skin acanthosis and inflammation. Hematoxylin and eosin–stained sections of ears from IL-22 KO(B) and wild-type littermate (A, C) mice injected subcutaneously with IL-23 (A, B) or PBS (C). IL-22 KO mice demonstrate a marked reduction in total ear thickness and submucosal inflammation with a corresponding reduction in epidermal thickness (B) versus wild-type mice (A). The graph in D illustrates this marked reduction in total ear thickness in IL-22 KO mice at multiple time points after injection (mean ± SD * p< .001). The graph in E illustrates a marked reduction in mean epidermal thickness in IL-22 KO mice. Reprinted with modifications from Nature (2007).
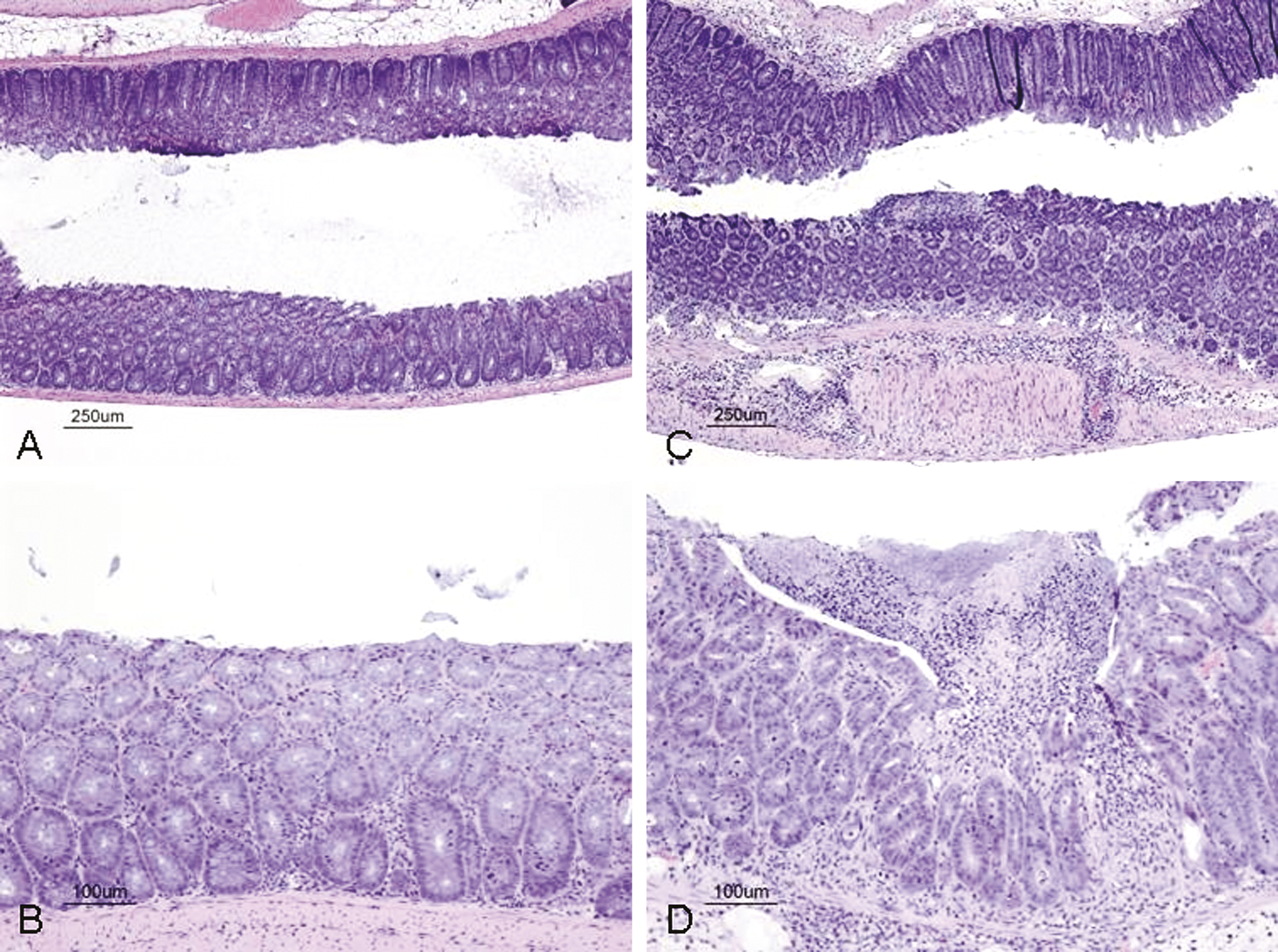
IL-22 deficiency renders mice susceptible to C. rodentium infection. Hematoxylin and eosin–stained sections of colons from IL-22 KO and wild-type mice eight days post-inoculation with C. rodentium. Panels A and B illustrate mild mucosal hyperplasia in wild-type mice, whereas panels C and D illustrate focal mucosal ulceration and transmural inflammation in IL-22 KO mice. Reprinted from Nature Medicine (2008).
Footnotes
The authors declared the following potential conflicts of interest with respect to the authorship and/or publication of this article: the authors are employees of Genentech, a member of the Roche group. The authors received no financial support for the research and/or authorship of this article.
Abbreviations
Acknowledgments
Data from a very large number of investigators, particularly those at Genentech, were used to generate this manuscript. Although there were far too many investigators to acknowledge all, we do want to single out several who were particularly helpful: Wenjun Ouyang, Yan Zheng, Susan Sa, Yulia Vugmeyster, Marna Williams, and Eric Stefanich. Additionally, Andrea Weir of CRL, David Hutto of Eisai, and Peter Bugelski of Centocor provided information that was very helpful in preparing this manuscript. We would also like to thank Jacqueline Tarrant of Genentech for critical review of the manuscript. For pathology evaluation of the twenty-six-week toxicity studies with rhuMAb Beta7, and for capturing the images used in preparing ![]() , we thank William Meir and Guenther Hoffman of Covance Labs, and Hajime Hiraragi of Genentech, respectively. Finally, we would like to acknowledge Mausumi Debnath and Aditi Roy of Anshin Biosolutions for compiling the first rough draft of this manuscript from the original PowerPoint slide presentation.
, we thank William Meir and Guenther Hoffman of Covance Labs, and Hajime Hiraragi of Genentech, respectively. Finally, we would like to acknowledge Mausumi Debnath and Aditi Roy of Anshin Biosolutions for compiling the first rough draft of this manuscript from the original PowerPoint slide presentation.