
Abstract
Autoimmune disease (AIDx) results from failure to sustain tolerance to self molecules. Dozens of AIDx involving one or multiple organ systems afflict 3% or more of people worldwide (>75% women). Predisposing factors for AIDx include genetic background, hormonal status, pathogens, and xenobiotic exposures. The incidence of AIDx is higher in individuals living in developed nations, including recent immigrants. Patients may have several AIDx simultaneously. Certain AIDx can prevent other AIDx. A history of AIDx raises the risk for developing hematopoietic neoplasia. Some common mechanisms for losing self-tolerance include reduced deletion or enhanced activation of autoreactive CD4+ T-helper (Th) lymphocytes, defective immunomodulation by CD4+ regulatory (Treg) and CD8+ suppressor (Ts) T-lymphocytes, dysregulated signaling (leading to a relative increase in pro-inflammatory cytokines), comparable structure between self-antigens and foreign molecules, or expression of new epitopes on previously hidden or xenobiotic-modified self proteins. Organ-specific AIDx is generally a cell-mediated (Th1 or Th17) process, while multi-organ AIDx also incorporates a robust autoantibody (Th2) component. Cytokine signatures of different AIDx overlap incompletely; for a given AIDx, different patients have divergent cytokine profiles. Newer anti-AIDx agents are based on our increasing knowledge of AIDx pathogenesis and usually attempt to reverse lymphocyte dysfunction, quell pro-inflammatory signaling, or restore self-tolerance.
The immune system is an intricate constellation of cellular and molecular elements that have evolved to guard the body against attack. Under normal conditions, the immune system exhibits tolerance (an inability to react) to molecules recognized as “self,” and thus does not respond to elements (whether carbohydrate, nucleic acid, or protein) that are expressed in endogenous tissues. When self-tolerance is lost, the immune system is deployed against one or more of the body’s own molecules. The civil war directed against autologous tissue resulting from the loss of self-tolerance is the hallmark of the autoimmune diseases (AIDx).
Theories of immune system recognition and AIDx development have evolved extensively over the past fifty years (briefly reviewed in Matzinger 2002). During the mid twentieth century, the “self–non-self” (SNS) model was the prevailing explanation for immune reactivity (Billingham, Brent, and Medawar 1953; Burnet 1959), culminating in a Nobel Prize in physiology or medicine in 1960. By this view, an AIDx develops when the body is faced with the task of repelling a foreign (“non-self”) threat. The key perils envisioned in this paradigm as means for invoking an immune response were external invaders (i.e., pathogenic microbes and parasites) and internal threats (e.g., neoplasia). Inadvertent attacks directed against “self” molecules were thought to result from immune system confusion in distinguishing between true foreign molecules and structurally similar “self” constituents. Three decades later, the “infection–non-self” (INS) model was devised (Janeway 1989) as a modified version of the traditional SNS scheme. In this scenario, the focus of the SNS discriminatory capacity resides in the requirement by antigen-presenting cells (APCs) that antigens be presented in combination with a co-stimulatory molecule before an immune response will be mounted; thus, resting APCs will be activated when one of their germ line-encoded pattern recognition receptors (PRRs) recognizes a microbial pathogen-associated molecular pattern (PAMP). The most recent hypothesis is the “danger” model (Matzinger 1994), in which the spur that activates quiescent APCs is the generation of one or more alarm signals by injured cells. The injury can result from many causes (neoplasia, pathogens, toxicants, trauma, etc.), since all can release elements that comprise a damage-associated molecular pattern (DAMP). However, programmed cell death would not activate APCs since apoptotic cells are scavenged before their “self” constituents are released to interact with nearby cells. Further adaptations to our understanding of immunity will arise in the future because the INS and danger models begin with divergent assumptions about what elicits an immune response and thus provide different predictions about the immune system’s responsiveness to foreign-but-harmless (i.e., fetuses) and self-but-harmful (i.e., certain mutations) materials. At present, the danger model appears to provide the more useful perspective of potential AIDx pathogenesis since its tenets encompass possible AIDx mechanisms that fall outside the SNS/INS model, such as the induction of cell death by excessive stress or toxicants as well as disruption in the extent and clearance of physiologic cell death (Matzinger 2002).
The various cellular, chemical, and protein components of the healthy immune system and its autoreactive counterpart have been well reviewed in many recent publications (D. R. Smith and Germolec 1999; Pollard 2006; Roitt et al. 2006; Janeway et al. 2007; Radbruch and Lipsky 2010) and therefore will not be considered in depth here. Instead, this mini-review briefly outlines the scope of AIDx and communicates recent themes in our understanding of AIDx pathogenic mechanisms that lead to the end of self-tolerance.
Scope of Autoimmune Diseases
Dozens of AIDx have been confirmed, and autoimmune mechanisms are suspected to contribute to the pathogenesis of nearly as many more chronic inflammatory conditions. Some AIDx affect a single organ (e.g., immune-mediated [Hashimoto’s] thyroiditis, the most common AIDx by a factor of ten; Jacobson et al. 1997), others damage multiple sites of a single organ system (e.g., immune-mediated vasculitis), and yet others disrupt multiple organs spanning many systems (e.g., systemic lupus erythematosus [SLE]). At least one AIDx has been identified for nearly every organ and tissue in the body. While some organs are attacked by multiple AIDx, the diseases affecting a single site often have distinct clinical and pathological presentations (Shih and Targan 2009; Westbrook, Szakmary, and Schiestl 2010), presumably as a result of divergent antigenic stimuli rather than anatomical and physiological differences among individuals. Burgeoning evidence suggests that other health conditions, including but not limited to amyotrophic lateral sclerosis (Niebroj-Dobosz, Dziewulska, and Janik 2006; Staines 2008), atherosclerosis (Ross 1990; Golovanova et al. 1998; Xu et al.1999), infertility (Fichorova and Boulanov 1996; Geva et al. 1997; Melner and Feltus 1999), post-menopausal osteoporosis (Pacifici 2007), and schizophrenia (Shinitzky et al. 1991; Spivak et al. 2009), may have an autoimmune component as well.
As a group, AIDx have been estimated to afflict 3–5% of people worldwide (Jacobson et al. 1997; Van Loveren et al. 2001). The actual prevalence is predicted to be even higher because the extent of less common AIDx is likely underestimated due to the scarcity of epidemiological data (Jacobson et al. 1997). To put the risk in perspective, the number of AIDx victims in the United States is slightly greater than the number of people with cardiovascular disease and nearly three times higher than the number with cancer (Patrick 2009). Approximately 75% of AIDx patients are women (Jacobson et al. 1997), and the risk of developing an AIDx is about tenfold higher for post-pubescent women than it is for age-matched young males (Beeson 1994). Nevertheless, certain AIDx are more frequent in males, especially the several autoimmune renal diseases (Beeson 1994). The pattern of AIDx differs by age, with insulin-dependent (type 1) diabetes mellitus (IDDM) and SLE more often affecting young people while scleroderma and pemphigus vulgaris are more common in older individuals (Beeson 1994). Clusters of AIDx tend to occur within certain families (D. R. Smith and Germolec 1999; Goronzy and Weyand 2009), indicating that genetic influences are a major factor in the pathogenesis of AIDx. Members of AIDx-prone families all may develop a particular organ-specific AIDx due to a genetic predisposition (Heward and Gough 1997). However, in some cases familial clusters of AIDx may present as a vulnerability to multiple different AIDx, thereby showing that the predisposition to AIDx sometimes represents a trend to enhanced auto-reactivity in general rather than a genetically mediated amplification in family members’ sensitivity to a specific auto-antigen.
Some individuals develop multiple AIDx (Rao and Richardson 1999; D. R. Smith and Germolec 1999), typically arising in one of two patterns. In the first kind, a patient may exhibit classic presentations for two distinct AIDx (e.g., rheumatoid arthritis [RA] and immune-mediated thyroiditis) and is considered to suffer simultaneously from both diseases (Punzi and Betterle 2004). Similarly, certain AIDx commonly occur together, such as idiopathic inflammatory myopathy with immune-mediated vasculitis or RA with SLE (Ginn et al. 1998; Lorber, Gershwin, and Shoenfeld 1994; Purrmann et al. 1992). In other instances, the spectrum of signs and symptoms does not match the usual presentation of any single AIDx but instead includes aberrations that are characteristic for two or even three AIDx (Koskinas et al. 1999; Boberg et al. 2011; Fietta, Delsante, and Quaini 2009). Such patients are diagnosed with “overlapping syndrome” because the clinical presentation is a mixture of features from multiple discrete AIDx. The extent of AIDx appears to be rising in developed nations, for reasons that are not fully understood. Environmental influences such as exposure to chemical pollutants and overmedication as well as chronic hormonal stimulation (“stress”) have been suggested as possible causes (Patrick 2009). Another likely explanation is reduced exposure to microbial and parasitic antigens due to enhanced hygiene and better pathogen-reducing measures (e.g., anti-infective therapies and vaccines), permitting the focus of immune surveillance elements to more readily shift toward “self” antigens (Bach 2001). The populations in countries with emerging economies do not yet possess these attributes of prosperity and thus have much lower incidences of AIDx.
The presence of an AIDx can impact the development of other chronic conditions. Some AIDx protect victims from acquiring other AIDx (e.g., multiple sclerosis [MS] reduces the likelihood of getting RA and vice versa; Sirota et al. 2009). Similarly, a prior history of AIDx has been linked to an elevated risk for developing lymphoproliferative (Turbyville and Rao 2010) and myeloproliferative (Kristinsson et al. 2010) neoplasia. These findings indicate that sustained autoreactivity in one leukocyte lineage may substantially impact the function of other leukocyte lineages as well.
Many immune-mediated diseases in animals have been investigated as potential models for understanding and treating human AIDx. Some animal models are spontaneous (H. R. Smith, Chused, and Steinberg 1984; Giarratana, Penna, and Adorini 2007), others are engineered (Boyton and Altmann 2002), while still others are induced (Bolon et al. 2011). The incidence and severity of immune-mediated disease in animals is influenced by a host of genetic (e.g., specific mutations, sex traits), physiologic (e.g., neuroendocrine circuits), and environmental (e.g., normal and pathogenic microflora, stress) factors. As with any question in translational science, data obtained with different animal models are quite variable both among species and among strains of a single species (Bolon et al. 2011; Farine 1997). Accordingly, the best animal model for exploring AIDx differs with the interests of the laboratory (e.g., a focus on conditions affecting a particular organ) as well as the expectations for the way in which data will be used (e.g., increased understanding of basic biological mechanisms vs. assembly of a suitable package for product registration).
Mechanisms of Autoimmunity
Basic Immunological Mechanisms
The distinction between “self” and “non-self” by the mature immune system depends on an intricate meshwork of sequential and well-regulated interactions between the afferent arm (APCs) and the efferent branch (effector cells and their products) to ensure that self-elements are tolerated. In general, such self-tolerance is mediated chiefly by constituents of the acquired immune system (i.e., highly specific but must first be built), in particular the complement of B- and T-lymphocytes with their membrane-bound, antigen-specific recognition molecules. However, certain elements of the innate immune system (ever-ready but nonspecific) also participate in the pathogenesis of AIDx.
Innate immunity mechanisms function as the first line of defense against foreign insults and the initial responders tasked with repairing injured tissue. The primary components encompass cells (e.g., macrophages, natural killer [NK] lymphocytes, and polymorphonuclear granulocytes) and soluble mediators (e.g., complement and cytokines). In general, cellular elements engulf foreign material and/or release powerful enzymes to digest it, while soluble factors act to hasten the readiness and facilitate the functions of the more specific acquired defenses. Members of the myeloid lineage, especially circulating and tissue-based macrophages, carry the primary burden in degrading and removing damaged cells.
The acquired immune system, including responses in AIDx, is driven primarily by signals derived from mononuclear leukocytes, particularly lymphocytes. Current dogma is that both spontaneous and induced AIDx are launched and perpetuated primarily by T-lymphocytes (Kong et al. 1989; Singer and Theofilopoulos 1990; Waldor et al. 1985). Cells of the CD4+ T-helper (Th) class are especially potent in this regard, releasing a broad spectrum of cytokines that promote the actions of other immune effector cells. Multiple classes of Th-lymphocytes have been defined. The most notable contributions to promoting AIDx appear to come from the Th1 class, which boosts immune cell activity; the Th2 class, which stimulates the humoral (antibody) response; and the Th17 class, which secretes factors to recruit and stimulate neutrophils. These master Th-cell phenotypes are activated by immediate proximity to APCs (commonly dendritic cells but on occasion also mitogen-stimulated B-cells; Shlomchik 2009) that express major histocompatability complex (MHC) type II as well as a co-stimulatory molecule (e.g., B7 [CD80/86]). Lymphocyte activation occurs only if the T-cell forms an immune synapse with an APC using three signals at once: (1) the primary T-cell receptor (TCR) binding to MHC II, (2) the T-cell co-stimulatory receptor (e.g., CD28) linking with the APC’s co-stimulatory molecule, and (3) APC-secreted cytokines interacting with T-cell receptors in a paracrine fashion (Gutcher and Becher 2007). Stimulation by a single receptor-ligand modality is not sufficient to prime the lymphocyte. A comparable receptor-mediated event in which surface-anchored immunoglobulin (Ig) serves as the B cell receptor is required for B-cell activation. In AIDx, autoreactive T- and B-lymphocytes engage in mutually assisted positive feedback to perpetuate the disease over time (Shlomchik 2009).
Extensive recombination within the genes encoding the T- and B-cell receptor proteins (TCR and Ig, respectively) modifies the scope of the global antigenic pool that will be recognized by the entire complement of lymphocytes. Mixing of the many gene elements for TCR and Ig occurs at random, so combinations that yield antigen-binding domains capable of reacting with endogenous self molecules are a fairly frequent outcome. Therefore, immune cells must be taught to ignore endogenous molecules (self-tolerance) during development and then persuaded to actively maintain their quiescence throughout life if AIDx is to be avoided.
Cellular Mechanisms of Autoimmunity
The hallmark defect in AIDx is the loss of self-tolerance, which renders immune cells unable to distinguish among “self” and “non-self” antigens. In this regard, the autoreactive immune system is functioning “within normal limits” to eliminate threats—except for its unfortunate choice of target. Therefore, the key to unraveling AIDx pathogenesis and to discovering new anti-AIDx therapeutic strategies is to understand the many possible ways in which the body normally prevents self-destruction and the various means by which civil war still may be commenced.
Normal individuals preserve self-tolerance in T-lymphocytes through many mechanisms (Table 1), all of which must be actively maintained throughout life (Kronenberg 1991; Mondino, Khoruts, and Jenkins 1996). Accordingly, the loss of self-tolerance may occur through multiple mechanisms (Table 2) (Boyton and Altmann 2002). The three main cell-oriented means for preventing autoimmunity are deletion (removal), anergy (relaxation), and suppression (restraint). The first option, deletion, involves irreversible pruning of self-reactive T-cells. This process of “central tolerance” (i.e., occurring in a core immune organ) occurs mainly in the thymus, the primary lymphoid organ for lymphocyte production. In the thymic cortex, naïve lymphocytes (Th0 class) that learn during development to ignore self elements as potential targets are positively selected for continued survival, while T-cell precursors that exhibit any affinity for self antigens are eliminated via apoptosis. The survivors migrate to the thymic medulla where they are exposed to self-antigens in the presence of MHC type I receptors (present on most body cells as a demonstration of “self” status) but not MHC type II receptors (the form on APCs) or co-stimulatory molecules. Any T-cells that will bind self-antigens with high affinity in the absence of MHC type II and a co-stimulatory signal are negatively selected and diverted into an additional round of TCR-mediated apoptosis. Together, these two rounds of programmed cell death confer central tolerance. If needed, additional apoptosis is undertaken in secondary lymphoid organs. The programmed cell death responsible for this central tolerance is controlled by interactions between Fas and Fas ligand (Nagata and Suda 1995; J. Wu et al. 1994). The absence of these molecules results in reduced pruning of autoreactive T-cells during development, which promotes systemic autoimmunity.
Strategies for maintaining immune system self-tolerance.

Selected mechanisms for self-tolerance breakdown in autoimmune diseases.

Other processes used to reduce autoreactive immune cell lineages take place in secondary lymphoid organs and/or sites of inflammation. Collectively, these mechanisms are termed peripheral tolerance as they occur downstream from the core (“central”) immune organs. The second cell-based alternative for preventing autoimmunity is anergy, an induced state of unresponsiveness that quenches the function of the autoreactive clones while leaving them alive. The main means by which T-cell anergy is induced is for an APC to present a self-antigen as a target in the absence of a co-stimulatory signal (Schwartz 2003). The anergic state induced by insufficient co-stimulation may be reversed later in life (Mondino, Khoruts, and Jenkins 1996; Schwartz 2003). The final cell-based approach is clonal suppression, where anti-self responses are quenched by factors derived from other leukocytes (e.g., CD4+ CD25+ regulatory T-cells [Treg] or CD8+ suppressor [Ts] cells). This repression may be mediated by cytokines with negative feedback activity (e.g., transforming growth factor-β [TGF-β]; Shull et al. 1992), by protein cross-linking to neutralize surface receptors with affinity for a given antigen, or by the genesis of anti-autoantibodies (anti-idiotypes) that recognize the antigen receptors on autoreactive lymphocytes (Yoshida and Gershwin 1993). Xenobiotics can interfere with T-cell suppression by modifying the cytokine profile produced by newly recruited leukocytes (Bird et al. 1998).
Breakdown of one or more of these central or peripheral, T-cell–based tolerance mechanisms is a common feature of AIDx. In particular, Treg cell function is decreased in many AIDx, which removes an outsized portion of the negative check on autoreactive T-cell clones (Flynn et al. 2007; Hsu et al. 2009; Wilde et al. 2010). However, production of autoreactive T-cells does not automatically condemn an individual to a full-blown AIDx. Autoreactive T-cells that exhibit low-affinity binding or specificity for self antigens are often not deleted in the thymus. They usually exist instead as anergic or suppressed (i.e., nonfunctional) memory cells in the peripheral lymphoid tissues. A small number of functional autoreactive T- and B-cells are found in the normal immune cell pool, as the production of autoantibodies in low amounts is frequently observed in healthy individuals (Jacobson et al. 1997). Therefore, the mere presence of autoreactive elements (an autoimmune response) should not be construed as conclusive evidence that a progressive anti-self reaction (autoimmune disease) has been launched (Kleinau, Lorentzen, and Klareskog 1995; Lleo et al. 2010). Instead, clinical AIDx usually does not follow an autoimmune response unless some other concurrent event precipitates and sustains tissue damage (Molina and Shoenfeld 2005).
Similar principles apply to B-lymphocyte self-tolerance. Most of these mechanisms take place after cells have left the bone marrow (i.e., as facets of “peripheral tolerance”). The simplest means for reducing the number of autoreactive B-cells is to induce anergy by exposing them to self-antigens in the absence of T-cell support (Bretscher and Cohn 1970). Anergy is especially likely if soluble self-antigen is bound by naïve B-cells as this event will down-regulate cell surface Ig expression, thereby decreasing the number of potential receptors to permit cell activation in the future. Such down-regulation may be transient, thereby allowing autoreactive cells to reenter the surveillance pool at a later time (Hodgkin and Basten 1995). Receptor crosslinking in B-cells with high affinity for self-antigens leads to suppression or deletion (D. A. Smith and Germolec 1999).
At least three additional cell-based measures for maintaining self-tolerance have been identified. These represent further aspects of “peripheral tolerance.” The first is by developing obstacles to stop immune effector cells from reaching segregated antigens. This mechanism usually involves the presence of a physical barrier to isolate an entire organ and all the cryptic epitopes within it. Examples of this barrier strategy are the brain and the testes. Breaching the barrier (generally by trauma or a nearby area of invasive inflammation or neoplasia) will lead to substantial inflammation, typically involving multiple leukocyte lineages, directed against the unmasked antigens. A second possibility is to maintain a stable population of circulating lymphocytes (Krupica, Fry, and Mackall 2006). The existence of this mechanism is implied by the observations that lymphopenia is a common precursor to AIDx (Marleau and Sarvetnick 2005) and that numbers of autoreactive T-cells that are both persistently activated and constantly circulating is high in some AIDx (Wilde et al. 2010). The putative mechanism is increased access of autoreactive T-cells to APCs due to the decreased competition from conventional T-cells (Goronzy and Weyand 2009). A third option is from a defect in pluripotent stem cells. This mechanism leads to functional deficits in both B- and T-cells, which permits an autoimmune response to begin (Borchers et al. 2000).
Molecular Mechanisms of Autoimmunity
The pathogenesis of AIDx is also driven by numerous mechanisms at the molecular level. These pathways all ultimately promote dysregulated intercellular communication in one form or another. Both innate immune cells and elements within the acquired immune system have been determined to be involved at the molecular level.
Innate immune mechanisms for autoreactivity
The induction of autoimmunity is often considered to be an acquired response, but innate immune cells play important roles in moderating self-tolerance (Waldner 2009; L. Wu and Van Kaer 2009). Activated dendritic cells can prime autoreactive T cells. Natural killer T (NKT) lymphocytes, a small subset of innate immune cells that recognize glycolipid antigens associated with the antigen-presenting molecule CD1d instead of MHC class I or II, can suppress or exacerbate autoimmunity depending on a constellation of animal-specific factors. While they have potent immunomodulatory properties, NKT cells are distinct from the CD4+ CD25+ Treg cells that participate in the adaptive (acquired) immune response.
In the innate immune system, three endosomal Toll-like receptors (TLR) have been identified as major participants in some AIDx (Trivedi and Greidinger 2009). These molecules, which are highly conserved across species, have evolved as receptors to recognize specific forms of microbial (viral) nucleic acid: TLR-3 for double-stranded (ds) RNA, TLR-7 for single-stranded RNA, and TLR-9 for ds DNA. Binding of the appropriate nucleotides induces a pro-inflammatory signal. Unfortunately, these TLR have also been shown to recognize certain human antigens. The expression patterns for these three TLR are often cell type–specific; B-cells bear TLR-7 and TLR-9, dendritic cells harbor either TLR-3 alone or both TLR-7 and TLR-9, and fibroblasts carry only TLR-3. These TLRs are thought to foment AIDx by directing the cells that express them to attack self molecules and enhance their expression of pro-inflammatory cytokines. In addition, initial B-cell activation may be undertaken by TLR (especially TLR-9) in the absence of T-cell support, so that B-cell clones break tolerance first (Shlomchik 2009). Activated B-cells then in turn activate naïve T-cells, which is necessary for initiation of a full-blown AIDx (Shlomchik 2009).
Acquired immune mechanisms for autoreactivity
Most alternatives for autoimmune induction and progression are tallied to the acquired immune system. This preponderance likely reflects both the specific nature of the defense responses by this immune effector arm and the greater resources spent to date on exploring in detail the biological pathways that control these responses.
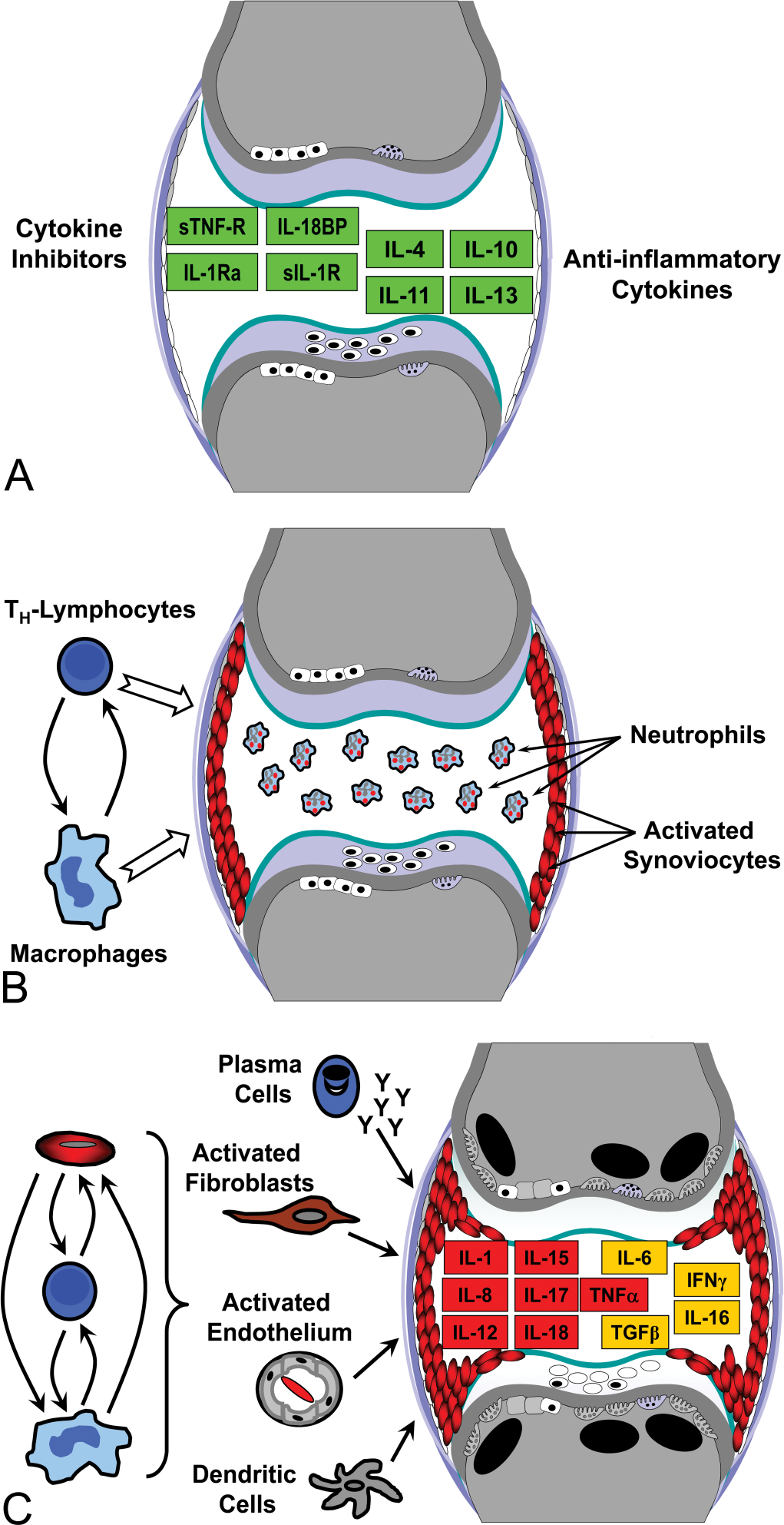
Schematic diagram of a diarthrodial joint (a common target site for autoimmune damage in many species, including humans). Induction of autoimmune disease (AIDx) depends on the balance between pro- and anti-inflammatory stimuli in a target organ. In health (A), tissues express a diverse population of immune-dampening molecules (denoted by green boxes), including anti-inflammatory cytokines such as interleukins (IL)-4, -10, -11, and -13 as well as inhibitors like receptor antagonists (IL-1ra), soluble binding proteins (IL-18BP), and soluble receptors (sIL-1R and sTNF-R for removing IL-1 and tumor necrosis factor-α [TNF-α], respectively). Joint architecture is within normal limits: cartilage lacunae (white ovals) contain chondrocytes (off-center black ovals); cartilage matrix (pale lavender) is intact; synovium (flat white ellipses) lining the joint capsule (curved, lavender, vertical lines) are in a resting state; and subchondral bone contains numerous osteoblasts (white rectangles with black nuclei) but few osteoclasts (pale lavender cells with one flat margin
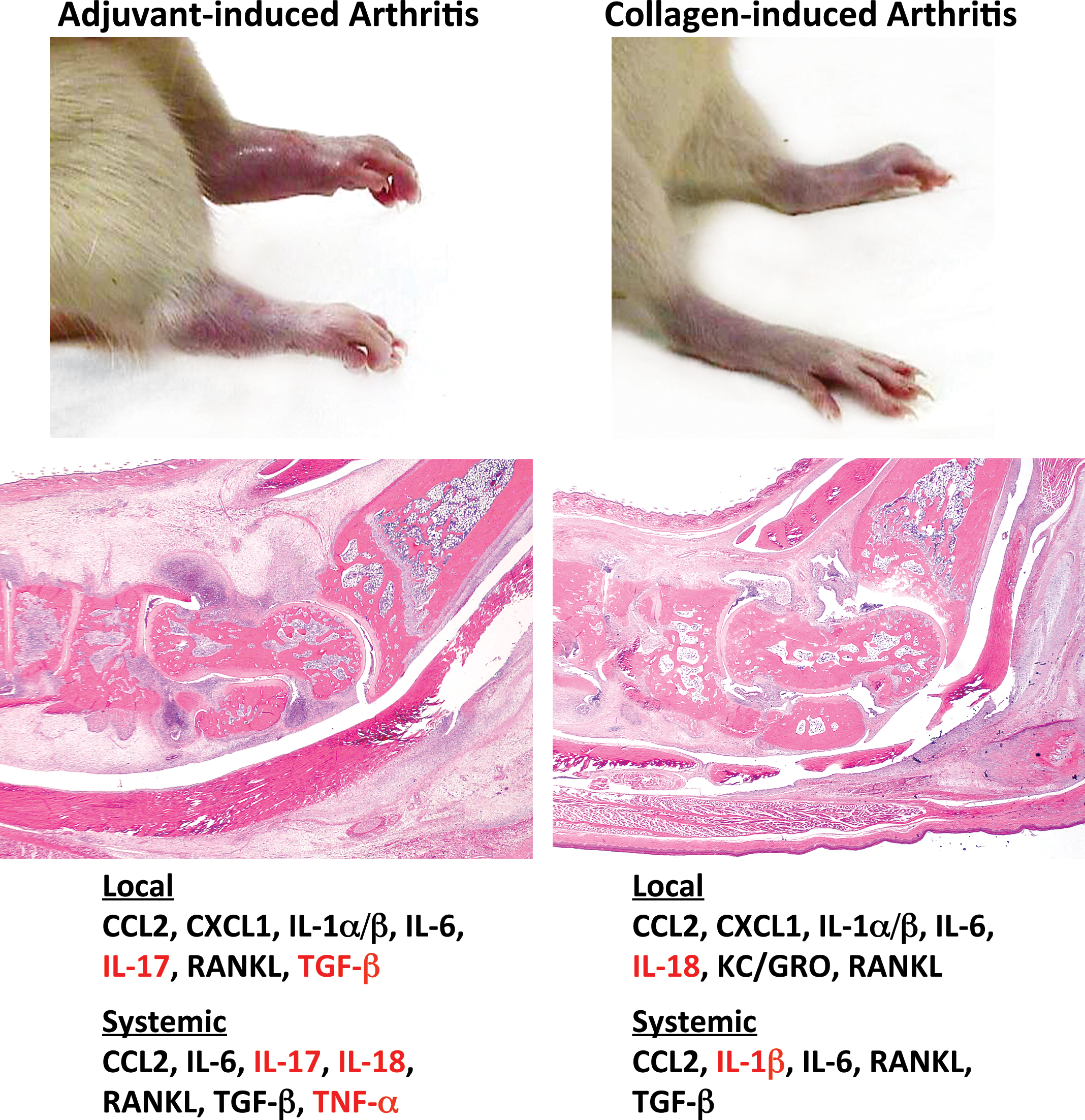
A given target organ may be affected by multiple autoimmune conditions, each having some unique clinical, pathological, and biochemical features. For example, adult Lewis rats develop more severe arthritis (indicated here by greater swelling and reddening of the tibiotarsal region [hock or “ankle”]) seven days following treatment with a single inoculation of heat-killed Mycobacterium than occurs after administration of multiple injections of collagen type II. One proposed explanation for this divergence is the variation in cytokine profiles between the models; whether measured locally (in joint extract) or systemically (in serum), some pro-inflammatory molecules (shown in red) are produced in only one model. Abbreviations are defined in the legend for Fig. 1, with these exceptions: CCL2, chemokine (C-C motif) ligand 2 (formerly MCP-1); chemokine (C-X-C motif) ligand 1 (formerly KC/GRO); RANKL, receptor activator of nuclear factor-κB. Gross images reprinted from (Bolon et al. 2011), while the cytokine signatures were reported in (Stolina et al. 2008, 2009).
Cytokine imbalances in the acquired immune system are well-recognized accomplices in the induction and progression of AIDx. Normal tissues express many different molecules to down-regulate the immune response, including both anti-inflammatory cytokines and soluble cytokine inhibitors (Fig. 1). These signals commonly are secreted by several leukocyte lineages, especially T-lymphocytes and macrophages (Fig. 1). However, B-lymphocytes as well as many non-immune but activated cells (e.g., endothelium, fibroblasts, synoviocytes) in inflamed tissues also produce cytokines (Fig. 1). In general, current thinking is that AIDx develops when the normal balance between pro-inflammatory and anti-inflammatory signals within a target organ is disrupted, leading to a chronic relative excess of local pro-inflammatory stimuli.
The exact mixture of pro-inflammatory chemokines and cytokines varies among different AIDx (Godessart and Kunkel 2001; Gutcher and Becher 2007). Traditionally, most pro-inflammatory signals in AIDx have been thought to be generated by CD4+ T-lymphocytes, but recent data have shown that mediators released by B-cells also play an important role (Youinou et al. 2009). However, the constellation of signals produced in a particular target organ differs as well, even among patients with a clinically equivalent presentation of AIDx. For example, the local and circulating profiles of pro-inflammatory mediators are different in induced rodent models of immune-mediated joint disease in which the anatomic pattern of joint involvement and the inciting agents are distinct (Stolina et al. 2008, 2009) (Fig. 2). This finding is consistent with the nature of AIDx, where multiple factors—genetic background, hormonal status, microbiome, and xenobiotic exposure, to name a few (Rao and Richardson 1999)—cooperate in launching the autoreactive process. More importantly, this principle derived from animal research is mirrored in human patients with comparable conditions. Human patients with RA have similar clinical and pathological presentations, but the AIDx is nonetheless a heterogeneous condition in which some patients respond only to interleukin (IL)-1 inhibition, others improve with tumor necrosis factor-α (TNF-α) blockade, still others recover following anti-IL-6 therapy, and some are not helped by quenching any of these “master” cytokines (Fitzgerald et al. 2005; Senolt et al. 2009; Patel and Moreland 2010; Turkstra, Ng, and Schuffham 2011). Clinically equivalent demyelinating AIDx have also been shown to have divergent profiles of T-cell–derived cytokines (Segal 2010). The complex character of cytokine dependence in such seemingly uniform conditions suggests that anti-AIDx regimens will continue to rely on two different strategies: relatively nonspecific, broad-spectrum immunosuppressive agents (like cyclosporine A or methotrexate) or target-specific entities (e.g., biologics or small molecules to inhibit single cytokines or a given leukocyte class) that have been empirically tested in each patient to assess their potential response.
The most common pro-inflammatory phenotypes in AIDx are attributed to enhanced activity of CD4+ T-helper lymphocytes. Three distinct phenotypes are especially important in this regard: Th1, Th2, and Th17. As a general rule, organ-specific AIDx are driven by cell-mediated processes designed to attack intracellular pathogens and thus develop when Th1 cytokines like IL-2 and interferon (INF)-γ predominate (Mosmann et al. 1991; Street and Mosmann 1991; D. A. Smith and Germolec 1999). In contrast, multisystem AIDx typically include a vigorous humoral (antibody-based) response and are characterized by raised levels of Th2 cytokines such as IL-4, IL-5, and IL-10; extensive autoantibody production and immune complex deposition; and cell targeting by complement-mediated lysis (Mosmann et al. 1991; Street and Mosmann 1991; D. A. Smith and Germolec 1999). The Th2 response usually is associated with higher induction of macrophages, which leads to exacerbated fibrosis in many AIDx (Fairweather and Cihakova 2009). Some AIDx, such as multiple sclerosis, are not easily classified as their cytokine signature exhibits both organ-specific and systemic characteristics. The Th17 phenotype contributes to the genesis of AIDx by producing IL-23 as a neutrophil chemotactic and activating factor (Wilde et al. 2010). Some Th cells appear to have a mixed Th1/Th17 phenotype as they can generate both IFN-γ and IL-17, but in general both Th1- and Th17-cells are produced in parallel because they overlap only partially in their functions (Dardalhon et al. 2008; Martin-Orozco et al. 2009). Importantly, Th17-cells can drive AIDx in the absence of a concomitant Th1 response (Haak, Gyulveszi, and Becher 2009; Damsker, Hansen, and Caspi 2010).
Dysregulated intracellular signal transduction is an important abnormality in many AIDx. Multiple different pathways have been implicated, a few examples of which are given here. Reduced cytotoxic T-lymphocyte antigen (CTLA)-4 expression in Treg-cells permits the inappropriate activation of naïve T-cells and promotes retention of autoreactive T-cells in organs (Jain et al. 2010). Altered amounts of signal transducers and activators of transcription (STAT)-3 impact the potential for autoimmunity in several fashions. First, STAT-3 directly controls the phenotype of naïve CD4+ T-cells, selecting between pro-inflammatory (Th17) and inhibitory (Treg) roles (Egwuagu 2009). In addition, STAT-3 also regulates other essential processes of T-cells, including cell growth, cell survival, and the transcription of pro-inflammatory genes (Egwuagu 2009). Deregulated signaling via nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB), a major modifier of gene transcription in cells of the innate and acquired immune systems, has been shown to be a player in several AIDx (Karin and Greten 2005). The purported mechanism is an increase in NF-κB activity in leukocytes due to the loss of inhibitory proteins like A20 (Coornaert, Carpenter, and Beyaert 2009). Aberrations in Fas and Fasl have already been noted as participants in various AIDx due to their importance in specifying lymphocyte survival (Nagata and Suda 1995; J. Wu et al. 1994; Teachey et al. 2008). The repression of this pathway may result from a direct mutation of Fas or Fasl or indirectly by effects on factors that inhibit Fas activation. Other critical signaling pathways implicated in induction of some AIDx are Notch, which is required for early T-lymphocyte commitment as well as TCR-mediated T-cell proliferation and activation (Teachey et al. 2008), and Wnt/β-catenin, elevation of which is linked to defective repair in multiple sclerosis lesions (Fancy et al. 2009). Over time, many other signal transduction defects are likely to be confirmed to participate in the induction and maintenance of AIDx.
Metabolic anomalies have been posited as a mechanism for AIDx. Again, many different ways have been suggested by which metabolic dysfunction may enhance the risk of autoimmunity. Altered phospholipid synthesis in the membranes of beta cells in pancreatic islets has been linked to early insulitis and eventual IDDM (Lindfors et al. 2009). A related factor in this and other AIDx seems to be the generation of reactive oxygen species (ROS) capable of damaging cell membranes. Indeed, ROS-induced injury has been implicated as a common mechanism for chemically-induced AIDx (Yoshida and Gershwin 1993). Aberrant protein degradation has been identified as an additional means for inciting autoreactivity. Numbers of circulating proteosomes are elevated in some AIDx (Wang and Maldonado 2006), suggesting that enhanced molecular destruction may heighten the likelihood that fragments of self-molecules will be presented as possible antigens. Furthermore, autoantibodies to proteosomes are found in some AIDx (Wang and Maldonado 2006), which likely will reduce the proteosome pool’s ability to regulate the levels of potential self-antigens in circulation. The general theme is that insults which damage macromolecules and/or membranes are likely to provide a substrate that can support an autoreactive immune response.
Microbial interference is suspected to be an essential driver in many AIDx. All animals, including humans, are actually an amalgam (“super-organism”) comprised of endogenous cells and a rich microbiome; in fact, approximately 90% of the cell mass in humans is estimated to be of bacterial origin (Turnbaugh et al. 2007). The basis for many AIDx seems to reside in the combined mass of human and bacterial genes (the “metagenome”), working in tandem to specify the genetic and macromolecular composition against which health and disease play out (Proal, Albert, and Marshall 2009; Rath et al. 1996). Several mechanisms have been posited by which microbes may initiate autoreactivity (Ercolini and Miller 2009). The first is bystander activation, or the nonspecific promotion of a generalized pro-inflammatory environment as a consequence of the anti-microbial immune response. For example, a virus-associated mouse model of IDDM is driven by elevated IFN-γ secretion in response to the infection, which in turn widely up-regulates MHC type II expression (von Herrath, Dyrberg, and Oldstone 1996). The increased ability to present antigens to lymphocytes raises the likelihood that APCs will promote the activation of lymphocytes that have a low affinity for a self-antigen. Another mechanism is molecular mimicry, in which an immune response to a microbial molecule results in an accidental attack on a structurally homologous self peptide. Examples of this way include rheumatic fever (an antibody-mediated heart AIDx resulting from the high degree of similarity between cardiac myosin and a β-hemolytic Streptococcus membrane protein; Dell et al. 1991; Guilherme et al. 2000) and thyroid-localized AIDx (antibody-mediated conditions arising from cross-reactivity between thyroid self-antigens and components of Yersinia enterocolitica; Luo et al. 1993, 1994; Wenzel et al. 1988). A third mechanism is polyclonal activation, where certain microbes produce super-antigens capable of nonspecifically cross-linking MHC II to TCR on numerous T-cell lineages primed for myriad epitopes (including autoreactive ones) at once, thereby leading to their unintentional stimulation and massive cytokine release (Friedman et al. 1991; Schiffenbauer, Soos, and Johnson 1998). Examples of this association include the pairings of coxsackievirus B with IDDM (Chehadeh et al. 2000), Klebsiella pneumoniae with ankylosing spondylitis (Ebringer and Wilson 2000), Mycoplasma arthritidis with arthritis (Tumang et al. 1991), and Proteus mirabilis with rheumatoid arthritis (Ebringer and Wilson 2000). Some microbial metabolites instigate autoimmunity by interfering with human gene expression (altered transcription, transcript repair, and translation) (Proal, Albert, and Marshall 2009).
Neo-antigen formation is thought to represent a primary means of AIDx induction. The conjugation of a hapten—usually a reactive metal (Kubicka-Muranyi et al. 1996) or small chemical (Christen et al. 1994; Christie 1993)—to an endogenous molecule changes the self-epitope’s conformation (Griem et al. 1998). Contortions in the cross-linked molecule generally will expose new epitopes, which may be recognized as new (i.e., foreign) antigens (Craft and Fatenejad 1997; Thomas and Messner 1986). Alternatively, the conjugated endogenous molecule may now have a much higher affinity for MHC II, which will process it to reactivate anergic cells (Ayer, Edworthy, and Fritzler 1993). Any resulting AIDx stemming from production of an immune response against a neo-antigen may persist after the hapten is cleared, presumably because the reaction is mainly against the endogenous portion of the complex.
Premature lymphocyte senescence has been suggested as a means for initiating AIDx (Weyand et al. 2009). The proposed mechanism is an accelerated (i.e., age-inappropriate) loss of telomeres in naïve T-cells (Koetz et al. 2000). Possible explanations for this loss include defective stem cells (Schonland et al. 2003) or an altered capacity for telomere repair (Fujii et al. 2009). By this model, the damage would occur years to decades in advance of the actual disease (del Puente et al. 1988; Nielen et al. 2004). This hypothesis implies a threshold level of chromosomal damage above which AIDx will develop automatically, although this premise has yet to be proven. However, senescence of the immune system with age has been linked with an increase in autoantibodies, shifting cytokine signatures, and changing T-cell subsets (Boren and Gershwin 2004). These proven age-related alterations could readily permit the activation of autoreactive pathways.
Physiological Contributions to Autoimmunity
Genetic background has been clearly shown to affect the onset and progression of AIDx (Gershwin 2007). The most firmly established genetic links to autoreactivity are for specific alleles within the MHC gene complex (Altmann, Sansom, and Marsh 1991; Theofilopoulos 1995). In humans, certain haplotypes are closely associated with specific AIDx: HLA-B27 with ankylosing spondylitis; HLA-DR2 with multiple sclerosis and SLE; HLA-DR3 with IDDM, myasthenia gravis, and SLE; and HLA-DR4 with IDDM, rheumatoid arthritis, and pemphigus vulgaris. Animals bearing human MHC genes can develop comparable AIDx (e.g., arthritis and colitis in HLA-B27-transgenic rats; Khare, Luthra, and David 1995; Rath et al. 1996). However, the presence of a particular MHC haplotype is not sufficient for initiating AIDx, and AIDx-linked MHC haplotypes are found in individuals who have no clinical disease. Certain non-MHC genes may also provide substantial contributions to autoreactivity, such as TCR haplotypes (Imberti, Sottini, and Primi 1993).
Hormonal status clearly plays an important immunomodulatory role in AIDx (Da Silva 1995; Elenkov et al. 1999). This ability is obvious given the greatly increased prevalence among women. Females are often more susceptible to xenobiotics that promote AIDx, including to prenatal treatments that predispose to long-delayed increases in AIDx (Mustafa et al. 2008, 2011). The exact means by which this enhanced vulnerability is mediated is still incompletely defined, but several factors have been identified. For example, females generally have more robust humoral and cellular immune responses than males (Rao and Richardson 1999). Furthermore, leukocyte trafficking within peripheral tissues differs between females and males and varies in relation to the degree of autoreactivity (Yung et al. 1997). Many immune effector cells, including T- and B-lymphocytes and macrophages, express estrogen receptors (Cutolo et al. 1995). Estrogen-exposed leukocytes exhibit alterations in adhesion molecule expression, apoptotic sensitivity, and cytokine production, all of which may augment the immune response (Cutolo et al. 1995). Hormonal rhythms appear to contribute to the circadian patterns of certain clinical signs and symptoms in some AIDx (e.g., joint stiffness and swelling in rheumatoid arthritis) (Cutolo and Masi 2005).
Stress hormones (e.g., glucocorticoids, catecholamines) also modulate autoreactivity. The impact of stress on the immune response is complex, having both amplifying and inhibitory effects. For example, increased stress hormone levels inhibit the release of pro-inflammatory cytokines like IFN-γ and TNF-α while stimulating the production of anti-inflammatory cytokines such as IL-4, IL-10, and TGF-β (Elenkov and Chrousos 2002). This shift in the cytokine signature from a cell-mediated to a humoral bias reduces the likelihood of a persistent Th1-driven up-regulation and lowers the potential for activation of autoreactive lymphocytes (Elenkov et al. 2005). This anti-AIDx tendency can persist for extended periods following an episode of acute stress (Harbuz et al. 2006). However, chronic stress eventually leads to a decrease in adrenal glucocorticoid release, which promotes the secretion of IL-1 and TNF-α with gradual entry into a state of raised immune responsiveness (Cutolo and Straub 2009; Elenkov and Chrousos 2002). A comparable increase in immune readiness occurs in individuals with low baseline epinephrine levels (Elenkov et al. 2008). Therefore, the final effect of stress hormones on autoimmunity depends on the nature (acute or chronic) and severity of the stress (Cutolo et al. 2000). An intriguing focus of recent research is the potential interaction of stress-induced physiological changes with sex-specific anatomical and functional variations in promoting the transition from enhanced autoreactivity to full-blown AIDx (Becker et al. 2007).
Toxicant Contributions to Autoimmunity
The incidence of AIDx in humans and animals may be altered by exposure to many different xenobiotics (Holladay 1999; Rao and Richardson 1999; D. A. Smith and Germolec 1999). Agents implicated in AIDx induction include such known immunotoxicants as chemotherapeutics, corticosteroids, polycyclic hydrocarbons, and polyhalogenated hydrocarbons. Enhanced autoimmune tendencies may occur following exposure during adulthood, but organisms exposed during the prenatal and/or perinatal developmental periods may experience even more pronounced and persistent effects (Holladay 1999).
Many mechanisms have been proposed to contribute to the genesis of toxicant-induced AIDx. For instance, transcription modifications in immune cells resulting from xenobiotic exposure have also been postulated as a frequent means for breaking tolerance (Rao and Richardson 1999). In particular, certain agents are thought to incite AIDx by modifying gene expression in cells participating in immune responses. Examples include agents that interact with cytokine receptors as agonists or antagonists and small molecule enzyme inhibitors. Gene modification can occur in many intracellular sites, including DNA itself (a direct effect) as well as receptors or downstream signaling elements (which act indirectly). For instance, increased methylation of certain nucleotides in T-cell DNA has been associated with enhanced autoimmunity (Quddus et al. 1993; Richardson et al. 1990), presumably because the added side chains prevent the attachment of transcription factors and thus suppress gene transcription. Structural changes to histones mediated by several enzymes (Bird et al. 1998; Pazin and Kadonaga 1997) have also been invoked as means by which gene expression can be repressed during autoimmunity. Gene alterations leading to elevated AIDx susceptibility can affect APCs and lymphocytes. The genetic damage may be in the mitochondria, not just the nucleus (Yu et al. 2009). The proposed mechanism is increased production of ROS leading to continual low-level cell death and auto-immunization. Presumably, ROS might also damage cell membranes, thus providing a second simultaneous insult. Extensive membrane damage might even lead to cell death, which in sufficient quantities would be capable of boosting the amount of endogenous molecules in circulation available to APCs for processing and presentation to autoreactive immune effector cells.
Other mechanisms have also been proposed whereby toxicants may incite AIDx. A major hypothesis (discussed briefly previously) is neo-antigen formation by conjugation of a hapten (metal or small molecule) to an endogenous antigen. Another recognized possibility is deficient immunoregulation due to disrupted T-cell development in the thymus. This phenomenon seems likely to result from either insufficient generation of Treg cells (i.e., reduced capacity for immune system down-regulation) or failure to repress or eliminate autoreactive Th cells (i.e., retained ability to mobilize an autoimmune reaction), or both. A third but indirect means by which xenobiotics may promote AIDx is endocrine disruption (Patrick 2009). In theory, antibiotic therapy and vaccination to modulate the composition of the microflora and/or the body’s response to it represent another route by which the incidence and severity of AIDx might be altered (Molina and Shoenfeld 2005).
Summary
Autoimmune disease remains a major medical concern throughout the world, the more so because the mechanisms responsible for its induction and progression are poorly understood. The primary cause of any AIDx is a diminished capacity for self-tolerance due to a failure in “central” processes (i.e., in primary lymphoid organs) and/or “peripheral” means (i.e., secondary lymphoid organs and/or inflamed tissues) for removing or repressing autoreactive immune cell lineages. Many cellular, molecular, and physiological mechanisms have been explored as potential explanations for the loss of self-tolerance. The data supporting such claims have been derived from animal experiments (using autoimmunized, chemically induced, or genetically predisposed subjects) and, in many instances, have yet to be confirmed as relevant to the human clinical setting.
Nonetheless, certain fundamental principles have been discovered. First, the autoreactive immune system appears to begin its attack based on a case of mistaken identity. In essence, early in AIDx the immune system is “normal” except for its choice of a self-antigen as its main target. Over time, the system adopts a systemic pro-inflammatory bias to maintain the positive feedback loop necessary to indefinitely maintain its assault. The second key concept is that essentially every organ and tissue is subject to at least one AIDx, either as a stand-alone organ-specific condition or as part of a more widespread multisystemic syndrome. In many cases, patients may be experiencing two or even more concurrent AIDx. The last main idea is that the primary drivers of AIDx are considered to be CD4+ T-lymphocytes, which modify the responses of other immune cells. The main CD4+ classes engaged in this regard are Th1 and Th17 cells, which help recruit and stimulate other immune effector elements. This role is normally counterbalanced by the opposing activity of CD4+ Treg cells, which exist to fine-tune immune system responsiveness. A reduction in the numbers and/or function of Treg cells will remove the check that normally reins in the exuberance of any potential immune reaction—especially that directed against one’s self.
Great progress has been made in recent years in moving toward more targeted anti-AIDx therapies (Mor 2007). The major thrust has been to devise biologics and small molecules directed against pro-inflammatory cytokines (St Clair 2009; Senolt et al. 2009; Wagner and Laufer 2006). However, other innovations have also entered the ring as novel anti-AIDx treatments, including hematopoietic cell transplants (de Carvalho, Pereira, and Gershwin 2009), Treg-cell expansion (Jaeckel et al. 2006), and even antibiotics to alter the composition of the intestinal microflora (Ochoa-Reparaz et al. 2009). These mechanism-based regimens hold great promise for selectively attenuating autoreactive immune responses. Nevertheless, for the present, the heterogeneity of autoimmune mechanisms coupled with the continued need for empirical testing to select the best therapies for AIDx patients indicates that considerable work remains to be done before the promises of translational research and personalized molecular medicine will truly conquer AIDx.
Footnotes
Acknowledgments
The author thanks Ms. Beth Mahler, illustrations editor for Toxicologic Pathology, for her invaluable assistance in optimizing the format of the figures in this article and Dr. Paul Snyder for reviewing the immunological concepts presented in this treatise.
The author(s) declared no potential conflicts of interests with respect to the authorship and/or publication of this article. The author(s) received no financial support for the research and/or authorship of this article.