
Abstract
The hypothesis of this study was that alterations in Fe distribution triggered by lipopolysaccharide (LPS) administration were affected in vivo by Fe overload. Lipopolysaccharide treatment by itself significantly decreased Fe content in serum and increased the blood NO-hemoglobin (NO-Hb) EPR signal and nitrotyrosine protein content in liver, as compared to values in control animals. Fe overload (produced by Fe-dextran ip administration) caused an increase, as compared to values in control animals, in Fe content in serum, and a significant enhancement in ferritin (Ft) content, Fe content in Ft, the labile Fe pool (LIP), and the protein carbonyl content in the liver. The simultaneous administration of LPS and Fe-dextran lead to a significant increase in the Fe content in serum, blood NO-Hb EPR signal, the content of Fe, Fe in Ft, LIP, protein carbonyl, and nitrotyrosine protein in liver, as compared to values in control animals. The data reported here indicate that the protective strategy against endotoxemia of sequestering serum Fe content is not fully operative under Fe overload conditions. However, the oxidative condition of the liver does not seem to be being affected, since endogenous mechanisms were able to regulate the amount of catalytically active Fe to the same levels observed after Fe-dextran administration, even in the presence of LPS, over the initial six-hour period.
Introduction
Liver is the main organ for Fe storage (Papanastasiou et al. 2000). Treatment of rats with Fe-dextran resembles hemochromatosis secondary to Fe-loaded anemias (anemias treated with repeated transfusions) and high-Fe oral intake (Powell et al. 1980). The ability of excess Fe to generate oxidative damage strongly depends on the cellular Fe distribution. Under physiological conditions, the amount of Fe within the cell is carefully regulated to provide an adequate level of micronutrient while preventing its accumulation and toxicity. Fe is transported and stored in specific proteins (transferrin, lactoferrin, ferritin [Ft], and heme proteins, among others; Galatro et al. 2007). Fe is sequestered in Ft, the main intracellular Fe-storage protein (Arosio and Levi 2002). The cytosolic labile Fe pool (LIP) is a transitory, catalytically active compartment that has been implicated in cell Fe homeostasis and in metal-induced cytotoxicity by radicals generated by the Haber-Weiss reaction (Haber and Weiss 1934). Moreover, this intracellular pool of low molecular weight Fe compounds acts as an intermediate between extracellular Fe and a wide variety of intracellular processes, and it is in equilibrium with stored Fe and Fe enzymes (Jacobs 1977). In Fe-rich conditions, Ft acts as Fe-sequestering protein, protecting cells against Fe toxicity, and under low-Fe conditions it acts as a source of Fe ions necessary for Fe-containing protein synthesis.
A number of studies have described the complex relationships between Fe and NO (Gow and Stamler 1998; Kagan et al. 2001). However, controversy remains as to the influence and significance of Fe on inflammatory NO production in vivo. Upon infection, macrophages phagocytose Fe, thereby preventing free radical injury and reducing Fe availability for invading microorganisms (Weinberg 2000). Fe-dextran administration enhanced infection in a dose-dependent manner and resulted in infection enhancement factors approaching 109 for virulent strains of N. meningitidis (Holbein et al. 1979). Moreover, according to Otterbein et al. (1997), lipopolysaccharide (LPS) induced hepatic injury that was not prevented by Fe-dextran treatment (which induced Ft), but protective effects were observed by an increase in hemoxygenase-1 activity. Zager et al. (2005) showed that Fe-dextran significantly enhanced LPS- or muscle injury–mediated TNF-α generation, at least forty-eight hours post-Fe injection in an organ-specific fashion, affecting kidney, heart, and lung, but not liver or spleen. Kemna et al. (2005) suggested the existence of a highly responsive LPS–IL-6–hepcidin axis linking innate immunity and Fe metabolism over the initial period (three to six hours) after LPS injection and showed that six hours after injection, the serum Fe level becomes significantly lower than in controls. In an experimental model of sepsis (Linares et al. 2003), oxygen-derived free radicals, as well as Fe level, remained unchanged in bile six hours post-LPS, but Fe content in bile significantly increased twenty-four hours after LPS administration. Thus, increases in both NO concentration and modification of Fe distribution in the body seem to be connected events. Galleano and Puntarulo (2008) employed a model of sepsis (4 mg/kg of LPS from E. coli serotype 0127:B8) including a pre-existent Fe overload condition (200 mg of Fe-dextran/kg) and showed that control rats showed a markedly decreased Fe content in plasma (66%) during the initial six-hour period post-LPS administration, whereas LPS administration produced a slight, nonsignificant decrease (14%) in Fe-overloaded rats. Moreover, Duvignean et al. (2008) showed that LPS injection led to oxidative damage to liver tissue and that the levels of labile Fe in the liver were significantly increased four to eight hours after the onset of endotoxic shock and that the temporarily concentration of labile Fe in the liver correlated with the temporary impairment of both mitochondrial function and tissue ATP levels.
Thus, since there is no clear agreement among the experimental data on the effect of endotoxemia on Fe-overloaded animals, the hypothesis of this study was that in a model of simultaneous administration of Fe-dextran and LPS, the excess Fe could affect the profile of distribution of Fe triggered as a consequence of endotoxemia in rat liver. In this model, serum Fe content, blood Hb-NO complexes, and the total Fe, LIP, and Ft content in rat liver were assessed. The damage to Ft itself was comparatively analyzed as the oxidation of the protein, and the decrease in the content of exposed Trp residues that occurred in vivo.
Materials and Methods
Experimental Preparations
Experiments were carried out on male Wistar rats that weighed 170–200 g. Agents were administrated as follows: 4 mg/kg LPS from Escherichia coli (serotype 0127:B8) ip and 500 mg Fe/kg as Fe-dextran ip. Samples were obtained 6 h after LPS and/or Fe-dextran. Dextran administration by itself did not show any significant effect over saline-injected animals on Fe content and NO production. Rat livers were excised and homogenized for thirty seconds in a blender with 40 mM potassium phosphate buffer (pH 7.4). Blood, obtained by cardiac puncture, was divided into 2 aliquots: an aliquot was immediately frozen in liquid nitrogen for electronic paramagnetic resonance (EPR) measurements, and the other was centrifuged (600 g, fifteen minutes) to separate serum. Lipopolysaccharide, Fe-dextran, thioglycolic acid, bathophenantroline, sodium dodecyl sulfate (SDS), bovine serum albumin (BSA), β-mercaptoethanol, bromophenol blue, ethylenediaminetetraacetic acid (EDTA), Chelex-100, deferoxamine (DF), and dinitrophenylhydrazine were purchased from Sigma-Aldrich Chemical Co. (St. Louis, MO, USA). The rest of the reagents were of the highest grade available. The buffers and the water used to prepare all solutions were passed through columns containing Chelex-100 resin to remove metal contaminants.
Determination of NO-Hemoglobin (NO-Hb) Complexes in Rat Blood
The EPR spectrum of NO-Hb in rats was determined at 77K in a Bruker (Karlsruhe, Germany) spectrometer ECS 106 with ER 4102ST cavity. Spectrometer settings were: field modulation frequency, 50 kHz; microwave frequency, 9.42; modulation amplitude, 4.75 G; microwave power, 10 mW; time constant, 164 ms; sweep width, 800 gauss; center field, 3300 gauss (Galleano et al. 2004).
Determination of Serum Fe Content
Serum Fe content was determined by a colorimetric method based on the reaction of ferrous ions with pyridyl bis-phenil-triazine sulphonate using a commercially available kit (Fer-Color, Wiener Laboratorios S.A.I.C., Rosario, Argentina).
Determination of Total Liver Fe Content
Total Fe content in rat liver homogenates was determined spectrophotometrically after reduction with thioglycolic acid measuring the absorbance at 535 nm in the presence of bathophenanthroline (Brumby and Massey 1967).
Determination of Liver Ft Content
Ferritin from rat liver was assayed for Western blot analysis. Rat liver samples were homogenized as described above and mixed with Tris-HCl buffer, pH 6.8 containing glycerol 10% (v/v), SDS 1% (w/v), β-mercaptoethanol 2.5% (v/v), and bromophenol blue 0.01% (w/v), and were separated by electrophoresis in 12% SDS-polyacrylamide gel under denaturating conditions. Gels were layered onto a 0.2-µm nitrocellulose membrane (BioRad, Hercules, CA, USA) to transfer the proteins by electroblotting. The nitrocellulose membranes were incubated with anti-horse spleen Ft primary antibody developed in the rabbit (1/5,000 dilution) and a horseradish peroxidase–conjugated goat anti-rabbit secondary antibody (Santa Cruz Biotechnology, USA) (1/5,000 dilution), followed by development of chemiluminescence using an Immun-Start HRP chemiluminescent kit (BioRad, Hercules, CA, USA). Band intensity was determined employing Scion Image for Windows (4.03, NIH).
Purification of Rat Liver Ft
Ferritin from rat liver was isolated according to Thomas et al. (1985) with modifications. The excised livers were homogenized in a blender using two volumes of extraction buffer (25 mM sodium acetate, 20 mM EDTA, pH 4.8) at 4°C. The homogenate was centrifuged for twenty minutes at 1,500 g at 4°C. The supernatant was filtered through a 53 µm nylon filter and added to an equal volume of 50% (w/v) saturated ammonium sulfate with continuous stirring for fifteen minutes and allowed to settle overnight. The pellet was dissolved in 25 mM sodium acetate buffer with 20 mM EDTA, pH 4.8, and centrifuged at 100,000 g for ninety minutes. The pellet was suspended in PBS (0.02 M phosphate buffer, pH 7.4, containing 0.1 M sodium chloride), and the nonsolubilized material was removed by centrifugation at 10,000 g for one hour. The supernatant was centrifuged again at 100,000 g for ninety minutes. The pellet containing the Ft was resuspended in 20 mM PBS buffer (pH 7.4) with 0.1 M NaCl, and the supernatant was then loaded onto a Sephacryl S-300 column (1.6 cm × 35 cm) and equilibrated in the same buffer. Fractions were collected, and proteins and Fe concentrations were assessed. The Fe-rich fractions were pooled and then concentrated through filters with 30,000 nominal molecular weight limit (Centricon YM30). The Ft pellets were dissolved in 40 mM potassium phosphate buffer, 0.15 M NaCl, pH 7.4 and stored a 4°C until use. Protein content in the sample was measured according to Bradford (1976) using BSA as standard. Total Fe content was determined as described above. Ferritin purity was verified by an SDS-PAGE (Stacking gel: 4% T, 75 V, twenty-two minutes. Resolution gel: 14% T, 150 V, sixty minutes). Prior to use, isolated Ft samples were incubated on ice in the presence of 10 mM EDTA for sixty minutes and passed through a Sephadex G-25 column equilibrated with 0.3 M NaCl (pH 7.0) to remove loosely associated Fe (Saito et al. 1985).
Fluorescence spectroscopy of isolated Ft was used to monitor for the intrinsic fluorescence of tryptophan (Trp) content in Ft (λex=285 nm, λem= 345 nm; Rousseau and Puntarulo 2009), employing Ft (20 µg/mL ≈ 0.05 µM) in 50 mM Hepes buffer (pH 7.0).
Determination of LIP in Rat Liver Homogenates
Determination of LIP was by EPR at 77K, according to Woodmansee and Imlay (2002), with modifications. Rat liver samples were homogenized in 10 mM Tris-HCl buffer, 120 mM KCl (pH 7.4), and 1 mM deferoxamine (DF). Samples were incubated at room temperature for ten minutes and then frozen with liquid nitrogen in a syringe. Electron paramagnetic resonance spectra were recorded under the following experimental conditions: 9.75 GHz, microwave frequency; 20 mW, microwave power; 50 kHz, modulation frequency; 4.759 G, modulation amplitude, 1600 G, centered field; 81.92 ms, time constant, and 800 G, sweep width.
Determination of the Content of Carbonyl Groups in Proteins
Carbonyl groups in proteins were derivatized as described by Levine et al. (1994). Samples were mixed with an equal volume of SDS (12% w/v) and then with two volumes of 20 mM dinitrophenylhydrazine dissolved in 10% (v/v) trifluoracetic acid. This mixture was incubated for twenty-five minutes at room temperature, and the reaction was stopped by adding 1.5 sample volumes of 2 M Tris-HCl/30% (v/v) glycerol. Proteins (3 µg per well) were loaded in 12% (w/v) acrylamide concentration mini-gels and run at room temperature under conditions of constant electrophoretic voltage (150 V) for one hour. For Western blot assays, proteins were electrotransferred to nitrocellulose membranes at 120 V for one hour. Blots were blocked with 5% (w/v) nonfat dry milk dissolved in PBS-T (10 mM potassium phosphate buffer pH 7.4, 150 mM NaCl, 0.1% [v/v] Tween 20), incubated overnight with primary antibody dissolved in blocking buffer (1/100), and washed several times with PBS-T. For carbonyl group detection, the primary antibody was rabbit anti-dinitrophenyl group (anti-DNP; Zymed, USA). Blots were then incubated for two hours with the secondary antibody (goat anti-rabbit IgG conjugated to horseradish peroxidase) (Santa Cruz Biotechnology, Santa Cruz, CA, USA) prepared 1/10,000 in PBS-T with 1% (w/v) nonfat dry milk, washed several times with PBS-T, and developed with a chemiluminescence detection kit (Bio-Rad, Hercules, CA, USA). Band intensity was determined employing Scion Image for Windows (4.03, NIH).
Western Blot Analysis for Assessing Nitrotyrosine Content in Liver Proteins
Rat liver samples were homogenized as described above, and they were mixed with an equal volume of sample buffer according to Laemmli (1970) and incubated for five minutes at 95°C. Proteins (30 µg per well) were loaded into 12% (w/v) acrylamide mini-gels, and electrophoresis was performed at room temperature under the conditions described above. After protein transference, membranes were blocked with 5% (w/v) nonfat dry milk dissolved in PBS-T, incubated overnight with the primary antibody dissolved in blocking buffer (1/200), and washed several times with PBS-T. Mouse anti-nitrotyrosine IgG (Chemicon International, Temecula, CA, USA) was used as the primary monoclonal antibody. Membranes were then incubated for two hours with the secondary antibody (goat anti-mouse IgG conjugated to horseradish peroxidase) (Santa Cruz Biotechnology, Santa Cruz, CA, USA) prepared 1/10,000 in PBS-T with 1% (w/v) BSA and washed several times with PBS-T. Western blot assays were developed by using a chemiluminescence kit (Bio-Rad, Hercules, CA, USA). Band intensity was assessed using Scion Image for Windows (4.03, NIH).
Statistical Analyses
All results are from experiments carried out in duplicate and replicated with at least three different preparations. Where indicated, values refer to mean ± SEM. Statistical tests (ANOVA) were carried out using Statview for Windows, version 5.0 (SAS Institute Inc., Cary, NC, USA).
Results
Treatment with LPS produced a significant decrease in serum Fe content (Table 1), as was previously reported (Galleano et al. 2008). Serum Fe levels increased dramatically in Fe-dextran–treated animals, independently of LPS administration (Table 1), suggesting a failure in the Fe-sequestering strategy under the high Fe levels present in these animals. The EPR spectrum of NO-Hb in the blood was detected at 77K, and an NO-Hb signal, showing a hyperfine splitting of nitrogen with a g value of 2.033, was seen in LPS and Fe-dextran-LPS–treated animals (Figure 1). A nonsignificantly different signal in the Fe-dextran-LPS group as compared to the LPS-treated animals was observed, as previously described (Galleano et al. 2004). Significantly lower signal was detected without an injection of LPS (Table 1). These data indicated that animals responded adequately to both treatments.
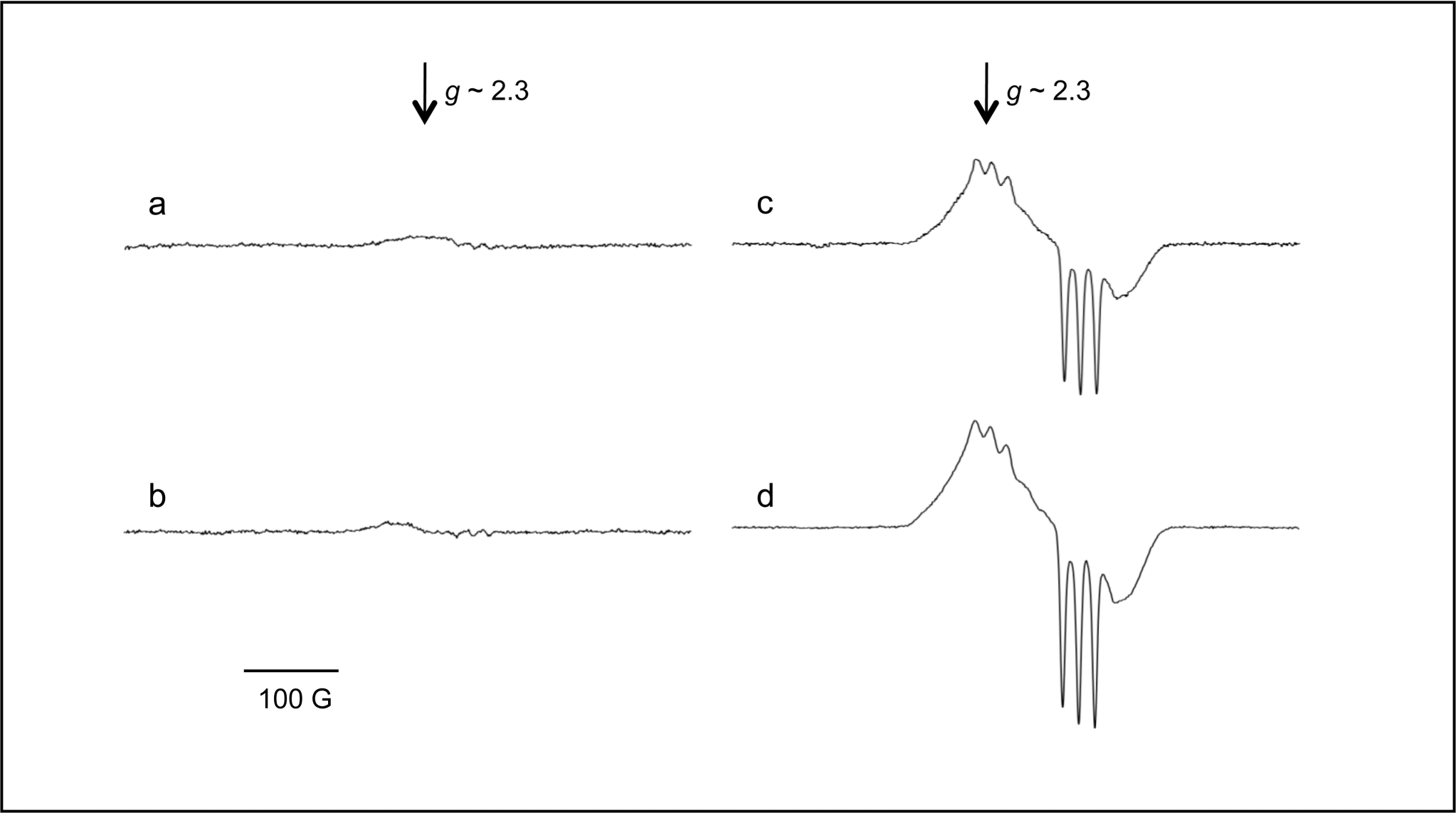
NO detection in rat blood as the signal from NO-Hb adduct at 77K. Typical electron paramagnetic resonance spectra of venous blood from rats after six hours of being injected with: (a) saline solution (Control); (b) Fe-dextran; (d) LPS; (e) Fe-dextran plus LPS are shown. Three or four animals per group were used in each experiment.
Serum Fe and NO content in rat blood.

*significantly different from values in control animals (p ≤ 0.01, ANOVA).
**significantly different from values in LPS treated animals (p ≤ 0.01, ANOVA).
a.u. stands for arbitrary units.
Three or four animals per group were used in each experiment.
Electron paramagnetic resonance detection of LIP by the signal of the DF-Fe3+ complex is understood as a reliable method (Tarpey et al. 2004). The typical EPR signal of DF-Fe3+ complex is shown in Figure 2A. A significant increase in the signal (45%) was observed in rat liver six hours after Fe-dextran administration, as compared to values in nontreated animals. However, LPS supplementation by itself did not affect the LIP content in the liver as compared to control animals, and the simultaneous administration of LPS and Fe-dextran did not significantly affect the LIP content detected in Fe-dextran–supplemented rats (Figure 2B).
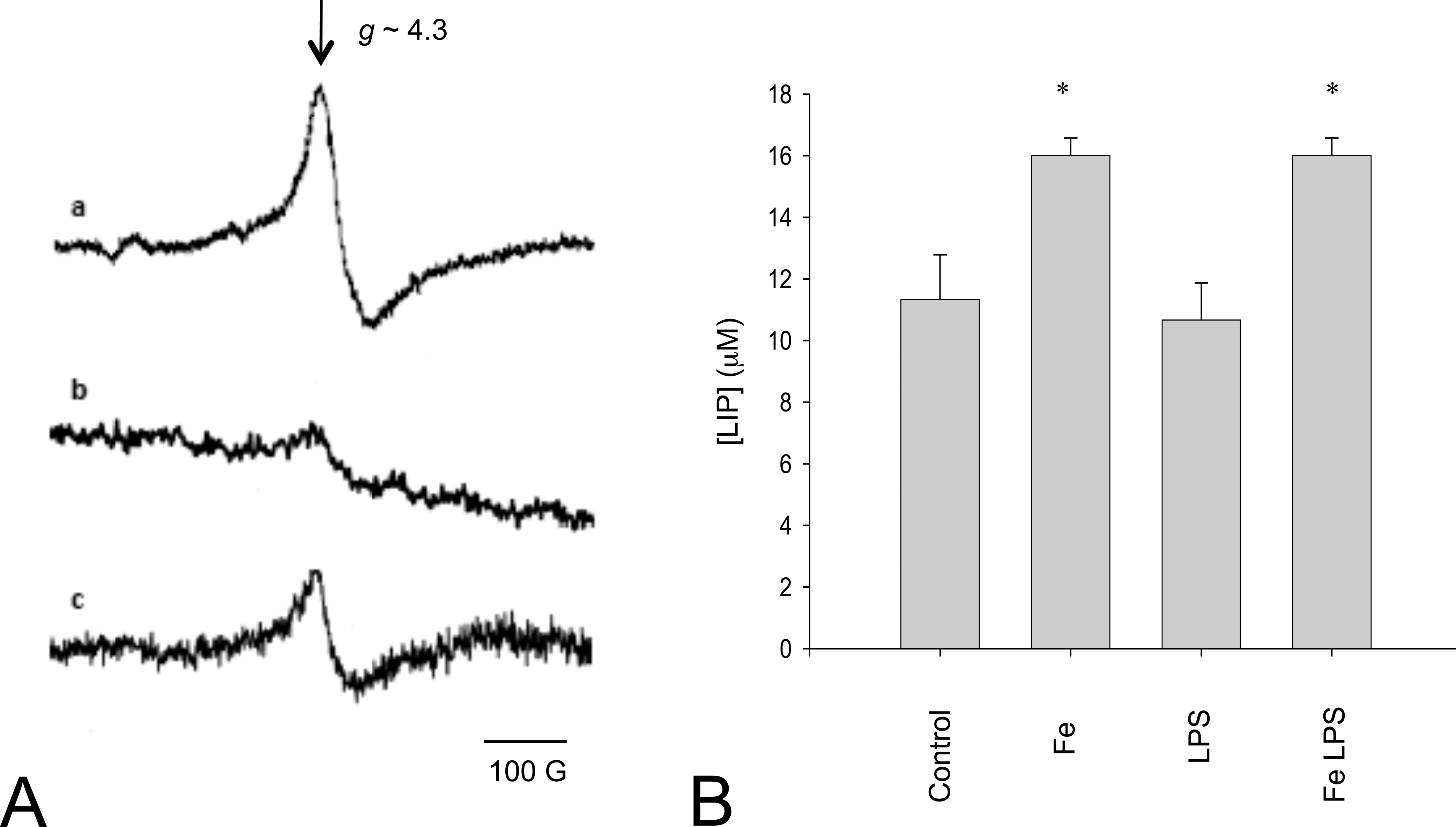
Labile iron pool determination by electron paramagnetic resonance. (A) Spectra from: (a) 50 µM standard Fe, (b) homogenates in 10 mM Tris-HCl buffer, 120 mM KCl pH 7.4 and 1 mM DF, from liver of saline solution injected rats (Control), and (c) liver homogenate from liver of Fe overloaded rats. Since spectra from liver of lipopolysaccharide (LPS)-treated rats and LPS and Fe-dextran–treated rats were not significantly different from spectra from control and Fe-dextran–injected rats, respectively, they are not shown. (B) Quantification of labile iron pool in liver of saline solution–injected rats (Control), and Fe-overloaded rats in the presence and absence of administration of LPS. Three or four animals per group were used in each experiment. *Significantly different from values in control animals (p ≤ .01, ANOVA).
Oxidative modifications on liver proteins were studied by Western blot employing anti-DNP antibodies, which allow identification of previously derivatized protein carbonyls (Figure 3A). Fe-dextran administration increased the content of carbonyl groups in liver proteins by 50% as compared to control livers, whereas LPS administration by itself did not affect the total amount of oxidized liver proteins. The administration of Fe-dextran and LPS increased carbonyl protein content, as compared to values in livers from LPS-treated rats, by 52% (Figure 3B).
Extent of protein oxidation in rat liver. (A) Protein carbonyl content was revealed by treatment with dinitrophenylhydrazine. Protein oxidation was determined by Western blot using anti-DNP antibodies in samples from liver of saline solution–injected rats (Control) and Fe-overloaded rats in the presence and absence of LPS. (B) Quantification of relative carbonyl content in liver from saline solution–injected rats (Control) and Fe-overloaded rats in the presence and absence of LPS. The exposed films were quantified with a densitometer, and the carbonyl content of proteins was expressed as a percentage of the corresponding values in control samples. Three or four animals per group were used in each experiment. *Significantly different from values in control animals (p ≤ .01, ANOVA).
One of the molecular footprints left by the reactions of reactive nitrogen species with biomolecules is the nitration of protein tyrosine residues. The levels of protein 3-nitrotyrosines were not increased by Fe-dextran administration but were increased by 133% and 100% by LPS supplementation in the absence and the presence of Fe-dextran, respectively (Figure 4). These results showed a correlation between NO level in tissues and the nitration of protein tyrosine, indicating that protein 3-nitrotyrosines are biomarkers of in vivo NO-dependent metabolism.
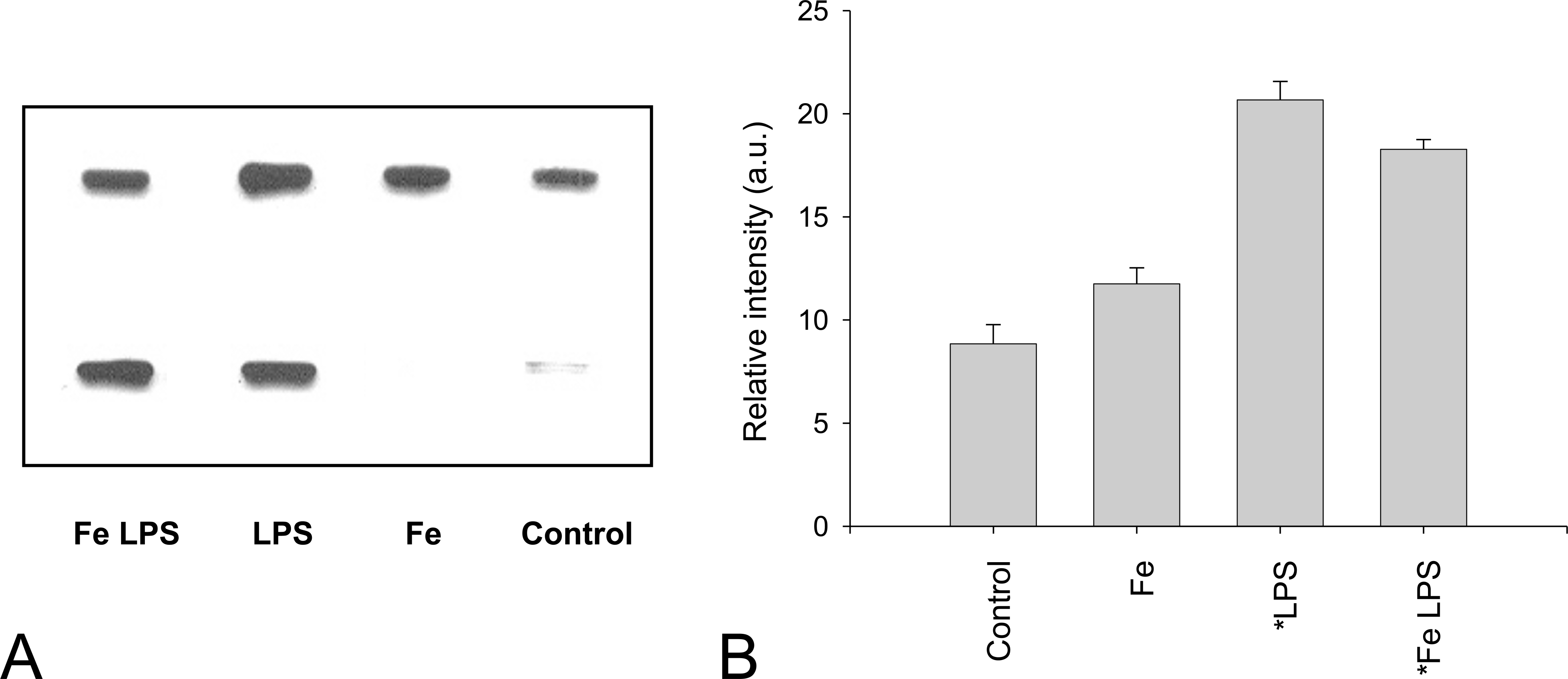
Nitrotyrosine content in rat liver proteins. (A) Typical Western blot assay. (B) Band intensity in samples from the liver of saline solution–injected rats (Control) and Fe-overloaded rats in the presence and absence of administration of LPS. Three or four animals per group were used in each experiment. *Significantly different from values in control animals (p ≤ .01, ANOVA).
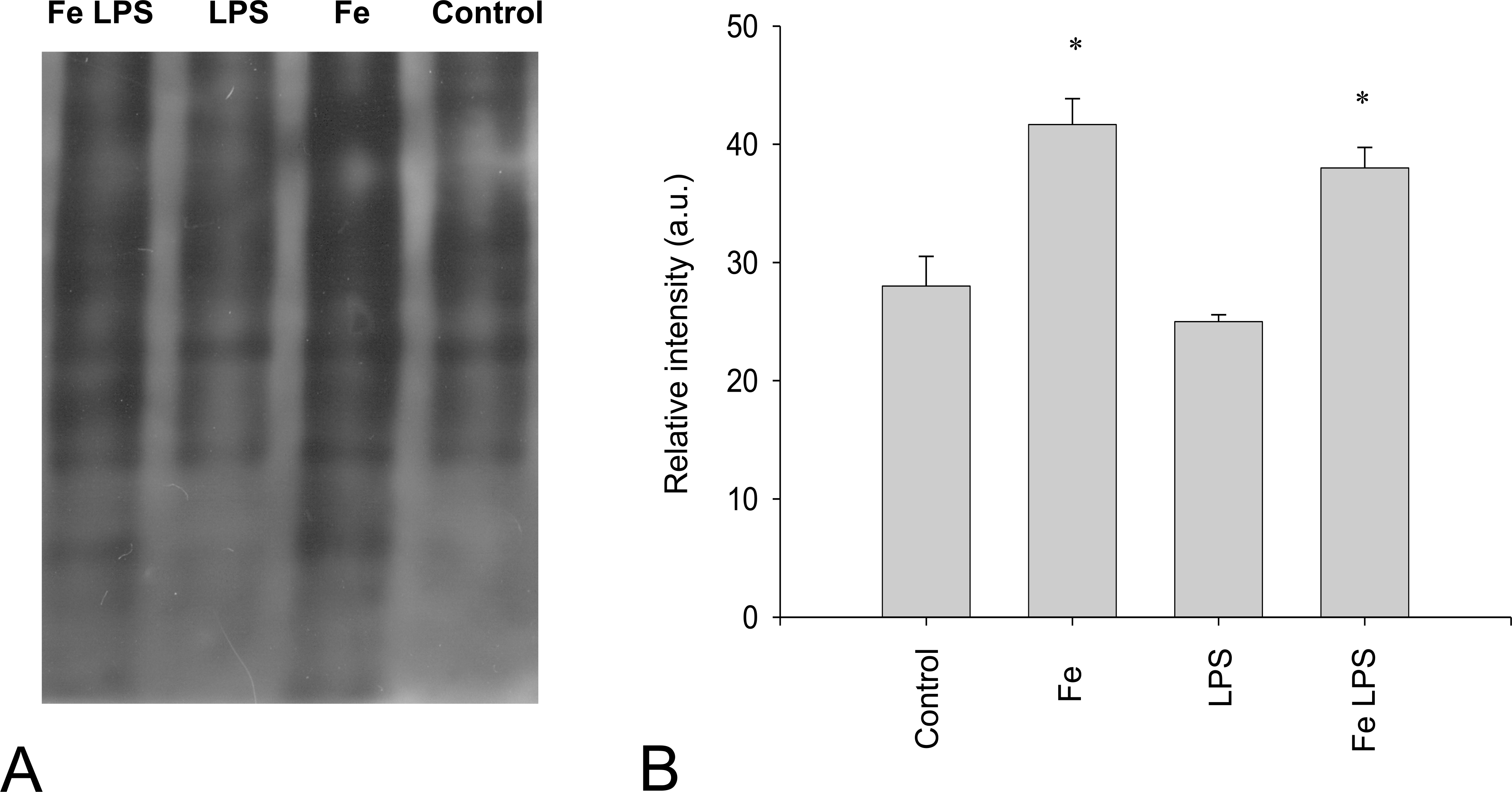
The Fe released from Ft can be incorporated to the cellular LIP, which could be responsible for catalyzing free radical–dependent alterations to lipids, proteins, and DNA (Galatro et al. 2007). On the other hand, NO chelates labile Fe in a form that decreases its potential to yield reactive intermediates, and NO reacts with and scavenges free radicals. However, NO is able to react with superoxide anion to generate peroxynitrite, which could exert serious, deleterious cellular effects (Radi et al. 1995). Thus, in this model with increases in both Fe and NO content, the scenario could be very complex and either be damaging (because of Fe-related formation of free radicals and peroxynitrite-dependent reactions) or a protective effect (because of NO “antioxidant properties”) could be postulated over the Ft protein, which seems to be a critical target. Analyses of the Ft peptides from rat liver, by Western blot, indicated that the proteins were composed of approximately 20 kDa protein subunits (Figure 5A). The content of Ft in rat liver of Fe-overloaded rats increased by 116% as compared to control livers, and in the presence of LPS, the Ft content in liver was increased by 122% after Fe-dextran administration as compared to the content in its absence (Figure 5B).
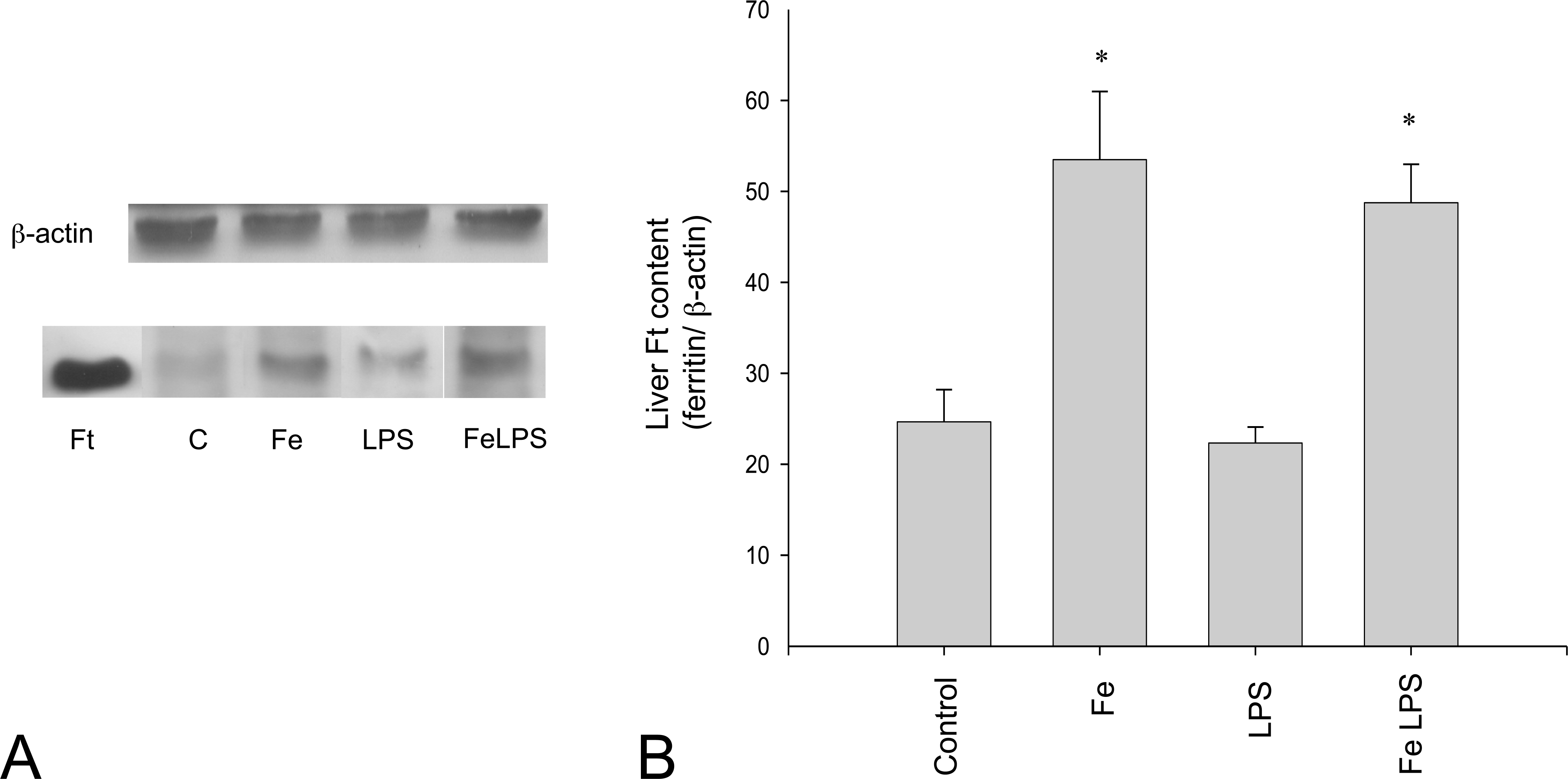
Analyses of Ft isolated from rat liver. (A) Western blot analysis showing the bands for horse spleen Ft (commercial) and Ft bands from protein isolated from liver of saline solution–injected rats (Control) and Fe-overloaded rats in the presence and absence of LPS. (B) Quantification of Ft content in liver of saline solution–injected rats (Control) and Fe-overloaded rats in the presence and absence of LPS. The expression of β-actin was detected with anti-β-actin antibody. Three or four animals per group were used in each experiment. *Significantly different from values in control animals (p ≤ .01, ANOVA).
Data in Table 2 show that the Fe content in the isolated Ft was increased by Fe overload by 26% and 33% in the absence and the presence of LPS, respectively, as compared to the appropriate controls. Previous in vitro studies suggest that the protein primary structure started to be affected during Fe upload, showing a decrease in the exposed Trp content (Rousseau and Puntarulo 2009). The results presented here show a decrease by 53% and 75% in liver Ft content of exposed Trp from rats treated with Fe-dextran in the absence and the presence of LPS, respectively (Table 2).
Effect of the simultaneous administration of Fe and LPS on the Fe and Trp content in isolated Ft from rat liver.
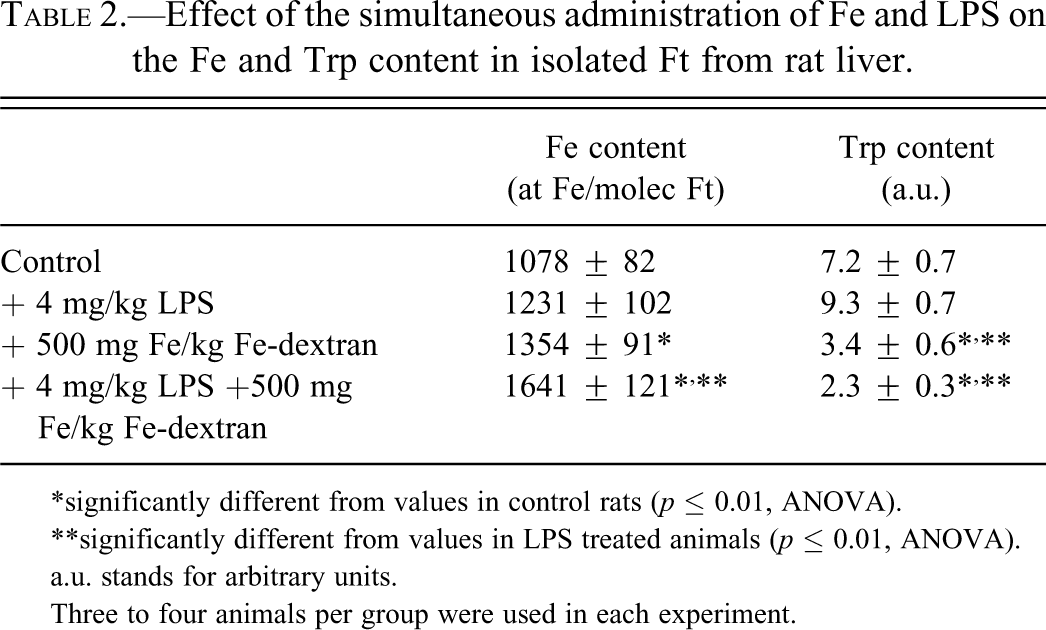
*significantly different from values in control rats (p ≤ 0.01, ANOVA).
**significantly different from values in LPS treated animals (p ≤ 0.01, ANOVA).
a.u. stands for arbitrary units.
Three to four animals per group were used in each experiment.
Discussion
Recent epidemiologic studies have highlighted the potential effect of Fe on microbial infections. Laboratory studies have focused on specific immune mechanisms that mediate Fe withholding from microbes constitutively and in response to infections, since specialized inflammation-regulated proteins chelate Fe, trap siderophores, and transport Fe or modulate its transport to alter its tissue distribution during infections (Ganz 2009). Moreover, it has been proposed that there is a combination of mechanisms to explain the increase in susceptibility to infection in patients with hemochromatosis (Khan et al. 2007), and the propensity of Fe-overloaded patients receiving a liver transplant to develop more fulminant disease presentation and a higher risk of disseminated disease resulting from a number of opportunistic infections (Singh and Sun 2008).
Immune effector cells are stimulated to release NO and ROS (Hampton et al. 1998), probably as a defense strategy, by attacking crucial Fe-containing enzymes of pathogens. However, a number of observations suggested that NO and oxidative stress may control Fe metabolism in immune effector cells from host tissues. Besides the potentially protective effect of NO on lipid peroxidation (Boveris et al. 2000), sepsis has been recognized as a situation associated to oxidative stress, and thus, Fe could aggravate that condition. An experimental model of sepsis including a simultaneous Fe overload condition has been analyzed in terms of oxidative Fe metabolism in the liver. This situation may be relevant to analyze a pathological condition of an acute Fe-overloaded patient subjected to infection and sepsis. Consistently with previous reports (Galleano and Puntarulo 2008), the data shown here indicate that LPS treatment increased the NO-Hb complex content in blood approximately nine-fold and decreased the plasma Fe content by 63% in control rats. However, when the animals were simultaneously subjected to a drastic Fe overload, the NO-Hb complex content in blood increased eight-fold, but the plasma Fe content was not significantly affected. These data strongly suggest that, in the early stages (six hours), this pathway of the defense strategy against pathogens was less operative in the presence of Fe overload, as compared to the response in animals with a physiological Fe content.
The liver also plays an important role for host defenses against infection by reacting in the presence of invading microorganisms and releasing several cytokines. Kupffer cells, which belong to the reticuloendothelial system in the liver and are intended to fulfill different functions than those of ordinary macrophages, showed the ability of further inducing iNOS in the presence of excess Fe (Hida et al. 2003) and releasing IL-6, IL-1β, and TNF-α into the plasma, which could be responsible for increasing NO production in rats preloaded with saccharated colloidal Fe. This observation is supported by in vivo reports indicating that an injection of Fe-dextran enhanced iNOS induction in the liver (Cornejo et al. 2001).
Thus, the role of the liver in the alteration of Fe metabolism seems to be a critical factor. Liver protein oxidation was increased by 50% by Fe overload, and by 60% after the simultaneous administration of Fe-dextran and LPS. Protein nitration was increased over two-fold by LPS administration, and it was not affected by Fe overload. Simultaneous supplementation with Fe-dextran and LPS did not increase the nitration degree observed in the presence of LPS. These data suggest that both treatments were independently effective in altering liver proteins, since Fe-dextran favored the oxidation process and LPS, via NO generation, the nitration process. Because of its role in Fe storage, the protein Ft seems to be a key factor that is affected by the simultaneous administration of excess Fe and LPS. The results presented here showed that both Ft content and Fe content in the Ft were significantly increased by Fe overload; however, endotoxemia did not affect these parameters. Moreover, the exposed Trp content of the protein was also decreased in vivo by Fe overload (53% and 75% in the absence and presence of LPS, respectively), suggesting a comparable deterioration of the Ft structure that could lead to a similar protein degradation rate, even in the presence or absence of endotoxemia.
However, since the Fe formation of the LIP is involved in the catalysis of ROS reactions, the effect of the treatments on this parameter seems critical. The alteration in the steady state concentration of the LIP after Fe exposure could be estimated by considering the following equation (equation 1), in which each term refers to the change in the concentration of Fe bound to each physiologically available Fe chelator in the cells. Under Fe overload conditions that lead to an increase in the LIP, not all the independent terms of the equation could follow the same profile of changing. Moreover, some of them could be substantially increased and others could be unchanged, depending on the availability of the Fe chelators in the cytosol.
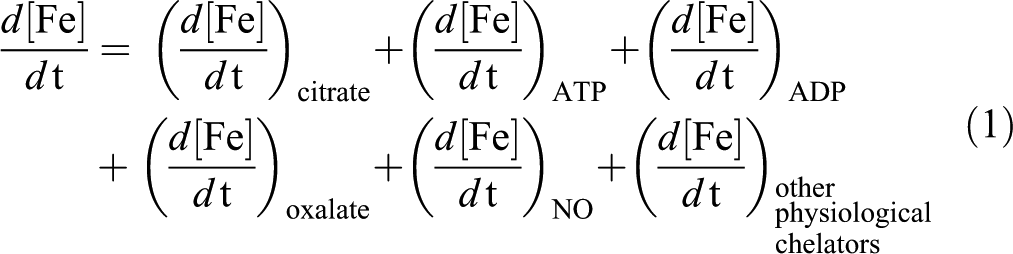
Since NO can readily diffuse across cell membranes (Radi et al. 1995), NO could favor Fe release from the cell, thereby avoiding its accumulation by the formation of the complexes summarized above that can leave the cell through the membrane. Previous reports indicated that acute Fe overload (Fe-dextran 500 mg/kg) significantly increased liver total Fe content (Galleano and Puntarulo 1992, 1994). Since the same increase in LIP content was measured (45%) when the rats were supplemented either with Fe-dextran or simultaneously with Fe-dextran and LPS, it could be suggested that the increased generation of NO did not affect liver availability of catalytically active Fe, as compared to the values in the absence of LPS, in spite of the lack of reduction of Fe content in plasma from animals subjected to both endotoxemia and Fe overload. However, the increase in the NO content after treatment with LPS can be responsible for changing the relative composition of LIP by favoring the generation of Fe-NO complexes. NO can bind to Fe and endogenous thiols and generate dinitrosyl-Fe, dinitrosyl-diglutathionyl-Fe or dinitrosyl-glutathionyl Fe complexes, among other nitrosyl-Fe complexes (Pedersen et al. 2007), as indicated in equation 2.
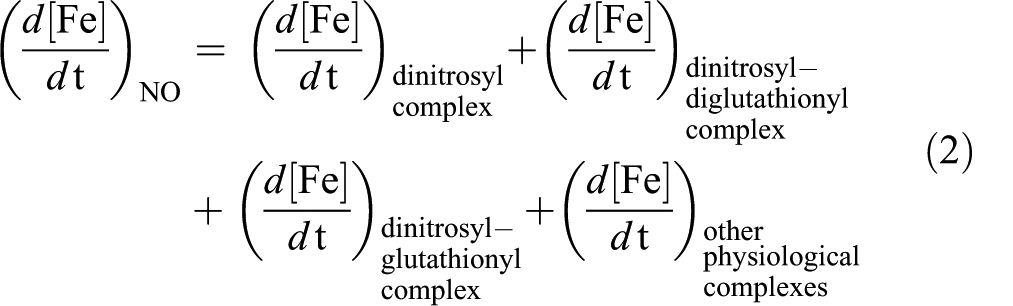
These complexes are supposed to be Fe transporters (Richardson and Lok 2008; Robello et al. 2009; Toledo et al. 2008), thus an increase in their generation after LPS treatment could be a viable mechanism to take Fe out of the liver cells. In this way, total Fe content of the LIP after both simultaneous treatments could be not significantly changed, as compared to Fe overload condition, but Fe release from the cell could improve. Only when this process is overwhelmed, a significant difference by LPS administration would be observed in Fe-overloaded rats. This assumption is consistent with previous reports in bile showing Fe increases after twenty-four hours of LPS administration (Linares et al. 2003).
Taken as a whole, the data reported here indicate that during the early stages of simultaneous administration of excess Fe and LPS, the protective strategy against endotoxemia of reducing plasma Fe content is not fully operative. However, the liver does not seem to be being affected since endogenous mechanisms were able to regulate the amount of catalytically active Fe at the same level observed in the absence of LPS administration, but probably changing the composition of the LIP. This change could, for longer periods, favor the mobilization of Fe to improve depletion mechanisms to avoid further damage. Further studies are required to elucidate the real nature of the early interactions of Fe and NO, in terms of the effect on the composition of the LIP in the liver and in blood, to assess the importance of Fe depletion in the complex mechanisms of sepsis control.
Footnotes
Abbreviations
Acknowledgments
This study was supported by grants from the University of Buenos Aires (B105), and CONICET (PIP 1171). S.P. and M.G. are career investigators from CONICET, and I.R. is a fellow at the University of Buenos Aires.