
Abstract
The role of estrogens during myocardial ischemia has been extensively studied. However, effects of a standard hormone replacement therapy including 17β-estradiol (E2) combined with medroxyprogesterone acetate (MPA) have not been assessed, and this combination could have contributed to the negative outcomes of the clinical studies on hormone replacement. We hypothesized that adding MPA to an E2 treatment would aggravate chronic heart failure after experimental myocardial infarction (MI). To address this issue, we evaluated clinical signs of heart failure as well as left ventricular (LV) dysfunction and remodeling in ovariectomized rats subjected to chronic MI receiving E2 or E2 plus MPA. After eight weeks MI E2 showed no effects. Adding MPA to E2 aggravated LV remodeling and dysfunction as judged by increased heart weight, elevated myocyte cross-sectional areas, increased elevated left ventricle end diastolic pressure, and decreased LV fractional shortening. Impaired LV function in rats receiving MPA plus E2 was associated with increased cardiac reactive oxygen species generation and myocardial expression levels of NADPH oxidase subunits. These results support the interpretation that adding MPA to an E2 treatment complicates cardiovascular injury damage post-MI and therefore contributes to explain the adverse outcome of prospective clinical studies.
Introduction
Aging is associated with an increased incidence of cardiovascular disease, especially in the transition phase to menopause. Observational studies suggested that combined hormone replacement therapy (HRT) including estrogen and a progestin would prevent menopause associated disorders and cardiovascular disease (Stampfer et al. 1991). However, clinical endpoint studies including the Heart and Estrogen/Progestin Replacement Study (HERS) and the Women’s Health Initiative (WHI) trial, which employed equine estrogens plus medroxyprogesterone acetate (MPA), failed to show a preventive effect of HRT on cardiovascular morbidity and mortality (Grady et al. 2002, 2000; Rossouw et al. 2002).
Considerable information has been collected on the cardiovascular function of estrogens and the estrogen receptor subtypes ER-α and ER-β (Mendelsohn 2002; Pelzer, Jazbutyte, et al. 2005; Hügel et al. 1999). But it remains unclear whether progestins, and in particular MPA, might aggravate chronic heart failure after myocardial infarction (MI) (Hulley 2000). Since progesterone receptors are functionally expressed in vascular cells and cardiac myocytes, progestins might either antagonize the protective function of estrogens or confer independent and unfavorable cardiovascular effects (White et al. 1995; Perrot-Applanat et al. 1988; Sitruk-Ware 2003). In addition, synthetic progestins such as MPA not only interact with progesterone receptors, but also activate androgen- (AR), mineralocorticoid- (MR), and glucocorticoid- (GR) receptors (Sitruk-Ware 2002, 2004; Wassmann et al. 2001). The effects of different synthetic progestins are thus likely to be more complex and variable than those of endogenous progesterone (Oishi et al. 2004; Simoncini et al. 2004). We have recently shown that adding MPA to 17β-estradiol (E2) treatment worsens renal injury due to excess MR activation (Arias-Loza et al. 2009). Very few experimental studies have focused on the effect of unopposed (estradiol) versus combined (estradiol plus MPA) HRT, and no one has addressed the important question of whether the addition of MPA might aggravate chronic heart failure (HF) and left ventricular (LV) dysfunction following MI.
We therefore tested the hypothesis whether a combined hormone supplementation of MPA and E2, but not E2 alone, would aggravate cardiac remodeling and dysfunction in ovariectomized (ovx) rats with chronic HF due to large MI. In addition, we tested whether the MR antagonist spironolactone (SPIRO) would antagonize the unfavorable effects of MPA on cardiac function after MI.
Methods
Animal Model
Female 8-week-old Wistar rats were obtained from Harlan Winkelmann (Borchen, Germany) and were randomized into seven treatment groups. Animals in groups 1 and 2 were sham ovx (intact), and animals in group 3, 4, 5, 6 and 7 were ovx. Ten days later, a ligature was placed around the proximal left anterior descendent coronary artery in all animals, but MI was induced only in groups 2 through 7 by tightening the ligature, as described before (Pelzer, Loza, et al. 2005). Pharmacological treatment was initiated immediately after ovariectomy. The following treatment groups were included: group 1, sham MI/intact ovaries; group 2, MI/intact ovaries; group 3, MI/ovx/placebo; group 4, MI/ovx/E2 (2 µg/kg BW/d); group 5, MI/ovx/ E2/MPA (E2 2 µg/kg; MPA 1mg/kg BW/d); group 6, MI/ovx/E2/MPA/SPIRO (SPIRO 80/kg BW/d); and group 7, MI/ovx/E2/SPIRO. Steroids were dissolved in ethanol and administered daily by subcutaneous injections using peanut oil as a carrier. SPIRO was dissolved in water and administered by gavage. Placebo-treated rats received peanut oil/ethanol injections. All surgical procedures were carried out under isoflurane anesthesia (isoflurane 1.5vol% supplemented by 0.5l oxygen per minute). Body weight, heart weight, uterus weight, and tibia length were measured following hemodynamic analysis. Relative heart weight was calculated from absolute heart weight, tibia length as previously described (Yin et al. 1982). Cardiac myocyte cross-sectional areas (CCSA) were measured using a staining for cellular membranes (Invitrogen, wheat germ agglutinin, Alexa fluor 594 conjugate) and manual tracing of LV cardiomyocytes located in the area remote to the infarct scar, using the NIH software Image J. Serum estradiol levels were measured by radio immunoassays (DPC-Biermann). This study was approved by the institutional animal research committee; the animals were kept under standard conditions at the animal facility of the University Clinic Wuerzburg.
Infarct Size
Infarct sizes were determined using mid-ventricular transverse sections (6 µM) fixed in Tissue-TEK OCT and stained with hematoxylin-eosin (HE) and picrosirius red (PSR). Infarct sizes were calculated as previously described, by manual tracings of ischemic areas in HE and PSR-stained sections, and are expressed as the percentage of infarcted versus total LV circumference (n = 3 sections/animal) (Pelzer, Loza, et al. 2005). The number of rats with minor infarcts measuring less than 30% of the LV circumference was not different among all treatment groups; animals with small infarcts were excluded from further analysis.
Hemodynamic and Echocardiographic Analysis
Echocardiography was performed 8 weeks post-MI using a Toshiba Aplio and a 15 MHz transducer under isofluorane anesthesia. 2D left-parasternal short-axis and transversal M-mode tracing were recorded according to published protocols (Pelzer, Loza, et al. 2005). Hemodynamic measurements were performed 8 weeks post-MI, under light isofluorane anesthesia and spontaneous respiration (Pelzer, Loza, et al. 2005). All measurements were performed by a single and trained observer blinded to treatment.
In situ detection of reactive oxygen species (ROS) by APF (3’- [p-amino phenyl] fluorescein) and DHE (dihydroethidium) staining. The fluorescent dye APF (Molecular Probes-Invitrogen) was used to visualize ROS generating enzyme activities in 25 µm cardiac cross-sections mounted on cover slides and incubated in a buffer containing CaCl2 (10 mM), MgCl2 (1.1 mM), KCl (5.4 mM), NaCl (140 mM), NaH2PO4 (4.2 mM), glucose (10 mM), NaHCO3 (23 mM), and APF (5 µM) for 1 h. Negative control slides were pretreated with superoxide dismutase (SOD; 1 µg/ml; Sigma) and catalase (1 µg/ml; Sigma) for 2 h. The reactions were stopped and samples were fixed by adding 4% paraformaldehyde in 100 mM/L PBS (pH 7.4). Digital images were obtained using a Nikon ACT 1 imaging system. Fluorescence intensities were quantified in 3 randomly selected fields of view in 3 sections from each animal using the Scion software (NIH).
In situ production of superoxide was evaluated by using dihydroethidium (DHE) according to published protocols (Socha et al. 2007). In brief, 25 µm heart sections were placed on slides, incubated in the presence of DHE (0.1 µM; D1168; Molecular Probes, Karlsruhe, Germany) in a light-protected humidified chamber (37°C, 20 minutes) and washed in PBS for 5 minutes before costaining with the DNA-specific dye SYTOX Green (Invitrogen, Karlsruhe, Germany). After PBS washing, sections were mounted with VectaShield mounting medium (Vector Laboratories, Otzberg, Germany); imaging was performed on a fluorescence imaging system (Keyence/Biozero, Neu-Isenburg, Germany). DHE staining was quantified using the Image J software (NIH).
Colocalization of APF and DHE Signals with the Microvascular Marker Platelet/Endothelial Cell Adhesion Molecule 1 (PECAM 1)
Following the ROS detection assays (DHE, APF) cardiac cross-sections were washed in PBS and incubated with nonidet P-40 and donkey serum. Afterward the sections were incubated O/N at 4°C with a primary antibody directed against PECAM 1 (Santa Cruz 1:50 rabbit polyclonal). Subsequently sections were incubated with donkey anti-rabbit IgGs Alexa Fluor 488 or Alexa Fluor 594 conjugate (1:300 dilution; Invitrogen). Directly adjacent sections, in which primary antibodies were omitted, served as negative controls.
Western Blotting Experiments
Cardiac protein expression was analyzed by Western blotting of crude cardiac extracts from the LV myocardium remote of the infarct. The following antibodies were used: anti-GAPDH (Chemicon 1:3,000 mouse monoclonal), anti-phospholamban (1:2,000 mouse monoclonal, Alexis), anti-phospho-phospholamban (Upstate 1:200 rabbit polyclonal), anti-p-67phox (BD Transduction Laboratories 1:500 mouse monoclonal), anti-SERCA 2a (1:1,000 rabbit polyclonal, Abcam), and anti-gp-91phox (1:500 BD Transduction Laboratories mouse monoclonal). LV extracts containing protease and phosphatase inhibitors were subjected to SDS PAGE and transferred onto nylon membranes; immunoreactive proteins were visualized by HRP-coupled secondary antibodies and ECL (Amersham). The ImageQuant software (Biometra) was used for densitometric analysis based on peak area.
Statistics
All data are expressed as mean + SEM. Multigroup comparisons were done by ANOVA tests followed by Students-Newman-Keuls post hoc pairwise testing. P-values < .05 were considered as significant.
Results
Infarct Size and Mortality
Extensive LV scar formation was detected in all animals subjected to experimental MI but not in sham operated rats. Mean infarct size was not different among all groups of infarcted rats and included 46–48% of the LV circumference (Table 1 ). Mortality rates within 48 hours post-MI were below 10% and not different between all treatment groups. Afterwards, only a single animal that received MPA plus E2 died 25 days post-MI.
Global, hemodynamic, echocardiography, and histological measurements.
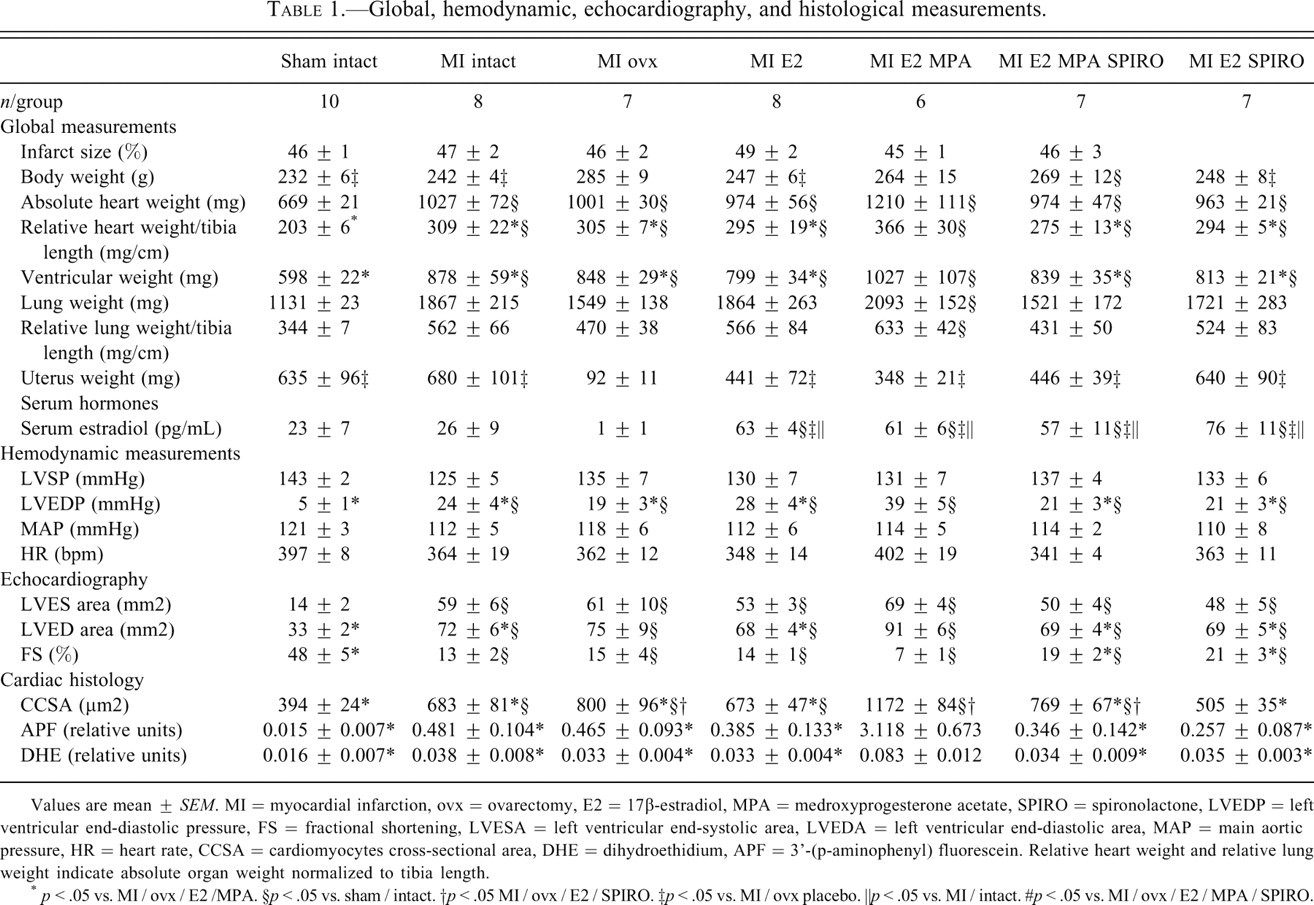
Values are mean ± SEM. MI = myocardial infarction, ovx = ovarectomy, E2 = 17β-estradiol, MPA = medroxyprogesterone acetate, SPIRO = spironolactone, LVEDP = left ventricular end-diastolic pressure, FS = fractional shortening, LVESA = left ventricular end-systolic area, LVEDA = left ventricular end-diastolic area, MAP = main aortic pressure, HR = heart rate, CCSA = cardiomyocytes cross-sectional area, DHE = dihydroethidium, APF = 3’-(p-aminophenyl) fluorescein. Relative heart weight and relative lung weight indicate absolute organ weight normalized to tibia length.
* p < .05 vs. MI / ovx / E2 /MPA. §p < .05 vs. sham / intact. †p < .05 MI / ovx / E2 / SPIRO. ‡p < .05 vs. MI / ovx placebo. ║p < .05 vs. MI / intact. #p < .05 vs. MI / ovx / E2 / MPA / SPIRO.
Global Measurements
Body weight was higher in ovx compared to intact rats but not altered by MI (Table 1). Hormone supplementation with E2, but not with MPA plus E2, attenuated the adipose phenotype of estrogen depleted rats. Uterus atrophy in ovx animals was attenuated by E2 and not significantly altered by MPA or SPIRO. In comparison to sham intact animals, lung weight and lung weight relative to tibia length were significantly increased only in animals receiving E2 plus MPA. Estradiol levels were lower in ovx rats compared to rats with intact ovaries. Supplementation of E2 resulted in serum levels usually observed during estrus (Table 1).
Cardiac Hypertrophy
Absolute as well as relative heart weight normalized to tibia length was elevated within all groups of infarcted compared to noninfarcted rats (Table 1). Estrogen supplementation had no effect on cardiac mass, whereas a combined treatment with MPA plus E2 resulted in a further increase of absolute and relative heart weight (Table 1). Addition of SPIRO to the regime E2 plus MPA prevented this additional increase in cardiac mass. Heart weight was not different between E2 treatment and the combination of E2 with only SPIRO. CCSA were increased in infarcted compared to sham operated rats, and this increase was augmented in the combined treatment with E2 plus MPA (Table 1 and Figure 1 ). Addition of SPIRO to the combination E2 plus MPA lowered CCSA to levels comparable to those of rats with intact ovaries after MI. Combination of E2 plus SPIRO resulted in a significant decrease of CCSA in comparison to ovx rats.
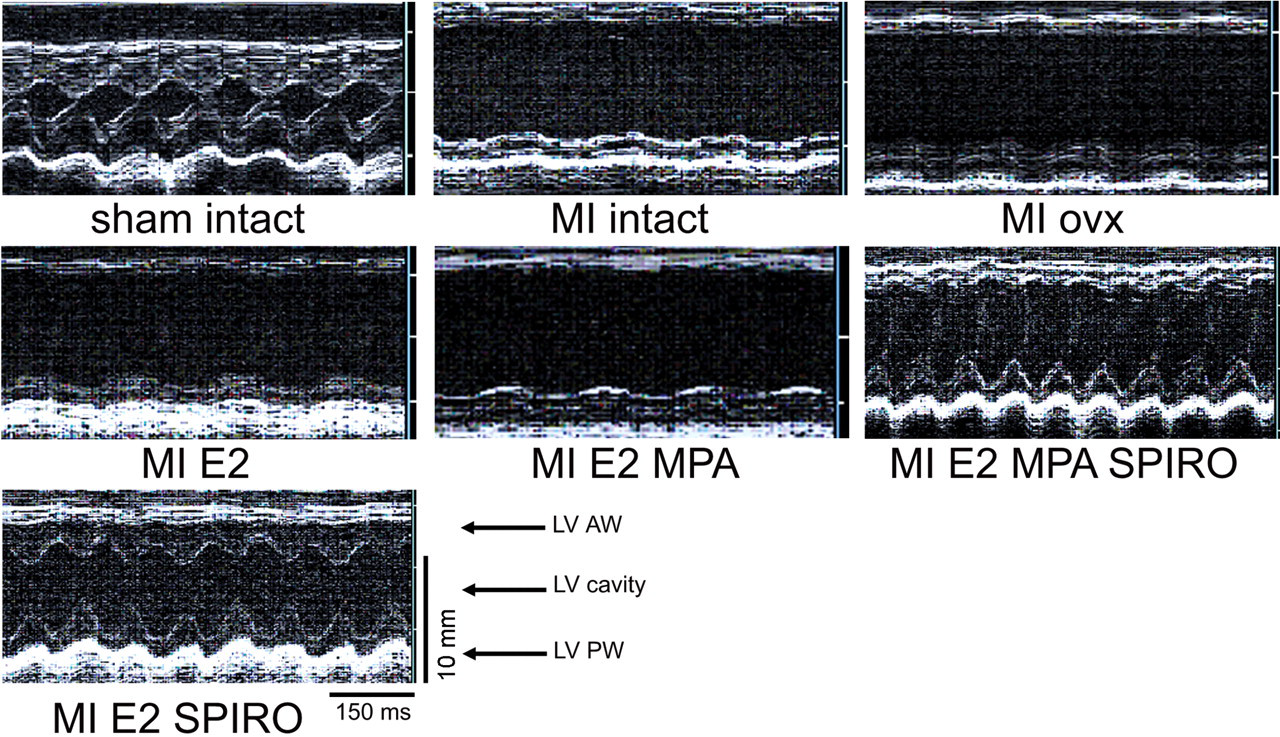
Representative cardiac transversal sections stained with wheat agglutinin. Left ventricle cardiac myocytes of the area remote to the infarct scar were observed by fluorescence microscopy. Myocardial infarction induced an increase in cardiomyocytes cross-sectional area that was further enhanced in animals receiving 17β-estradiol plus MPA. MI = myocardial infarction; ovx = ovariectomy; E2 = 17β-estradiol; MPA = medroxyprogesterone acetate; SPIRO = spironolactone.
Hemodynamic Analysis
LVEDP was higher among infarcted compared to noninfarcted rats. Combined treatment with MPA plus E2 further increased LVEDP (Table 1). Mean blood pressure and heart rate were not different among all groups.
Echocardiography
At a similar heart rate, LV end-diastolic and end-systolic areas were enlarged and LV fractional shortening (FS) was impaired in infarcted compared to sham operated rats (Table 1 and Figure 2 ). Hormone supplementation with E2 or treatment with E2 plus SPIRO did not alter LV dimensions. However, combination of E2 with MPA resulted in further LV dilatation as evident by increased LV end-diastolic and end-systolic dimensions. LV fractional shortening was severely impaired in infarcted compared to sham operated rats and lowest in rats receiving MPA plus E2 (Table 1 and Figure 2). There was no difference regarding LV end-diastolic area and FS between animals receiving E2 or E2 plus MPA and SPIRO.
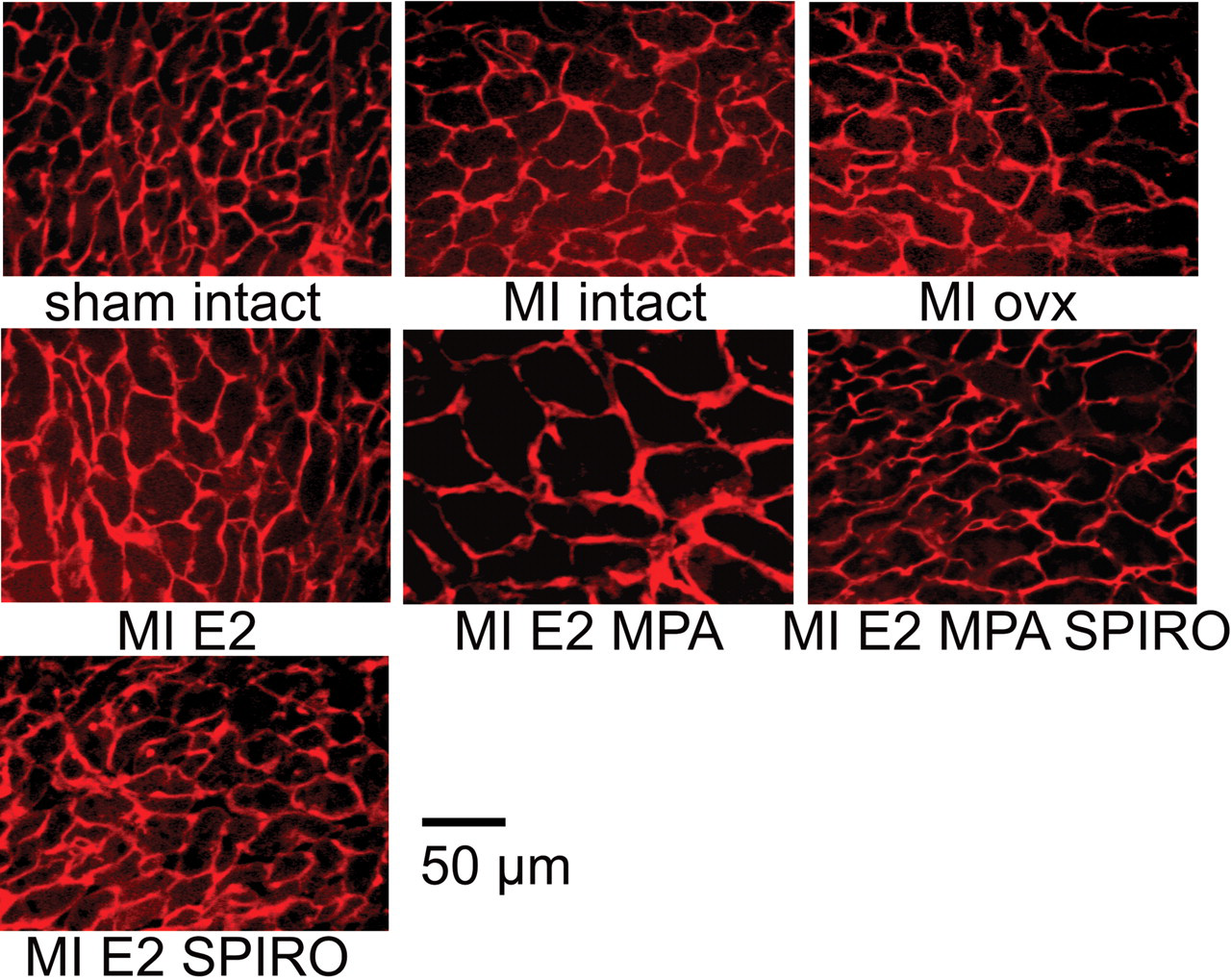
Representative M-mode echocardiograms. Myocardial infarction resulted in ventricular dilatation and a complete loss of anterior wall motion (MI / ovx + E2) that was not observed in noninfarcted rats (sham / intact). Treatment with 17β-estradiol plus MPA aggravated left ventricular dilatation and contractile dysfunction in infarcted rats (MI / ovx + MPA + E2). Groups receiving spironolactone presented improved cardiac contractility. LV = left ventricle; AW = anterior wall; PW = posterior wall.
LV ROS Generation
ROS generating enzyme activities were lower in LV cross-sections from rats not subjected to MI in comparison to rats receiving MI (Table 1; Figures 3 and 4 ). LV ROS generation activity, detected by APF, was significantly increased in cardiac sections of rats receiving the combination MPA plus E2 (Figure 3). Adding SPIRO to the combined treatment E2 plus MPA lowered LV ROS signal. No fluorescence signal was detected in any of the control sections pretreated with SOD and catalase. Combining the ROS generation assays with immuno-fluorescence for the established endothelial cell marker PECAM 1 revealed a consistent colocalization of DHE and APF staining with microvascular endothelial cells, which may have been the major source of LV ROS generation (Figure 5 ).
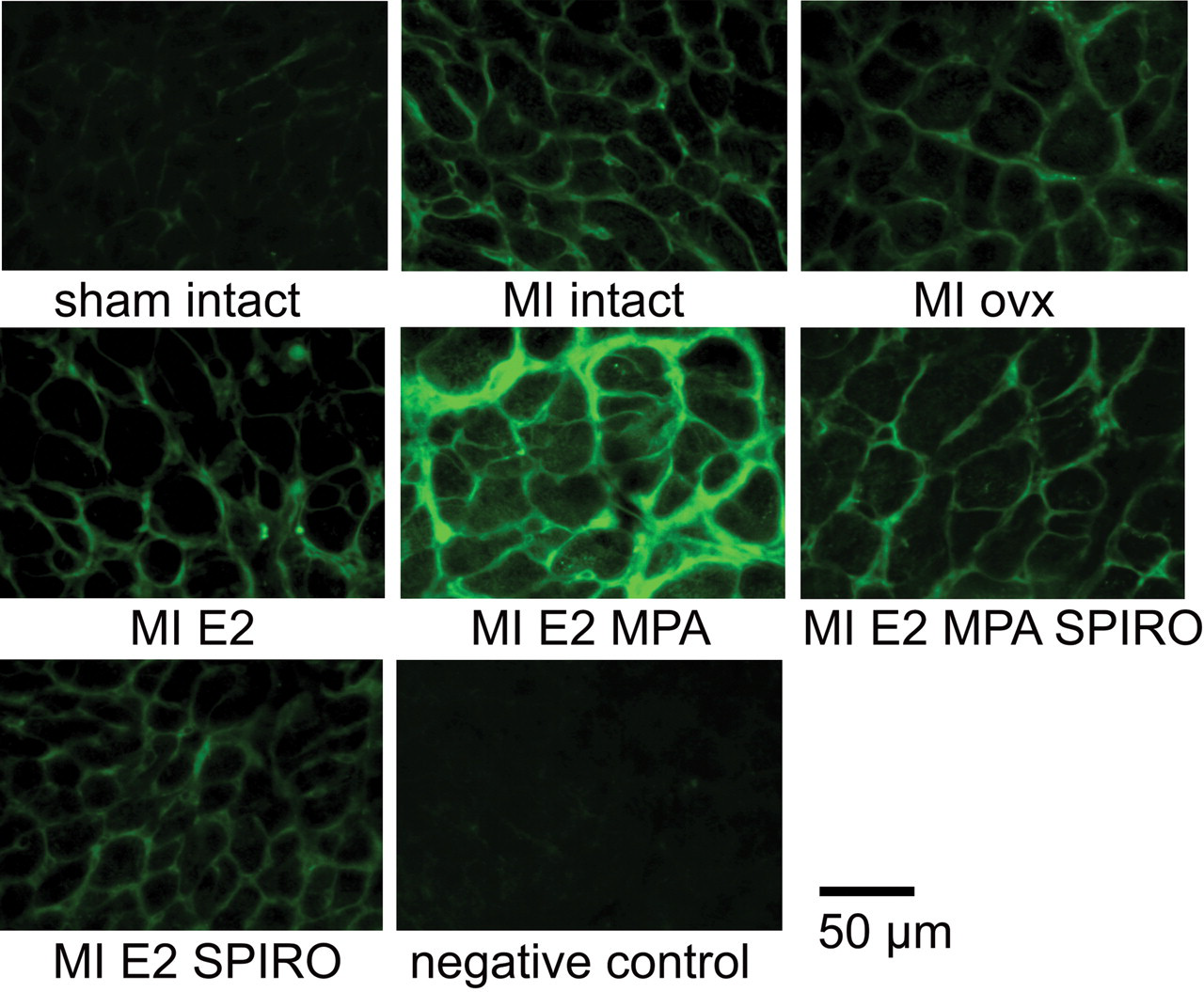
Left ventricular ROS generation detection by APF staining. Cardiac cross-sections from the remote left ventricular myocardium illustrate ROS generating enzyme activities among the different groups of noninfarcted and infarcted rats. ROS generation was highest in cardiac sections from rats that received combined treatment with MPA plus 17β-estradiol. No fluorescence signal was detected in control sections pre-incubated with catalase and SOD (negative control).
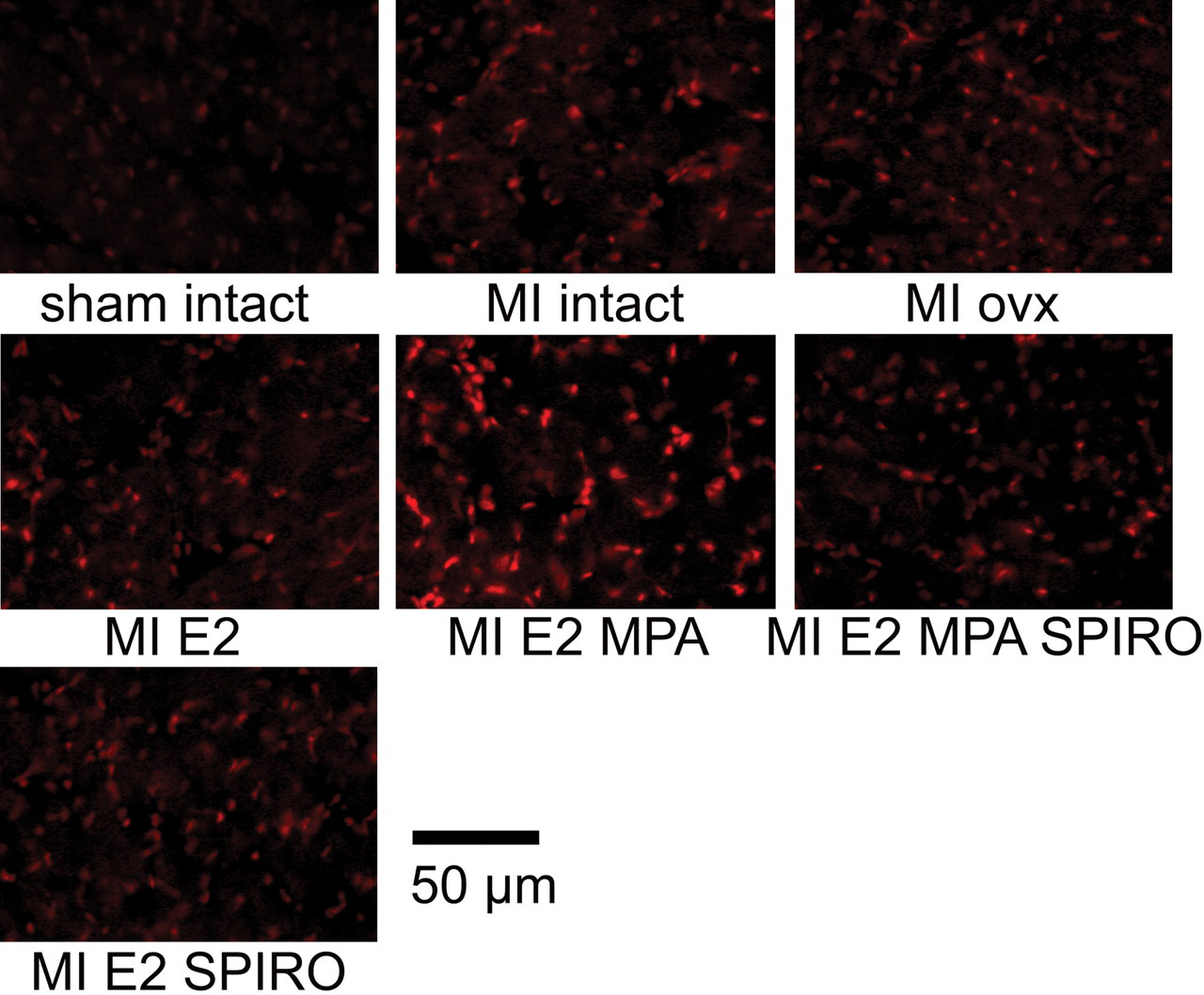
Representative cardiac sections stained with the fluorescent dye DHE. Increased cardiac reactive oxygen species generation was presented in rats treated with MPA plus 17β-estradiol. Addition of spironolactone to the regime MPA plus 17β-estradiol resulted in similar to 17β-estradiol DHE staining intensity.
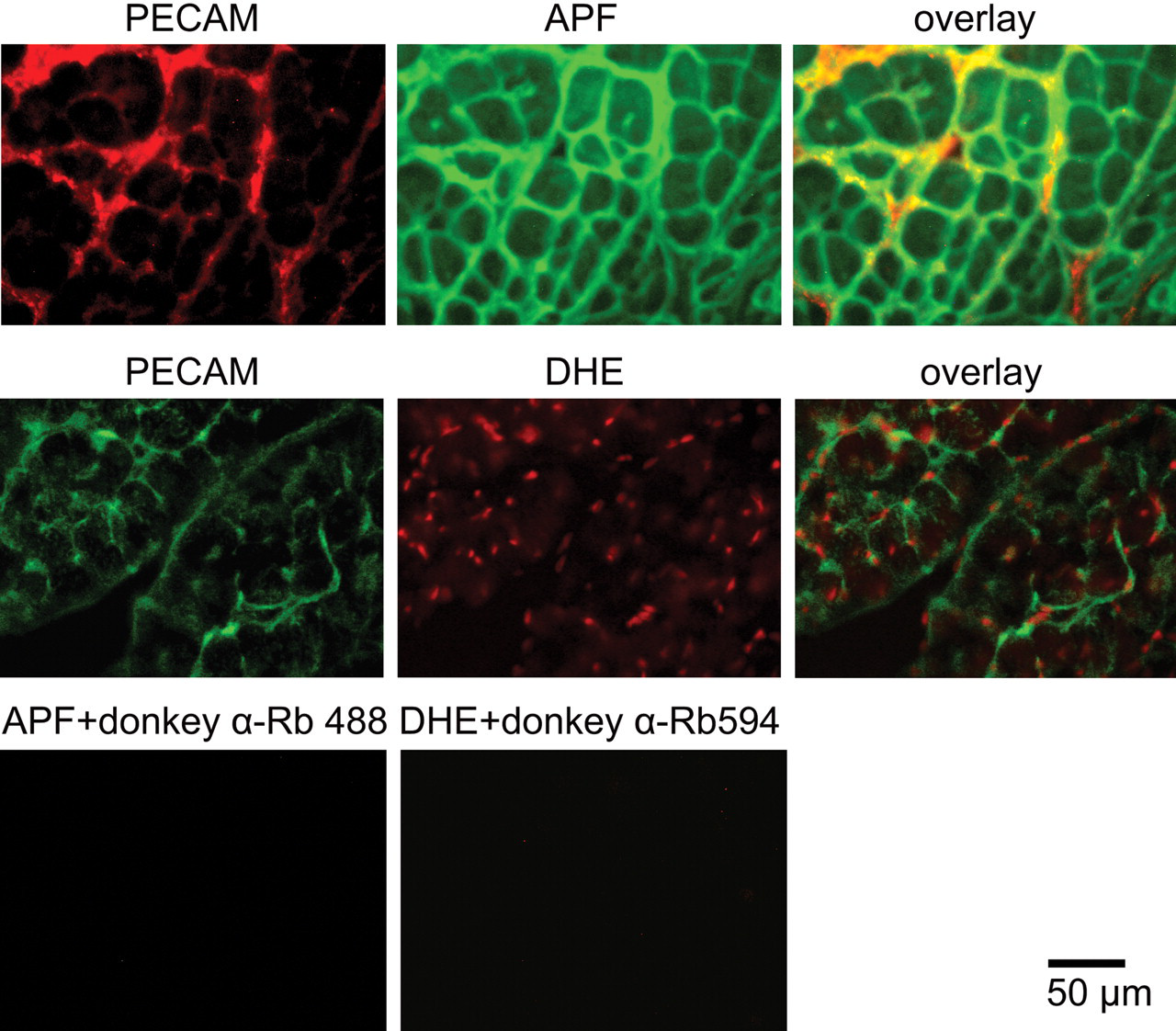
Colocalization of cardiac ROS generation with the microvascular endothelial cell marker (PECAM 1). Cardiac cross-sections from the remote left ventricular myocardium illustrate (APF) colocalization with the microvasculature marker PECAM 1. Consistently cells with a positive DHE staining (red nucleous) present predominantly a green cytoplasm (PECAM 1). Both results show that the microvasculature could be the main source of ROS in the heart sections of MPA plus E2 treated rats. Negative controls consisted in the absence of the primary antibodies and pretreatment with catalase and SOD.
Cardiac Gene Expression
LV expression levels of the regulatory NADPH subunits gp91phox and p67phox were elevated in rats with MI; this elevation only reached significance in rats treated with MPA plus E2 (Figure 6A and 6B). The extent of phospholamban phosphorylation was lower in ovx rats treated with placebo or with MPA plus E2 compared to all other groups (Figure 7C). Protein levels of SERCA 2a and total phospholamban were not altered within all treatment groups.
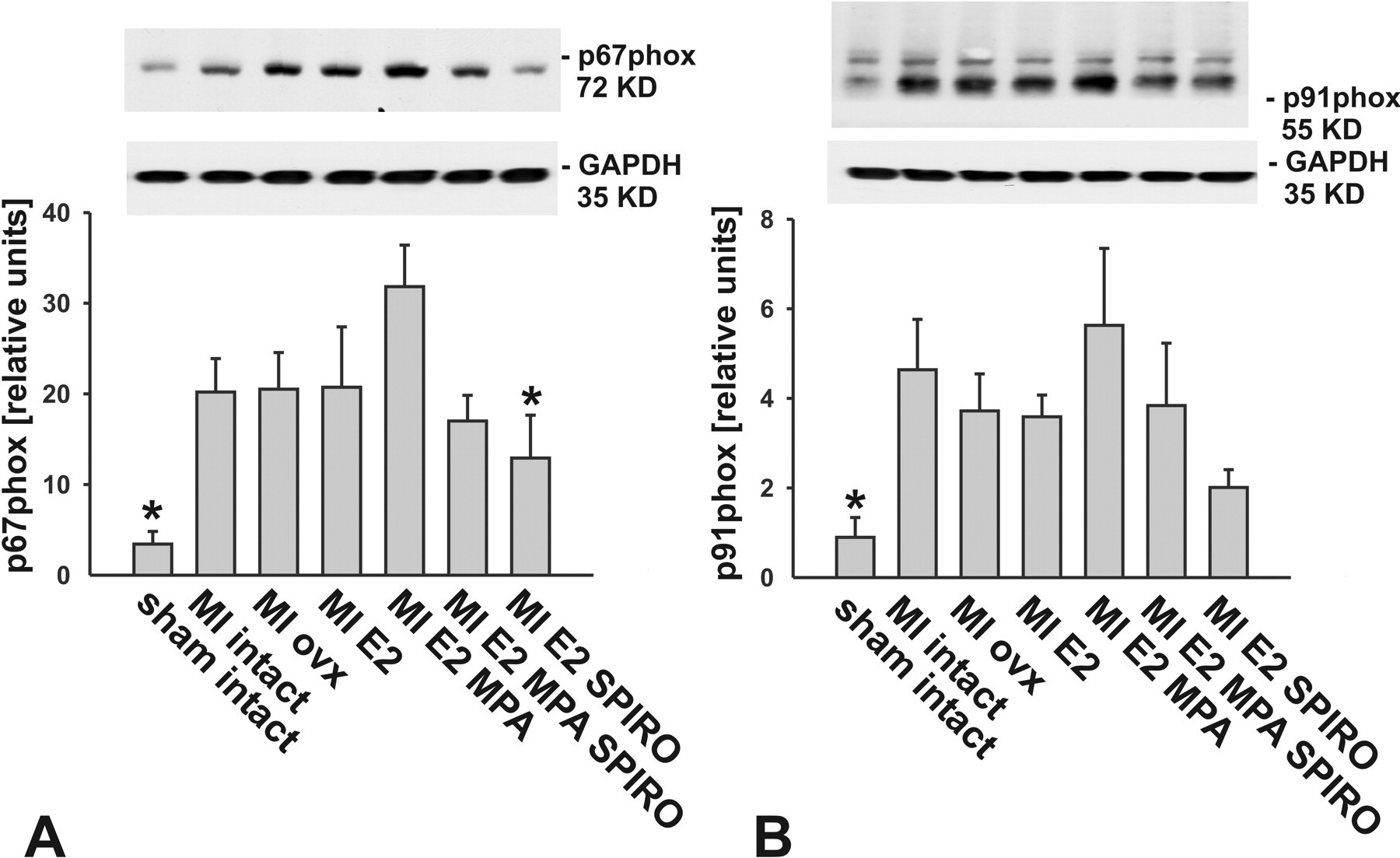
Left ventricular NADPH oxidase subunits protein expression. Left ventricular p67phox (panel A) and gp91phox (panel B) protein levels were increased in the remote myocardium of infarcted rats treated with MPA plus 17β-estradiol compared to sham intact rats (n = 6; *p < .05 vs. myocardial infarction rats treated with 17β-estradiol plus MPA).
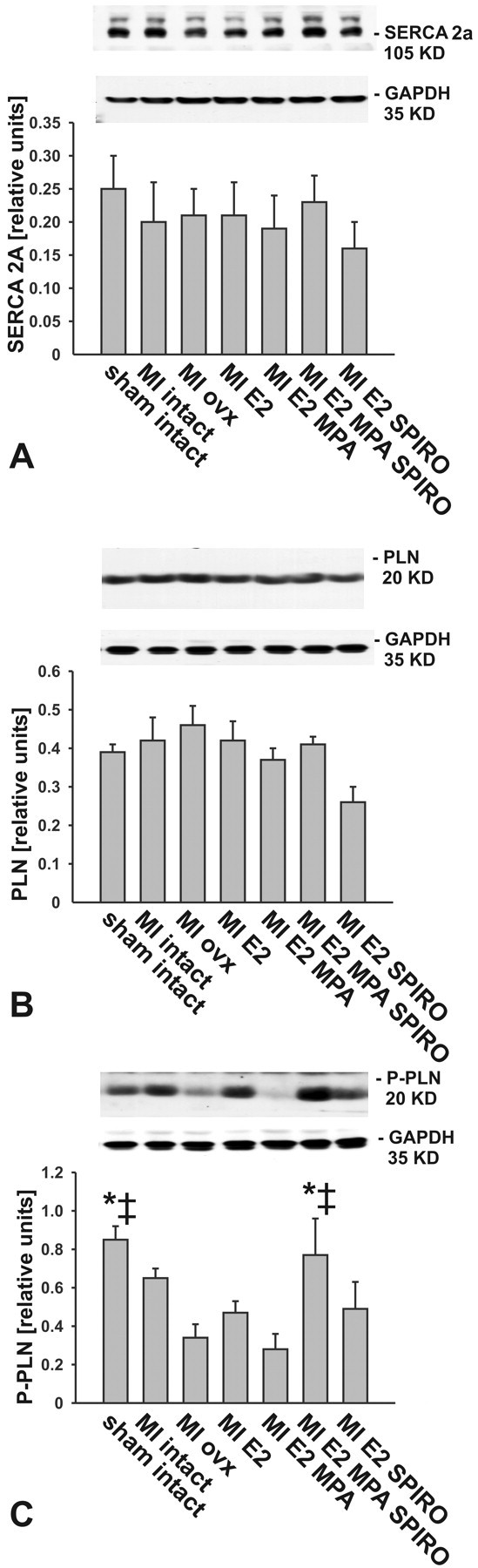
Representative Western blots and bar graphs illustrate SERCA and phospholamban protein expression. While SERCA (panel A) and total phospholamban (panel B) were no different among all groups, phosphorilation of phospholamban (panel C) was significantly lower in animals receiving myocardial infarction and treatment with either placebo or 17β-estradiol plus MPA (n = 6; *p < .05 vs. myocardial infarction rats treated with 17β-estradiol plus MPA; ‡ = p > .05 vs. myocardial infraction rats treated with placebo).
Discussion
The major findings of this study can be summarized as follows: (1) Combined treatment of MPA plus E2, but not estradiol alone, exacerbates LV remodeling and further impairs LV function following MI. (2) The unfavorable effects of the combined treatment with MPA are linked to enhanced ROS generating enzyme activities and were accompanied by an increased expression of NADPH-oxidase subunits gp91phox and p67phox. (3) Addition of SPIRO to the combination of E2 plus MPA attenuated myocardial oxidative stress, cardiac hypertrophy, and LV dysfunction.
Despite considerable advances in diagnosis and management of cardiovascular disease, acute MI continues to be a major public health problem. In 2006, MI had a prevalence of approximately 2.6% in women; in addition, coronary heart disease caused 1 of every 6 deaths in the United States (AHA Writing Group Members 2010). Therefore, the analysis of factors that may have an influence upon MI and heart failure development is of high relevance. Chronological hormone replacement therapy is started in postmenopausal women at an age in which the risk for cardiovascular disease raises in women; thus, analysis of possible cardiovascular effects of HRT regimes is of high relevance. Estrogen supplementation alone has repeatedly been shown to confer protective effects on the vasculature and the myocardium in experimental studies (Mendelsohn and Karas 1999). But to control endometrial proliferation, HRT in women with an intact uterus requires the combination of estrogens and progestins. Experimental animal studies conducted without progestin cotreatment do not mimic combined HRT and could thus not answer the question of whether MPA might have contributed to the unfavorable outcome of the HERS and WHI trials. There is increasing evidence suggesting that addition of MPA to a HRT results in harmful effects on the cardiovascular system by either ablating the beneficial roles of estradiol or by conferring unfavorable effects through itself. In primates, the cotreatment of estradiol with MPA inhibited the vasodilatory effects of estrogen responses by 50% (Williams, Adams, and Klopfenstein 1990; Williams et al. 1994). We previously have shown in a hypertension and heart hypertrophy animal model (aldosterone salt treatment) that treatment of MPA plus E2 led to a complete attenuation of the beneficial cardiovascular effects of estradiol, while intact ovarian hormones played a protective role (Arias-Loza et al. 2006). However, cardiovascular effects due to hemodynamic stress induced by hypertension cannot be extrapolated to the events occurring after an ischemic challenge. Infarct size is of pivotal importance for the extent of cardiac remodeling and chronic HF following MI. Booth and Lucchesi (2007) and Jeanes et al. (2006) previously reported that an acute addition of MPA to an E2 treatment resulted in an extension of the necrotic zone after a cardiac ischemic effect. Nonetheless, those findings were limited to the events occurring during the first 4 hours after reperfusion injury. Thus, the important questions of whether and to what extent MPA might impair the development of chronic HF in rats with large MI and potential mechanisms that may underlie such an effect were not previously addressed. Our results show that long-term treatment (8 weeks) with MPA plus E2 after MI did not alter infarct size (no statistical difference by including or excluding small infarct sizes). Therefore, chronic effects of MPA plus estradiol treatment are not limited to the early events following MI but also affect the ventricular remodeling and function after healing of the infarct area.
Impaired LV function and mean infarct size following MI were not altered by treatment with E2, findings that closely match observations made by Hügel et al. (1999), who reported that the restoration of physiological estrogen serum levels neither reduced infarct size nor improved LV function in ovx rats with large MIs. Together, these data do not only indicate that estrogen supplementation is unlikely to improve LV function post-MI, but also suggest that the addition of MPA and not estradiol treatment alone might have caused the deterioration of cardiovascular events observed in the HERS trials. Beer et al. (2007) reported that high-dose treatment with E2 resulted in a complete retrieval of LV remodeling and a full restoration of LV function in rats with large MI. Although the underlying mechanisms were not determined, it is unlikely that these are solely estrogen receptor mediated because full estrogen receptor activation is achieved already by physiological doses of estradiol (Hügel et al. 1999).
The development of HF following MI in humans and rodents is closely linked to structural remodeling of the remote and noninfarcted myocardium, which further deteriorates impaired LV function. According to these criteria, combined treatment with MPA plus E2, but not E2 alone, enhanced LV remodeling as evidenced by increased cardiac hypertrophy LV intra-cavitary dimensions, impaired LVFS, and elevated LVEDP. Adding MPA to E2 thus aggravated several key features of cardiac damage and dysfunction previously associated with alterations in cardiac calcium homeostasis and Ca2+ protein transporter expression (MacLennan and Kranias 2003). SERCA 2a plays a critical role in the cardiac cycling contractions by transferring Ca2+ from the cytoplasm to the lumen of the sarcoplasmatic reticulum, which leads to relaxation. The activity of SERCA 2a and therefore relaxation is inhibited upon interaction with PLN (Sun et al. 2005; Mørk et al. 2007). Reduction of the expression and/or the activity of SERCA 2a has been found in various forms of experimental and clinical heart failure (Armoundas et al. 2007; Meyer et al. 1995). In addition, an increase of PLN in its non-phosphorylated state results in a decreased SERCA 2a to PLN ratio, which has been associated with defective Ca2+ handling and cardiac contractility in models of HF (Hajjar et al. 1997; Ito et al. 2000). Previously it has been shown that estrogen-depletion increases PLN inhibition of SERCA 2a, which predicts reduction of LV contractility (Ren et al. 2003; Bupha-Intr and Wattanapermpool 2006). In our current settings, phosphorylation of PLN, which prevents inhibition of SERCA 2a, was significantly reduced in ovx rats with MI receiving placebo or MPA plus E2. Thus, it is possible that decreased PLN phosphorylation in ovx rats and in animals receiving combined HRT might have contributed to LV dysfunction. However, this is probably not the only reason for the impaired contractility observed in the MPA plus E2 treated group, because ovx rats receiving only placebo also presented a decrease in PLN phosphorylation, whereas their cardiac function was less impaired. Alternative mechanisms that could explain the further deterioration of LV function in rats treated with MPA plus E2 may have played a role.
Importantly, synthetic progestins such as MPA bind and transactivate not only their cognate receptor but also the MR, the AR, and the GR (Sitruk-Ware 2004; Schindler et al. 2003; Thomas, Liu, and Vats 2006). Excessive, ligand-dependent MR activation has repeatedly been shown to impair cardiac remodeling and chronic HF. Therefore, we speculated that the partial GR and MR-agonist activity of MPA might have contributed to the aggravation of heart failure observed in MPA treated rats. In line with this concept, the MR-antagonist SPIRO not only attenuated cardiac hypertrophy and dysfunction, but also reduced ROS generation in rats receiving the combination E2 plus MPA.
Chronic HF following MI is known to be associated with an increased ROS generation that worsens structural damage and promotes LV dysfunction (Maack et al. 2003; Josephson et al. 1991). In agreement with this concept, enhanced LV ROS generation in rats receiving MPA plus E2 following MI was associated with a progressive deterioration of LV function. The source of ROS generation in the failing myocardium has not been clearly identified, but activation of the myocardial NADPH oxidase appears to be a major source (Bendall et al. 2002; Grieve et al. 2001). The enzymatic activity of the NADPH oxidase is modulated by the location and amount of its subunits, amongst others gp91phox and p67phox. Increased expression of gp91phox (Nox 2), p40phox, p47phox, and p22phox is found in the failing heart (Gao 2004; Zhang et al. 2009). In particular, gp91phox containing NADPH oxidase (Nox2) has been proven to contribute to the development of cardiac contractile dysfunction and interstitial fibrosis in response to pressure overload (Grieve et al. 2006). Accordingly, increased NADPH activity via enhanced gp91phox and p67phox expression and further aggravated LV dysfunction post-MI provides a likely explanation for increased oxidative stress in rats treated with the combination E2 plus MPA. Calcium handling and oxidative stress-related proteins were proven to be crucial for the development of HF; therefore, it is logical to assume that the rate of myocardial contraction has an effect on the generation of oxidative stress and vice versa. Indeed, Liu et al. (2010) have proven that rabbits with HF present both an increased NADPH oxidase activity and a decreased SERCA2a to PLN ratio. Treatment with the NADPH oxidase inhibitor apocynin not only reduced oxidative stress but also recovered expression and activity of SERCA2a, thereby ameliorating cardiac dysfunction (Liu et al. 2010). With our experimental design with rats being treated with MPA plus E2, is not possible to prove or rule out an additive effect of the decrease in phosphorylation of PLN and an increase of oxidative stress on cardiac dysfunction; however, this could be an interesting hypothesis to follow in further studies.
We have shown that in hearts of rats with MI, the fluorescence pattern observed for DHE and APF is predominantly located in the non-cardiomyocyte compartment of the LV. Considering that cardiomyocytes represent only 30% of all myocardial cells and that NADPH oxidase subunits are expressed not only in cardiomyocytes, but also in vascular cells, we suspect that the main source of ROS detected in our sections was originated in the microvasculature (Zak 1973; Gorlach et al. 2000; Pagano et al. 1997; Mohazzab et al. 1997). Accordingly, combining either APF or DHE with immuno-fluorescence staining for the endothelial cell marker PECAM1 shows a consistent colocalization of ROS with the microvasculature. Considering that microvasculature damage and endothelial cell dysfunction play an important role in the pathophysiology of HF (Giannattasio et al. 2001; Landmesser et al. 2004; Luscher 2001), one can speculate that the impairing effects of the MPA plus E2 treatment involve the microvasculature.
In summary, we have shown that chronic treatment with MPA plus E2 after MI aggravates chronic HF, LV dysfunction, and oxidative stress. These observations contribute to explain the unfavorable outcome of clinical endpoint studies that employed combined estrogen and MPA treatment to prevent the development of cardiovascular disease. Alternative HRT combining E2 with progestins that are devoid of MR and/or GR agonist activity might thus confer more neutral and eventually even favorable effects that might improve the safety of HRT in women with preexisting heart disease.
Footnotes
T. P. received support from the Interdisciplinary Center for Clinical Research “IZKF” Würzburg. P. A. A.-L. received support from the German Academic Exchange Service (DAAD) and the IZKF Würzburg. T. P. has received financial support from the Schering AG Berlin that relates to the evaluation of novel steroid hormone receptor ligands.
Acknowledgments
The authors would like to thank Dr. Kai Schuh and Dr. Thomas Renne (Department of Pathobiochemistry, University of Wuerzburg) for their assistance in fluorescence microscopy.