
Abstract
The advent of molecular targeted therapies offers the hope of therapeutic advance in the fight against cancer. However, this hope is tempered by recent findings that certain targeted therapies may have unique side effects. The Hedgehog (HH) pathway is a potential target for treatment of several cancers, including basal cell carcinoma and a subset of medulloblastoma. Recent clinical trials in adults have shown responses to HH pathway inhibition in both basal cell carcinoma and medulloblastoma. However, concerns have been raised about the use of HH pathway inhibitors in children because of the role the HH pathway plays in development. Indeed, young mice treated with the HH pathway inhibitor HhAntag developed severe bone defects, including premature differentiation of chondrocytes, thinning of cortical bone, and fusion of the growth plate. In an effort to lessen the severity of bone defects caused by HhAntag, we treated young mice simultaneously with HhAntag and parathyroid hormone–related protein (PTHrP), which functions downstream of Indian Hedgehog to maintain chondrocytes in a proliferative state. The results show that whereas treatment with PTHrP causes a significant increase in trabecular bone, it does not prevent fusion of the growth plate induced by HhAntag.
Introduction
The Hedgehog (HH) pathway regulates proliferation and differentiation in many tissues during embryonic and postnatal development. Deregulation of HH pathway signaling and aberrant pathway activity have been linked to cancer. Germline and sporadic mutations that sustain HH pathway activity result in basal cell carcinoma (BCC) and medulloblastoma (MB) (Dellovade et al. 2006; Hahn et al. 1996; Johnson et al. 1996), and elevated HH pathway activity has been observed in stromal cells from a broad range of cancers (Theunissen and de Sauvage 2009; Yauch et al. 2008). These observations have encouraged development of HH pathway inhibitors for the treatment of cancer (Curran and Ng 2008; Romer and Curran 2005; Scales and de Sauvage 2009). We used a mouse model of MB to demonstrate proof-of-concept that a Smoothened (SMO) inhibitor was capable of eliminating spontaneous brain tumors (Romer et al. 2004).
Medulloblastoma is the most common malignant pediatric brain tumor, with peak incidence at seven years of age. In 70% of cases, MBs arise in children under sixteen years of age (Louis et al. 2007). Current treatments, involving a combination of surgery, radiation, and chemotherapy, have a relatively high five-year survival rate of over 70%. However, the success rate is significantly reduced in patients younger than three years of age. In addition, the side effects of therapy, particularly in the youngest patients, often result in significantly impaired neurological development and reduced quality of life. Therefore, there is a great need for new therapies to ensure that survivors of MB have a full and productive life. Approximately one-third of MBs exhibit enhanced Hedgehog pathway activity, which in half of these tumors arises as a consequence of mutations in Patched1 (PTCH1), a transmembrane protein that functions as a receptor for HH (Murone et al. 1999). Patched is localized to the primary cilium and inhibits the accumulation of the constitutively active transmembrane protein SMO within the cilium (Rohatgi et al. 2007). In the presence of HH ligands, PTCH1 is displaced from the cilia and SMO is able to accumulate and activate downstream target genes including the transcription factor GLI1. Several studies used cell-based screens to identify small molecule inhibitors that block pathway activity by binding to SMO. Several of these screens have been used as lead compounds for drug development (Frank-Kamenetsky et al. 2002; Williams et al. 2003). Each of the compounds identified functions by binding to the same site on SMO (Frank-Kamenetsky et al. 2002; Rominger et al. 2009).
After testing the ability of several of these inhibitors to downregulate the HH pathway in cultured cells, we chose HhAntag for in vivo studies (Romer et al. 2004). Using the mouse MB model Ptc1+/-p53-/- , we found that treatment with HhAntag completely eliminated spontaneous tumors and tumor allografts (Romer et al. 2004; Sasai et al. 2007; Sasai et al. 2006).
Medicinal chemistry approaches improved several features of the early compounds, including enhanced pharmacodynamics in humans, and several of these compounds have entered Phase I clinical trials (Robarge et al. 2009; Tremblay et al. 2009; Wong et al. 2009). A phase I trial was initiated in April of 2007 to test the HH pathway inhibitor GDC-0449 in locally advanced and metastatic solid tumors. Over half of the patients with BCC tumors showed positive responses within four months of treatment initiation, which led to a Phase II trial (Von Hoff et al. 2009). GDC-0449 was also used in a single adult patient with advanced metastatic MB (Rudin et al. 2009). Just two months after treatment initiation, PET scans showed a dramatic reduction in disease. However, the patient relapsed because the tumor had acquired resistance because of a mutation in SMO that prevented binding of GDC-0449, rendering it ineffective (Yauch et al. 2009).
Although treatment of adult mice with the SMO inhibitor HhAntag did not reveal any serious side effects, P10 mice treated for as little as two days developed permanent bone defects (Kimura et al. 2008). Three months after removing the drug, mice exhibited reduced weight and size, abnormal bone structures, shortened bone length, and a significant loss of proliferating chondrocytes in the femur and tibia. Proliferating chondrocytes differentiated prematurely into hypertrophic chondrocytes, resulting in accelerated invasion of the growth plate by osteoclasts and osteoblasts, premature trabecular bone formation, and bone dysplasia (Kimura et al. 2008). In addition, loss of HH pathway activity inhibited growth of incisor teeth in adult mice. These defects occurred even when a lower dose of HhAntag (25 mg/kg) was administered that was not sufficient to eliminate tumors (Kimura et al. 2008; Romer et al. 2004).
Indian Hedgehog (IHH), one of the three HH ligands in vertebrates, regulates the differentiation of chondrocytes and osteoblasts during endochondrial bone development (Chung et al. 2001; Karp et al. 2000; Maeda et al. 2007). During this process, growth plate chondrocytes proliferate in precise columns before differentiating into hypertrophic chondrocytes. After a period of growth, the hypertrophic chondrocytes enter apoptosis and vascularization, and bone deposition ensues (Erlebacher et al. 1995). Indian Hedgehog delays the differentiation of proliferating chondrocytes into hypertrophic chondrocytes in part through the activity of PTHrP (Karp et al. 2000). PTHrP is expressed in the periarticular perichondrium, a region surrounding the cartilage that gives rise to osteoblasts lining the surface of cortical bone. PTHrP binds to and activates the parathyroid hormone (PTH)/PTHrP receptor (PPR) in both the proliferating and prehypertrophic zones, thus restraining differentiation (Lanske et al. 1996; Lee et al. 1996; Vortkamp et al. 1996). In an effort to ameliorate the effects of HhAntag on bone growth, we co-treated mice with PTHrP. The results obtained indicate that whereas exogenous PTHrP was able to stimulate trabecular bone growth in young mice, it failed to prevent fusion of the growth plate caused by HhAntag.
Materials and Methods
Mouse Husbandry and Dosing
Wild-type FVB pregnant female mice were obtained from either Harlan Sprague Dawley, Inc. (Indianapolis, IN) or The Jackson Laboratory (Bar Harbor, ME). Animals were housed and maintained according to the regulations set forth by the Institutional Animal Care and Use Committee (IACUC). All procedures performed were approved by the IACUC at The Children’s Hospital of Philadelphia. P10-old mice were treated with 100 mg/kg HhAntag twice daily by oral gavage for four or six days, or treated with 20, 50, or 100 µg/kg PTHrP (1-37) twice daily by IP injection for four, six, or fourteen days.
Chemicals
HhAntag was obtained from Genentech, Inc. (South San Francisco, CA) and is available under a material transfer agreement (http://www.gene.com/gene/about/collaborations/contracts.jsp). PTHrP (H-5494: pTH-Related Protein [1-37; human, rat]) was obtained from Bachem (King of Prussia, PA).
Histology
For Figures 1 and 3, mice were perfused with 4% paraformaldehyde (PFA) in 1× PBS. For Figure 2, mice were post-fixed in 10% formalin. Hind limbs were dissected and decalcified in 14% EDTA solution for ten to fourteen days at 4°C. Limbs were embedded in paraffin and cut into 5-µm sections. Sections were stained with hematoxylin and eosin (H&E) according to standard protocols and imaged on a Nikon Eclipse 90i microscope using a Nikon DS-Fi1 camera.
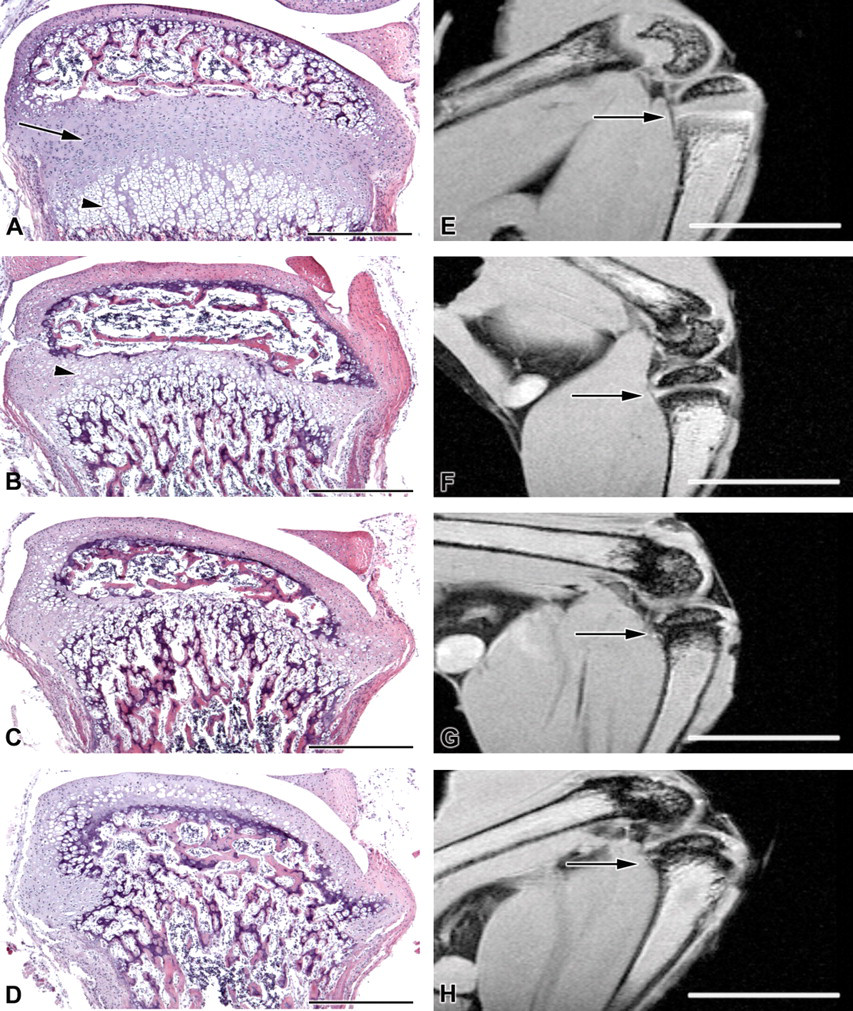
HhAntag treatment causes severe and irreversible bone defects. Wild-type mice were treated with 100 mg/kg HhAntag twice daily for six days starting at P10. Hematoxylin and eosin images (A–D) and magnetic resonance images (E–H) of the right tibia of untreated (A, E), six days treatment with no rest period (B, F), six days treatment with two days rest (C, G), and six days treatment with four days rest (D, H). Arrow in A represents proliferating and prehypertrophic chondrocytes. Arrowhead in A represents hypertrophic chondrocytes. Arrows in (E–H) represent the growth plate region of the tibia. Scale bars in A–D represent 500 µm. Scale bars in E–H represent 5 mm.
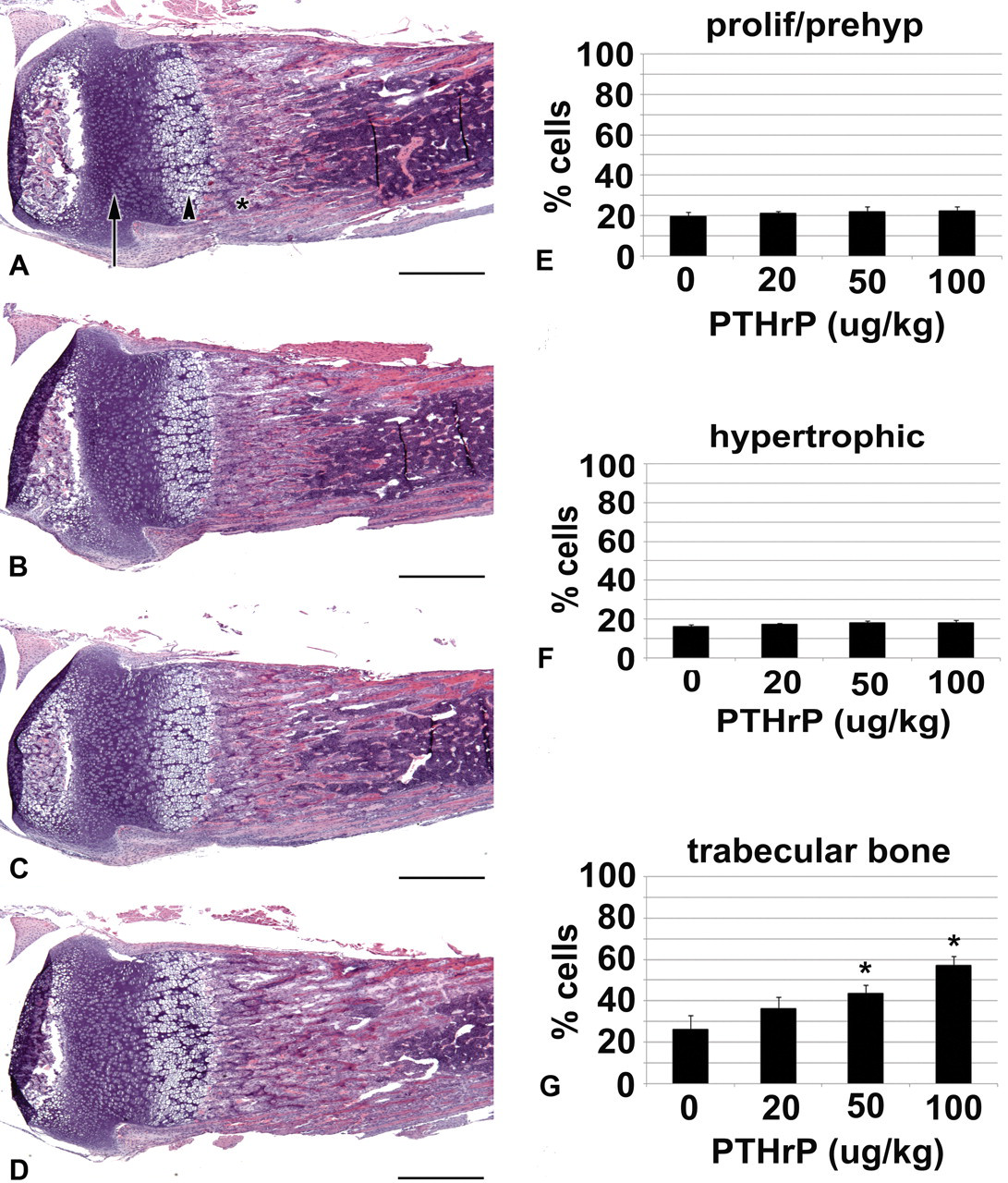
Administration of PTHrP alters bone morphology. Tibia from P14 wild-type mice, untreated (A), or treated with 20 µg/kg PTHrP (B), 50 µg/kg PTHrP (C), or 100 µg/kg PTHrP (D) from P10 to P14, twice daily. Arrow in A shows proliferating and prehypertrophic chondrocytes. Arrowhead in A shows hypertrophic chondrocytes. Asterisk in A shows trabecular bone. Scale bars in A–D represent 500 µm. The percentage of proliferative/prehypertrophic chondrocytes (E), hypertrophic chondrocytes (F), and trabecular bone (G) within a defined region of the right and left tibias were calculated. The graphs show a representative from each dose category. Standard deviation was calculated from the average of four limbs from two mice for each dose category. p value was determined by Student t test; *p < .001.
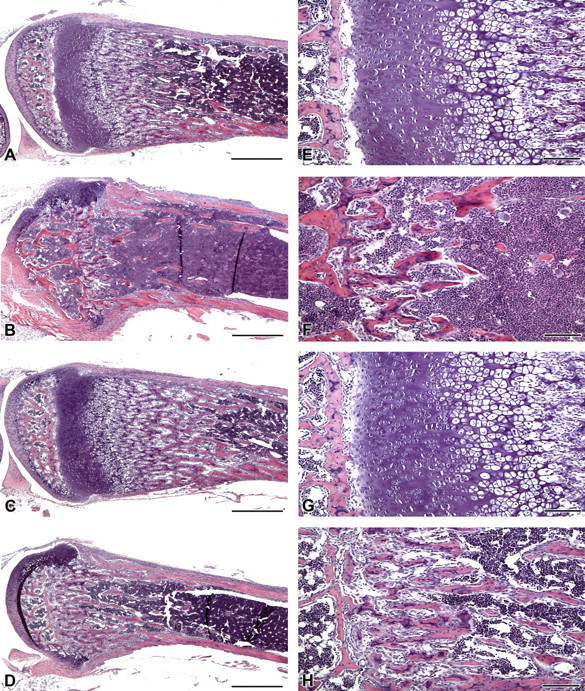
PTHrP treatment does not reduce the bone defects caused by HhAntag treatment. Tibia from wild-type P20-old mice, untreated (A, E), treated with 100 μg/kg PTHrP and 100 mg/kg HhAntag twice daily from P10 to P16 and sacrificed four days later (B, F), treated with 100 μg/kg PTHrP alone from P6 to P20 (C, G), and treated with 100 μg/kg PTHrP from P6 to P20 and 100 mg/kg HhAntag from P10 to P16 and sacrificed four days later (D, H). (E–H) Higher resolution image of the growth plate region shown in A–D, respectively. Scale bars in A–D represent 500 μm. Scale bars in E–H represent 100 μm.
Magnetic Resonance Imaging
After perfusion with 4% PFA, the right hind limbs from untreated and HhAntag-treated mice were dissected. Magnetic resonance images (MRI) of the excised mouse limbs were acquired with a Bruker Avance DMX 400 9.4T vertical bore magnet using the Paravision software. Each limb was inserted into a short, 10-mm tube and immersed in fomblin (Fomblin Y [8], Sigma Aldrich, St. Louis, MO). Two-dimensional gradient echo images were acquired in the sagittal orientation, making sure that the central slice contained the femur and the tibia. The field of view (FOV) was 1.4 cm in the vertical direction and 1 cm in the horizontal direction, with the readout in the vertical direction. Fourteen consecutive 0.25-mm slices were acquired in an interleaved manner. The matrix size was 256 × 192, yielding an in-plane resolution of 55 × 52 µm. The timing was as follows: repetition time (TR) 332 ms, echo time (Te) 4.3 ms with a 60° flip angle. The number of excitation (NEX) was sixteen, resulting in a thirty-four-minute scan.
Calculations and Statistical Analyses
To determine the percentage of proliferative/prehypertrophic chondrocytes, hypertrophic chondrocytes, and trabecular bone within the PHTrP treated bones, each limb from two mice for each dose was sectioned. Three sections from each limb, spaced 25 µm apart, were stained and imaged on a Nikon Eclipse 90i microscope, as described above. Each image was printed (11 cm × 16 cm), and a defined region was outlined (3 cm × 11 cm). The top of the 11-cm edge was placed at the start of the proliferating cells. The 3-cm region was placed in the horizontal center of the bone. Each region of cells was marked to separate proliferative/prehypertrophic chondrocytes, hypertrophic chondrocytes, and trabecular bone. After demarcation, the area for each type of cell was measured and divided by the total area to obtain a percentage. The graphs in Figure 2 show one of the two right hind limbs analyzed for each concentration of PTHrP tested (untreated, 20, 50, and 100 µg/kg). Standard deviation from four limbs was obtained and graphed (two limbs from two different animals). p Values were determined using a Student t test, with a significance set at p < .001.
Results
Postnatal HH Pathway Inhibition Causes Severe and Irreversible Bone Defects
Previously, we found that inhibition of the HH pathway in young mice resulted in a significant reduction in proliferating chondrocytes, leading to growth plate closure after four days of treatment (Kimura et al. 2008). To develop an assay in which we could test the effects of PTHrP treatment in HhAntag-treated mice and to better understand the severity of the phenotype, we treated mice with 100 mg/kg HhAntag twice daily for six days starting at P10 and analyzed bone morphology immediately, two days and four days post-treatment (Figure 1). Mice exposed to the antagonist failed to thrive during the period of treatment, and they lost weight. After cessation of treatment, the mice began to gain weight, but they remained smaller than their untreated littermates. Both H&E staining and MRI of the growth plate region showed that HhAntag treatment caused a significant loss of proliferating chondrocytes (Figure 1A–D) and premature growth plate closure (Figure 1E–H, arrows). Trabecular bone invaded the region formerly occupied by the proliferative/prehypertrophic and hypertrophic chondrocytes (Figure 1A, arrow and arrowhead respectively, compared to Figure 1B). The severity of bone defects increased with longer exposure to HhAntag. Mice treated with the antagonist for only four days showed less severe defects than those treated for six days (data not shown).
Administration of PTHrP in Wild-Type Postnatal Mice Causes an Increase in Trabecular Bone
Indian hedgehog activates PTHrP, thus preventing the differentiation of proliferating chondrocytes into hypertrophic chondrocytes. Therefore, we hypothesized that addition of exogenous PTHrP during HhAntag treatment might overcome the loss of IHH, preserving a pool of proliferating chondrocytes that would prevent premature growth plate closure. To determine the appropriate dose required to safely elicit a response in young mice, we treated P10 mice with 0, 20, 50, or 100 µg/kg PTHrP (1-37) twice daily for four days. At all doses tested, there was no change in the zones of proliferating and hypertrophic chondrocytes (Figure 2A–D). However, there was a significant increase in the amount of trabecular bone in mice treated with 50 or 100 µg/kg PTHrP compared to untreated mice (Figure 2A, C, and D). These results indicate that IP injection of 50-100 µg/kg PTHrP twice daily is sufficient to alter bone morphology in postnatal mice.
PTHrP Treatment Does Not Reduce the Defects Caused by HhAntag Treatment
Although PTHrP treatment did not increase the zone of proliferating chondrocytes, it did alter the amount of trabecular bone formation, which shows that it did have an effect on bone tissue (Figure 2E, G). Therefore, we chose to treat mice with 100 µg/kg PTHrP and 100 mg/kg HhAntag simultaneously to determine whether exogenous PTHrP could overcome the loss of proliferating chondrocytes in HhAntag-treated mice. Mice were treated with 100 µg/kg PTHrP and 100 mg/kg HhAntag twice daily from P10–P16. Four days after treatment, the mice were sacrificed. Mice treated with PTHrP showed a loss of chondrocytes and subsequent growth plate closure similar to those treated with HhAntag alone (Figure 3B, F).
We hypothesized that providing an excess of PTHrP to bone tissue before and after treating with HhAntag might help protect proliferating chondrocytes. Therefore, mice were treated with 100 µg/kg of PTHrP twice daily from P6–P20. Half of these mice were also treated with 100 mg/kg HhAntag twice daily from P10–P16. Mice treated with PTHrP only showed an increase in trabecular bone, consistent with our treatment of mice from P10–P14 (Figure 3C, G). However, neither pretreatment nor post-treatment with PTHrP alleviated the defects seen in HhAntag-treated mice (Figure 3D, H). These results indicate that exogenous PTHrP cannot compensate for the loss of IHH in developing bone.
Discussion
The well-known toxicities of most cancer chemotherapeutics has led to the development of alternative approaches that target the specific signaling pathways altered in tumor cells. It was hoped that these molecular-targeted therapies, because of their high degree of selectivity, would exhibit much reduced toxicity. However, in many cases these agents block signaling pathways that are particularly active in critical developmental processes. The HH pathway plays a major role during embryogenesis by establishing proper body patterning and regulating the development of many organs (McMahon et al. 2003). It has been suggested to play a broad role in cancer, either in tumor cells or through effects on stromal cells (Dellovade et al. 2006; Hahn et al. 1996; Johnson et al. 1996; Theunissen and de Sauvage 2009; Yauch et al. 2008). In the case of MB, a subset of tumors is caused by activation of the HH pathway and there is great interest in the therapeutic potential of SMO inhibitors. However, concerns have been raised about treating young children with SMO antagonists, since postnatal bone growth depends on IHH. Here we show that the bone defects caused by HhAntag increase in severity with prolonged exposure. In an effort to protect developing bone from the effects of HH pathway inhibition, we treated young mice with PTHrP, hoping to prevent a complete loss of proliferating chondrocytes and therefore lessen the severity of bone defects in mice with reduced HH pathway activity. Unfortunately, addition of PTHrP did not alleviate these defects. Instead of protecting proliferating chondrocytes and delaying chondrocyte differentiation, PTHrP treatment led to an increase in trabecular bone. This increase in trabecular bone supports the conclusions of previous studies, suggesting that PTHrP functions as an anabolic agent (Stewart et al. 2000; Xue et al. 2005). Furthermore, supplying PTHrP before and after HH pathway inhibition did not prevent the loss of proliferating chondrocytes and growth plate fusion. One caveat to this conclusion is that we cannot be sure that enough active PTHrP actually penetrated into the growth plate. To address the role of PTHrP directly in the growth plate, it would be necessary to target expression of the hormone to proliferating chondrocytes during bone development using a transgenic approach. Doing so would allow us to assess the effect of autocrine signaling on chondrocyte proliferation during treatment with HhAntag. However, this step is beyond the scope of the present study and it would not address the problem of how to alleviate the effects of SMO inhibitors on bone development in young children.
Although disappointing, these results are consistent with recent genetic analyses of a constitutively active PPR in Ihh mutant mice (Maeda et al. 2009). In that study, a conditional Ihh knockout mouse was generated that expressed a constitutively active PPR specifically in chondrocytes (Maeda et al. 2009). The authors showed that expression of the PPR was able to transiently maintain a growth plate. However, by P14, the growth plate was completely absent. In addition, expression of PPR was not able to prevent the loss of proliferating chondrocytes observed in Ihh mutants. These results support the suggestion that Ihh plays several PTHrP-independent roles in the regulation of chondrocyte differentiation (Kobayashi et al. 2005; Mak et al. 2008).
Endochondrial bone development requires the convergence and interaction of multiple signaling pathways, including the HH, Wnt, BMP, and FGF pathways (Adams et al. 2007; Day et al. 2005; Minina et al. 2001; Ornitz and Marie 2002). The complexity of this process poses a great challenge for the use of SMO inhibitors in children. Altering one component to compensate for another is not straightforward and would likely disrupt normal bone development. Therefore, it is likely that the use of SMO inhibitors in the youngest MB patients will be very problematic.
Footnotes
Acknowledgments
We thank Dr. Suzanne Wehrli for assistance with MRI and members of the Curran lab for comments on the manuscript.
The author(s) declared no potential conflicts of interests with respect to the authorship and/or publication of this article. The author(s) received no financial support for the research and/or authorship of this article.