
Abstract
Neoplasms of the nervous system, whether spontaneous or induced, are infrequent in laboratory rodents and very rare in other laboratory animal species. The morphology of neural tumors depends on the intrinsic functions and properties of the cell type, the interactions between the neoplasm and surrounding normal tissue, and regressive changes. The incidence of neural neoplasms varies with sex, location, and age of tumor onset. Although the onset of spontaneous tumor development cannot be established in routine oncogenicity studies, calculations using the time of diagnosis (day of death) have revealed significant differences in tumor biology among different rat strains. In the central nervous system, granular cell tumors (a meningioma variant), followed by glial tumors, are the most common neoplasms in rats, whereas glial cell tumors are observed most frequently in mice. Central nervous system tumors usually affect the brain rather than the spinal cord. Other than adrenal gland pheochromocytomas, the most common neoplasms of the peripheral nervous system are schwannomas. Neural tumors may develop in the central nervous system and peripheral nervous system from other cell lineages (including extraneural elements like adipose tissue and lymphocytes), but such lesions are very rare in laboratory animals.
Introduction
Many pathologists view the diagnosis of nervous system neoplasms with a degree of trepidation. As these neoplasms occur infrequently in both the spontaneous setting (mostly observed in aging animals) or when induced by carcinogenic stimuli, diagnostic experience may be limited. However, as with tumors in other tissues, diagnosis is aided considerably by key morphological features as well as other factors including sex, tumor location, and age at tumor onset (or, in most experimental settings, the day of death from the tumor).
The results described in this publication derive mainly from the data of Weber (1994), as well as from ongoing observations of brain tumors in rodents, mainly Wistar (WIST) rats, during the past twenty years (predominantly at RCC Ltd./Harlan Laboratories Ltd., Itingen, Switzerland). This article is not intended to establish diagnostic criteria that might compete with the international recommendations for the nomenclature of mouse and rat neural tumors, which will be published in the near future (International Harmonization of Nomenclature and Diagnostic Criteria for Lesions in Rats and Mice [INHAND], Nervous System Working Group). In fact, the diagnostic criteria described here for classifying neural neoplasms are equivalent to those being considered in the most recent INHAND drafts.
Factors Affecting the Prevalence of Spontaneous Neural Neoplasms
Species Effects
The prevalence of spontaneous nervous system (NS) tumors varies widely among species. Rats are the laboratory animal species in which NS tumors occur most frequently. In contrast, central nervous system (CNS) tumors in mice (Fraser 1971; Harlan Laboratories, Historical control data in CD-1 mice, unpublished; Morgan 1984; NTP 2004b) (Table 2 ) and hamsters (Deerberg et al. 1987; Ernst et al. 1989) are generally rare or reported in single cases only. Spontaneous NS tumors occasionally occur in dogs as spontaneous lesions (Harlan Laboratories, Historical control data on histological lesions in purebred beagle dogs, unpublished) but have not been reported to be induced under experimental conditions except by virus inoculation (Brisman et al. 1979; Shimura et al. 1985). Spontaneous NS neoplasms are extremely rare in monkeys (Takayama et al. 2000).
Incidence of central nervous system neoplasms in different mouse strains.
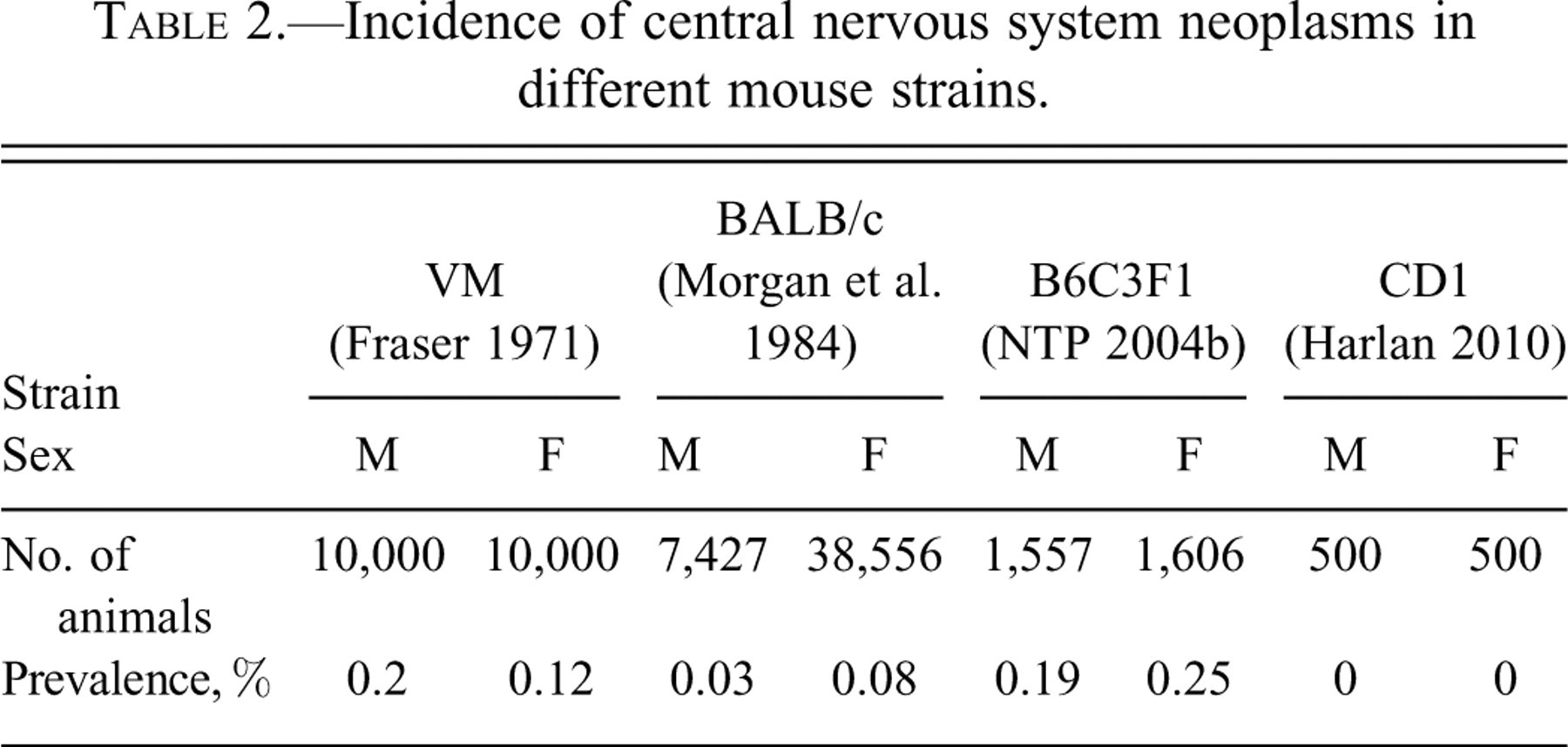
Strain Effects
Strain-specific differences in the incidence of NS tumors have been recorded in rats. Although the incidence did not diverge significantly among strains (Table 3 ), some strains reproducibly develop fewer neural neoplasms overall (e.g., Fisher 344 [F344]). The most common CNS tumors in Sprague-Dawley (SD) and WIST rats were granular cell tumors (derived from a specialized meningeal cell lineage), with incidences of approximately 1.5% in males and 0.7–0.8% in females that survived to the end of conventional two-year carcinogenicity bioassays. The next most frequent NS tumors in rats were gliomas as a class (including astrocytoma, oligodendroglioma, and glioma, not otherwise specified [NOS]), with incidences <1% in both sexes for both SD and WIST strains.
Incidence of central nervous system neoplasms in different rat strains.
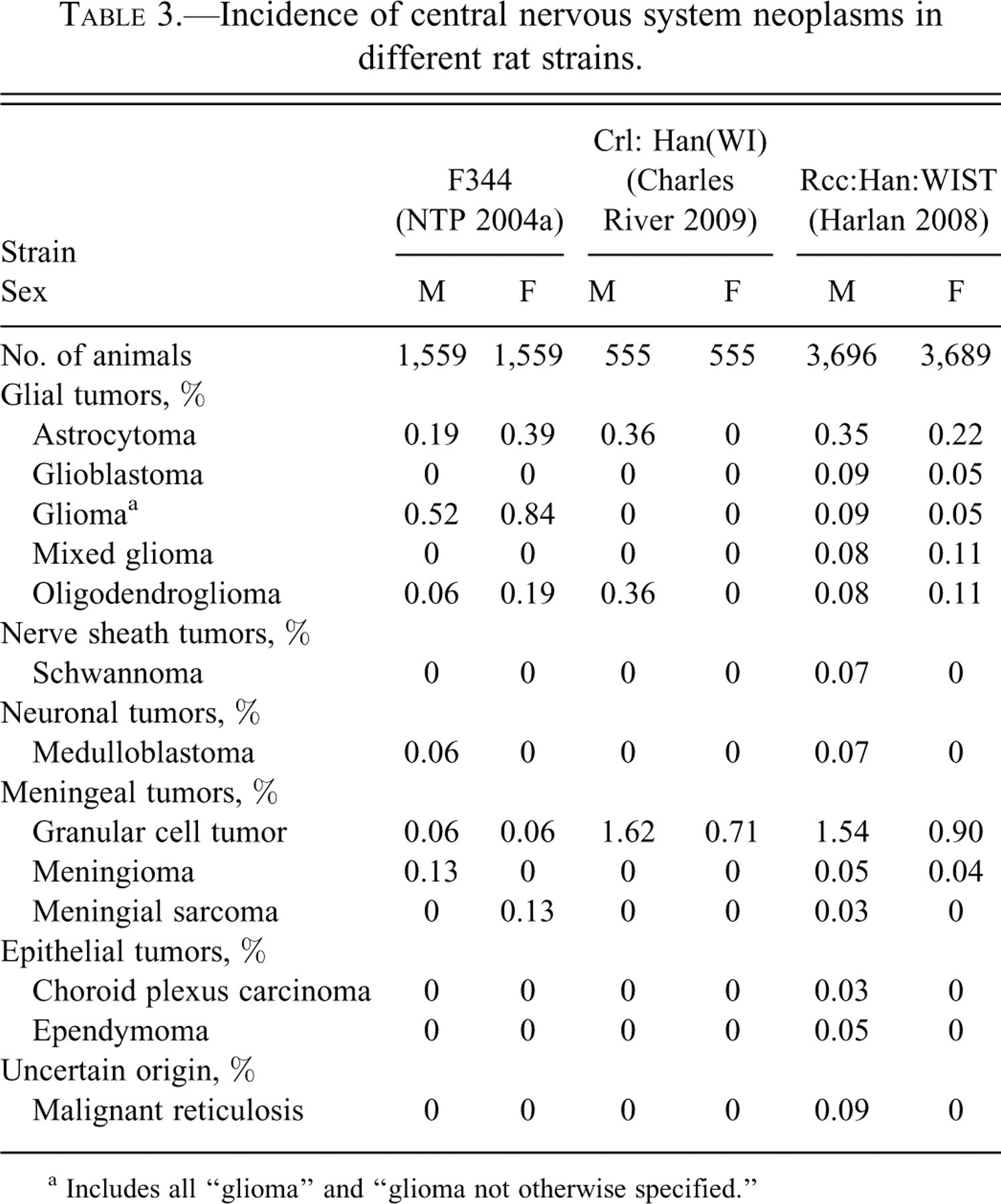
a Includes all “glioma” and “glioma not otherwise specified.”
Prevalence of central nervous system tumor types among 5480 RccHan:WIST rats per sex.
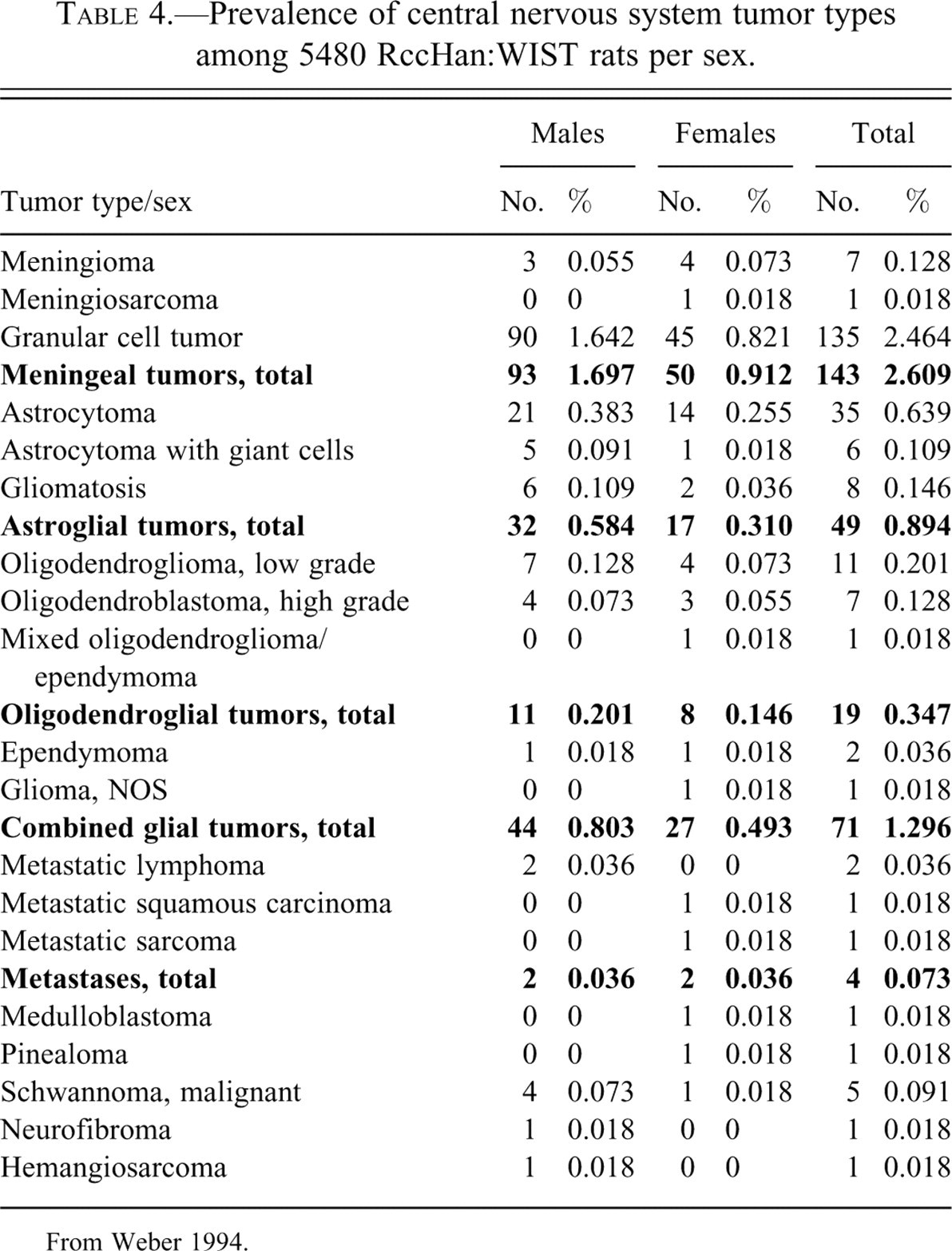
From Weber 1994.
Location Effects
Nervous system tumors may arise from many different cell types, including glia (glioma); neurons in the parenchyma (ganglioneuroma, medulloblastoma, neuroblastoma) or peripheral ganglia (paraganglioma, pheochromocytoma); meninges (meningioma); nerve sheaths (neurofibroma, neurofibrosarcoma, schwannoma); pigment-producing neural crest cells (melanoma, nevus); and neuroendocrine tumors (carcinoid; Table 1 ). Spontaneous NS tumors in RccHan:WIST rats generally arise in the brain (2.04% in 11,705 animals drawn from carcinogenicity studies) rather than the spinal cord (Weber 1994). However, spinal cord tumors do occur (2.14% of all spinal cord lesions in one study; Weber 1994) and can be quite varied (i.e., including astrocytoma, neurofibroma, infiltrating malignant lymphoma, and metastatic sarcoma in the same study; Weber 1994).
General overview of neoplasms originating from neural cells.

Sex Effects
Sex-related differences in the incidence of NS tumors in laboratory animal species are not well established. However, our investigations in RccHan:WIST rats has shown that NS tumors of all types are more common in males than females. The observed male-to-female ratios for different neoplasms in this strain are 1.9 for granular cell tumors, 1.9 for astrocytomas (all types combined), 1.4 for oligodendrogliomas (all types combined), and 1.6 for mixed gliomas.
Analytical Bias
The number of sections examined during the neuropathology analysis can have a profound impact on the reported tumor incidence. In a standard examination consisting of four coronal brain sections from 400 rats per sex, the frequency of NS neoplasia in Shoe:WIST rats was 1.5%. A more extensive evaluation (thirty sections per brain) discovered over three times as many tumors (6.75%; Weber 1994).
The Impact of Time on Tumor Biology
Determining the day of onset and growth curves for spontaneous NS tumors is not possible. However, the general time frame (early or late onset, slow or rapid progression) can be estimated from the day of death or premature termination. For example, in a large cohort of rats (Weber 1994), a Kruskal-Wallis analysis comparing the last day on study with tumor type (glioma or meningioma) revealed that oligodendroglial tumors developed first, followed by astrocytomas, granular cell tumors, and finally meningiomas. Within the astrocytoma class, relatively circumscribed neoplasms with binucleated giant cells were observed before diffusely spreading tumors (astryocytic gliomatosis) and low-grade astrocytomas. For all calculated values, the differences were statistically significant (Figure 1 ).
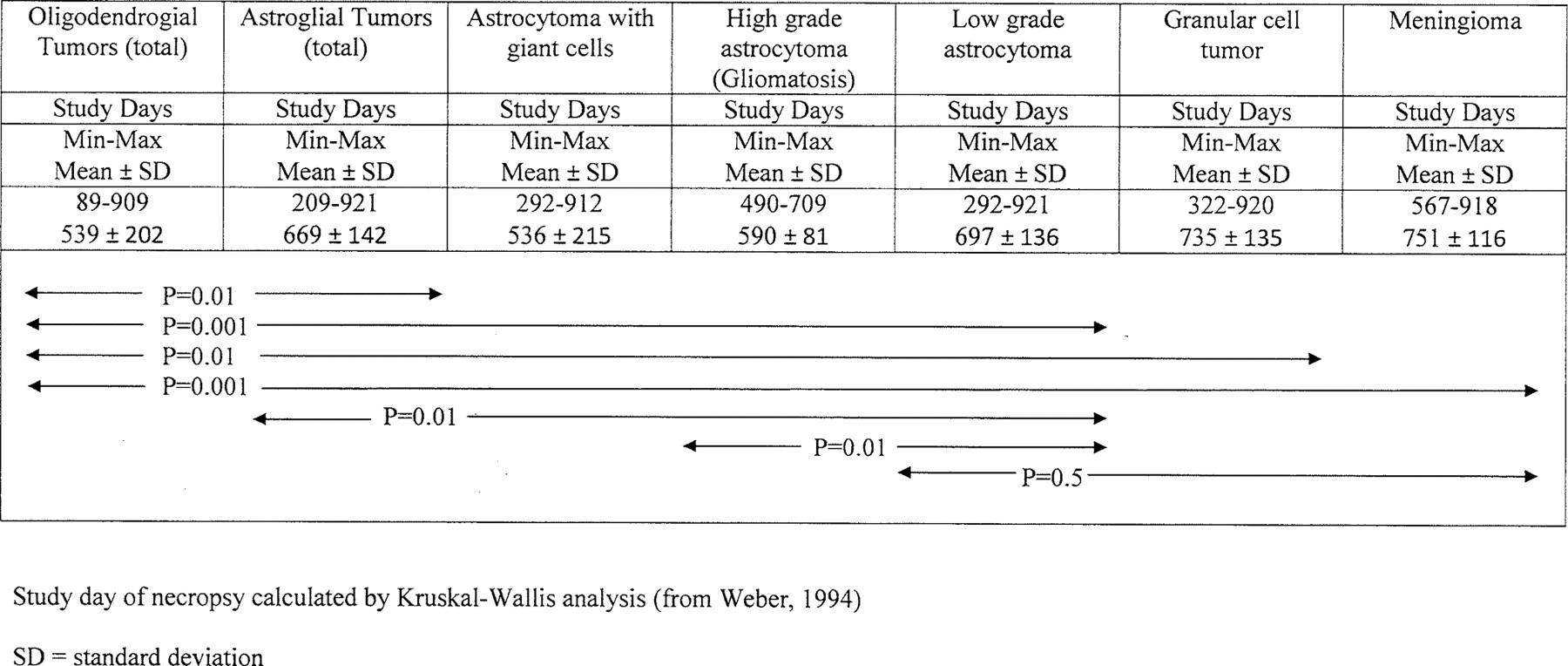
Different time course in the appearance of brain tumors in RccHan:WIST rats.
The question of glial cell tumor progression has been broadly discussed, but there have been no consistent conclusions (Bach et al. 2010a; Krinke et al. 2000). Numerous studies have shown that it is difficult to predict the biological behavior of NS neoplasms using such classic indicators of malignancy as infiltrating or destructive growth, tendency to metatasize, cellular immaturity or pleomorphism (e.g., high nuclear–cytoplasmic ratios), high mitotic index, atypical mitotic figures, incomplete or disorganized stroma, and so on. In the CNS, there are no true borders to indicate invasive growth, with the exception of the pituitary and pineal glands and the meninges. Intracranial metastases are difficult to establish on single sections, and extracranial metastases, observed in humans mainly with glioblastoma multiforme, are rare events for NS tumors (Bates 1973; Figueroa et al. 1999; Vieira dos Santos et al. 2005; Wasita et al. 2010). Although the extent of aggressive growth for different gliomas has been discussed in an attempt to define malignant versus benign phenotypes, clear behavioral outcomes cannot be established for tumors that spread throughout the parenchyma of the CNS. Such neoplasms are better classified as “low grade” or “high grade,” respectively, rather than “benign” or “malignant.” In contrast to gliomas, malignancy can be established more easily in the case of choroid plexus or meningeal tumors in the CNS, as well as peripheral nervous system (PNS) tumors in general. Extracranial metastases of some nonglial tumors have been observed in domestic animals, such as meningioma in dogs (Geib 1966; Perez et al. 2005; Schulmann et al. 1992), but have not been reported from spontaneous rodent NS tumors, except for one teratoma in a rat (Reindel et al. 1996) and, as noted in our studies, some malignant meningiomas or malignant schwannomas.
Architectural Features of Neural Tumors
The most useful features in the diagnosis of NS tumors are the microscopic structure of the masses combined with the cytoarchitecture of the neoplastic cells. Subcellular features combined with biochemical and molecular characteristics (which can be investigated using special stains, histochemistry, immunohistochemistry [IHC], and in situ molecular methods) are useful tools to confirm the primary diagnosis. With respect to the tumors found in laboratory animals, it must be remembered that tumors induced experimentally using carcinogens or genetically engineered mutations may have features—and behaviors—that are quite different from those observed in “spontaneous” NS neoplasms.
Discussions of NS tumor morphology often include the concepts of “primary” and “secondary” tumor architecture. Examples include the secondary structures of Scherer, proposed in the 1940s (reviewed in Pfeiffer and Kleihues 1999), and the three structural levels of Zülch (1956). The three levels of Zülch encompass not only features within the neoplasm proper but also structural changes imposed on and by the adjacent tissues.
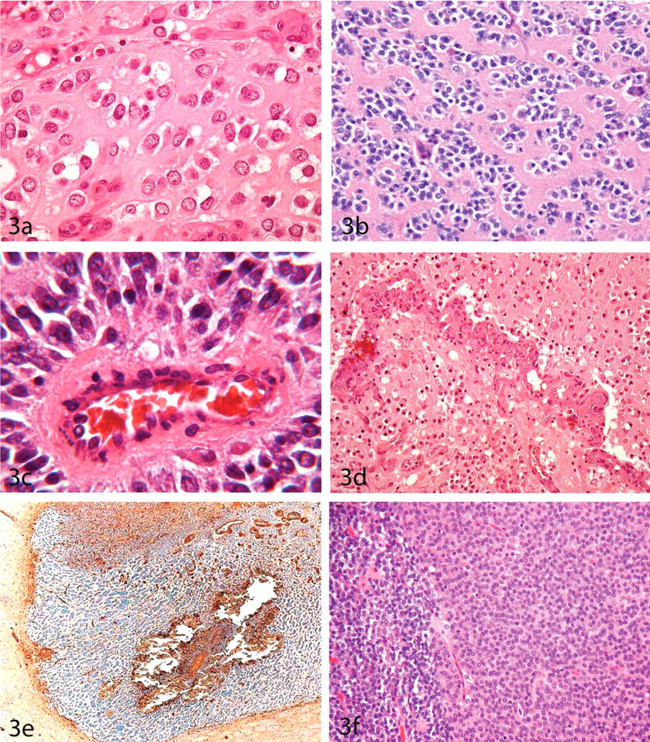
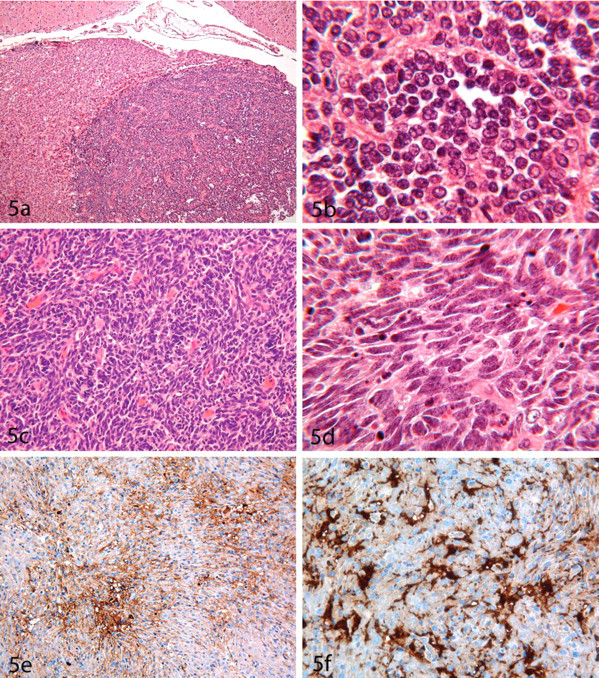
Level 1 structures relate to the functions and/or properties of the involved cell type and are independent of the neoplasm’s size and location or the presence of regressive changes. These features, which are often the most pathognomonic for specific tumor types, include such microscopic phenotypes as nuclear palisades (i.e., phalanx, or “parade positions”; Figure 7e ), papillae (Figure 4e ), and psammoma bodies (Figure 6a ). Slightly less pathognomonic structures of this class include radiating crowns with the formation of nuclear free spaces (Figures 3c, 4d ); a honeycomb appearance (Figure 2f ); combinations of true ependymal rosettes (i.e., a halo of cells surrounding an empty lumen) or ependymal pseudorosettes (i.e., concentric arrangement of tumor cells encircling small blood vessels), and epithelial tubules (Figure 4c); cell whorls or cell clusters (Figure 2a); pseudopalisade formation (i.e., cells arranged around a necrotic area or around blood vessels, in contrast to palisades [also called “phalanx” or “parade” position] where cells form parallel bands of nuclei; Figure 2c); parallel bands of cells called “fish swarm” structures (Figure 5c ); perivascular pseudorosettes (radial clusters of cells encircling a small blood vessel); Homer-Wright rosettes (a circular arrangement of tumor cells around central, pale eosinophilic areas containing neurofibrils, but no lumen; Figure 5c); and Flexner-Wintersteiner rosettes (a spoke- and wheel-shaped formation of tumor cells with short cytoplasmic extensions that surround a central lumen).
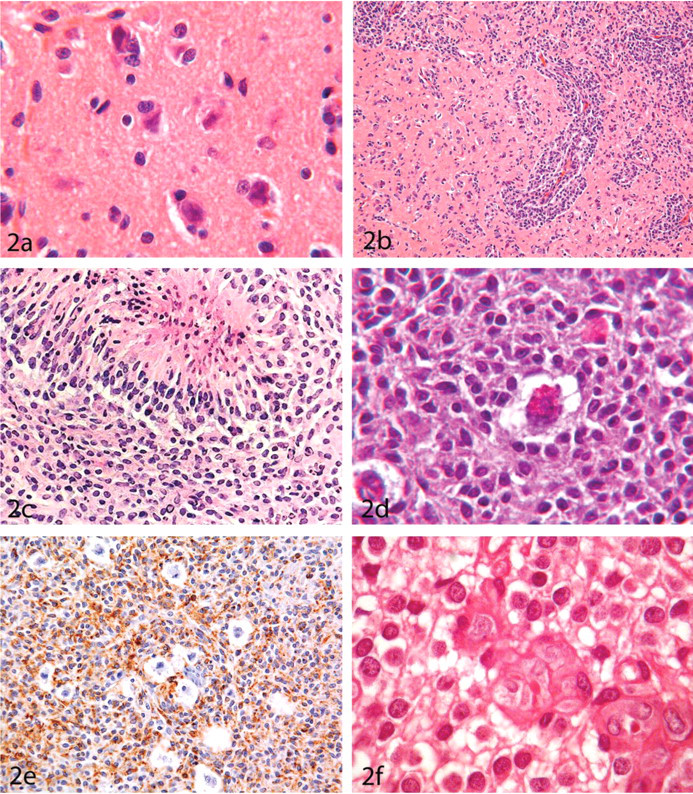
(A) Low-grade astrocytoma in the brain of an RccHan:WIST rat. A pattern of tumor cell satellitosis and neuronophagia is often present at the periphery; hematoxylin and eosin (HE), ×1,000. (B) High-grade astrocytoma in the brain of an RccHan:WIST rat, exhibiting diffuse infiltration of the brain; HE, ×100. (C) High-grade astrocytoma in the brain of an RccHan:WIST rat. Neoplastic cells are arranged in pseudo-palisades around a necrotic focus; HE, ×200. (D) Astrocytoma with granular, binucleated giant cells in the brain of an RccHan:WIST rat. The cytoplasm of the neoplastic astrocytes was periodic acid-Schiff (PAS) positive. In the giant cells, PAS-positive staining was observed in cytoplasmic granules but not the cytoplasm; PAS, ×1,000. (E) Astrocytoma with granular binucleated giant cells in an RccHan:WIST rat, demonstrating cytoplasmic reactivity of the neoplastic astrocytes but not the giant cells with antibodies against ED1; formaldehyde-fixed, paraffin-embedded section subjected to indirect immunoperoxidase immunohistochemistry, ×200. (F) Low-grade oligodendroglioma, showing the typical “honeycomb” appearance; HE, ×400.
Low-grade (A, D) and high-grade (B, C) oligodendrogliomas in the brain of the RccHan:WIST rat. Neoplastic cells are arranged in short rows embedded in an eosinophilic (A) and periodic acid-Schiff–positive (B) ground substance, and form pseudorosettes around capillaries (C). A typical pattern in this neoplasm is the formation of convoluted vessels (D). (E) High-grade oligodendroglioma in the brain of a CD(SD) rat showing a negative reaction of the neoplastic cells for the microglial marker Ricinus communis agglutinin-1. (F) Pituicytoma in the pituitary gland of an RccHan:WIST rat almost completely replacing the pituitary gland. Remnant cells of the pars anterior are seen on the left side. (A) Hematoxylin and eosin (HE), ×400; (B) Periodic acid-Schiff, ×200. (C) HE, ×1,000. (D) HE, ×100. (E) Formaldehyde-fixed, paraffin-embedded section subjected to indirect lectin histochemistry, ×40. (F) HE, ×100.
(A) Pituicytoma infiltrating the brain (same tumor as Figure 3F); hematoxylin and eosin (HE), ×200. (B) Mixed glioma in an SD-OFA rat brain consisting of dense areas of astrocytoma and oligodendroglioma; HE, ×40. (C) Ependymoma (presumptive) in a Shoe:WIST rat. There are ependyma-like tubes lined by ciliated epithelial cells; HE, ×100. (D). Ependymoma growing within the pituitary gland of an approximately fifteen-year-old domestic cat, demonstrating ependymal tubes and nuclear-free spaces; HE, ×100. (E) Choroid plexus papilloma in a Shoe:WIST rat, showing typical formation of papillae and branching connective tissue; HE, ×40. (F) Choroid plexus carcinoma of an RccHan:WIST rat forming dense masses of neoplastic cells within the ventricle and infiltrating the neuropil; HE, ×200.
(A) Pinealoma in an RccHan:WIST rat located between the brain hemispheres; hematoxylin and eosin (HE), ×100. (B) Pinealoma (a higher magnification of the one in Panel A). The karyoplasm of the tumor cells shows finely stippled chromatin. The cell cytoarchitecture resembles that of a seminoma; HE, ×1,000. (C) Medulloblastoma in the cerebellum of an RccHan:WIST rat. The tumor is composed of densely packed cells forming whorls. Homer-Wright rosettes (i.e., radial positioning of tumor cells around a central eosinophilic zone composed of fibrillary material [often composed of neurofibrils]) are visible but are not as definitive as in the corresponding tumors in humans; HE, ×100. (D) Medulloblastoma (higher magnification of the one in Panel C). Nuclei of the neoplastic cells are typically carrot shaped; HE, ×1,000. (E) Primitive multipotent neuromyoblastoma (Triton tumor) within the lateral brain base of an RccHan:WIST rat. A positive reaction to desmin is present; formaldehyde-fixed, paraffin-embedded section subjected to indirect immunoperoxidase immunohistochemistry, ×100. (F) Same tumor as in Panel E. Neoplastic cells express actin; formaldehyde-fixed, paraffin-embedded section subjected to indirect immunoperoxidase immunohistochemistry, ×200.
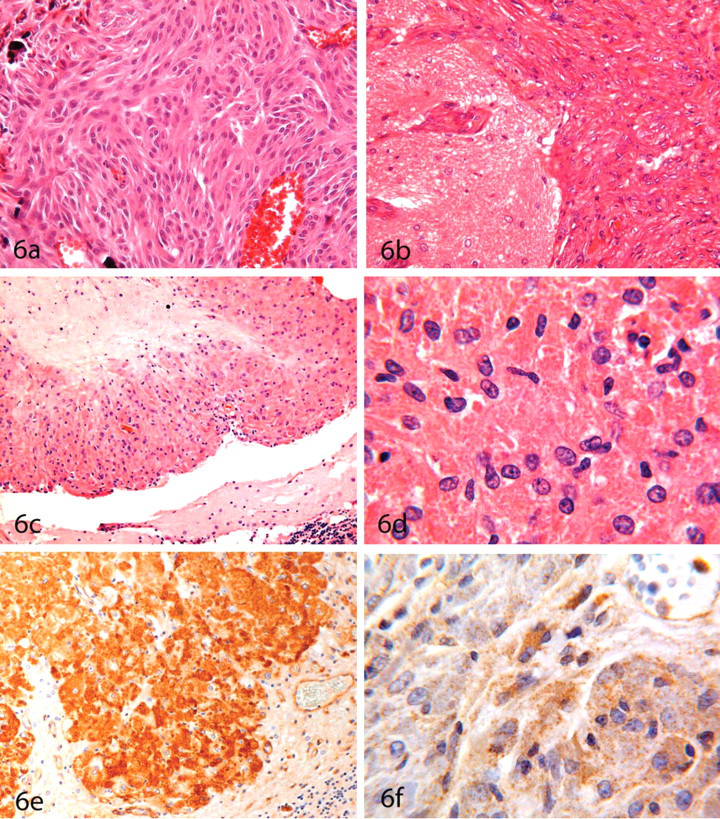
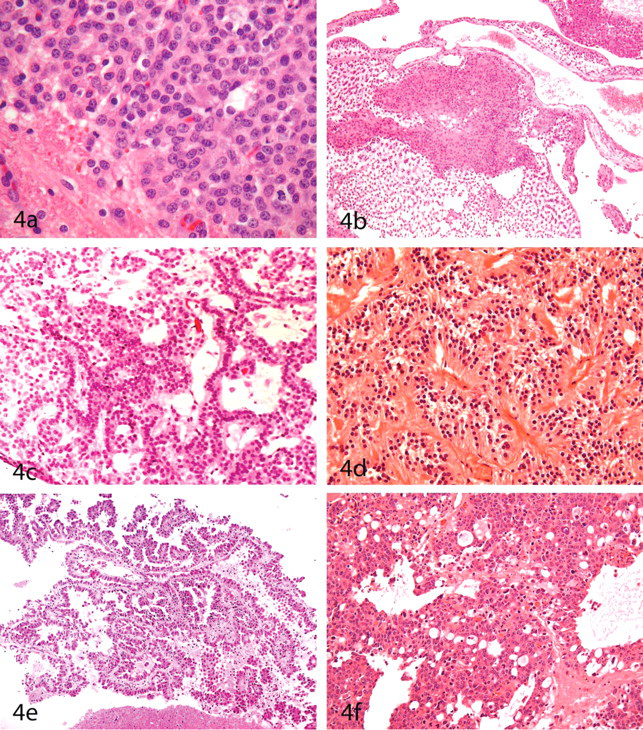
(A) Fibroblastic meningioma located in the cerebellum of an RccHan:WIST rat. The tumor contains mineralized foci (psammoma bodies); hematoxylin and eosin (HE), ×400. (B) Malignant meningioma (i.e., meningiosarcoma) in an RccHanTM:WIST rat exhibiting infiltration into the brain; HE, ×400. (C, D) Granular cell tumor on the dorsal surface of the cerebellum of an RccHan:WIST rat demonstrating the typical population of large cells packed with cytoplasmic granules (i.e., Type II cells). (C) HE, ×200. (D) HE, ×1,000. The granules react with the lectin RCA1 (Ricinus communis agglutinin-1) (E), and the granules, and to a lesser extent the cytoplasm, are labeled by antidesmosome antibodies (F). (E) Formaldehyde-fixed, paraffin-embedded section subjected to indirect lectin histochemistry, ×400. (F) Formaldehyde-fixed, paraffin-embedded section subjected to indirect immunoperoxidase immunohistochemistry, ×1,000.
(A) Granular cell tumor in an RccHan:WIST rat. A high proportion of the perinuclear cytoplasm reacts positively for vimentin. Formaldehyde-fixed, paraffin-embedded section subjected to indirect immunoperoxidase immunohistochemistry, ×1,000. (B, C) Craniopharyngioma in an RccHan:WIST rat. The neoplastic tissue, consisting of ciliated and goblet cells producing large quantities of mucus (B) and expressing pan-cytokeratin (C), may be confused with a metastatic carcinoma. (B) Hematoxylin and eosin (HE), ×400. (C) Formaldehyde-fixed, paraffin-embedded section subjected to indirect immunoperoxidase immunohistochemistry, ×200. (D) Hemangiosarcoma at the base of the brain of an RccHan:WIST rat; HE, ×40. (E) Schwannoma in an RccHan:WIST rat, demonstrating several arangments of neoplastic cells: “phalanx position,” cell-rich regions (Antoni Type A) and cell-poor areas (Antoni Type B); HE, × 400. (F) Neurofibroma located in the cauda equina of an RccHan:WIST rat; HE, ×200.
Level 2 structures are features of the neoplasm that arise in response to the resistance of the surrounding tissue. These are a consequence of the characteristics of the tissue being infiltrated and regressive changes within the tumor. Examples include satellitosis, Antoni Type A (i.e., densely packed cells) and Type B (i.e., loosely packed tissue with regions of regressive change characterized by formation of pseudocystic spaces or edematous foci and frequently appearing to be less well differentiated and more malignant) areas, and pseudopalisading.
Level 3 structures evolve in the parenchyma of the tumor and in surrounding tissues as a result of the interaction between the adjacent parenchyma and tumor tissue, and hence are reactive in nature. These characteristics develop after regressive processes that mainly affect the formation, density, and position of blood vessels (e.g., the glomerular-like vascular patterns in oligodendrogliomas; Figure 3d). Other regressive changes include cyst formation, edema, fibrosis, hemorrhage, mineralization, necrosis, pyknosis, neuronophagia, hyaline inclusions, and Rosenthal fibers (i.e., cytoplasmic inclusions in astrocyte processes that contain a hybrid of ubiquitin and, possibly, partially degraded glial fibrillary acidic protein [GFAP] fragments).
Our historical database of RccHan:WIST rat tumors contains two entities composed of multiple cell lineages. “Combination tumors” arise when a single neoplasm develops from two different cell types. The combination masses in our collection include mixed gliomas (generally astrocytes mixed with oligodendrocytes) and the amalgamation of a granular cell tumor with pituitary adenoma. In contrast, “collision tumors” are lesions in which two distinct tumors originating from two cell lineages intersect and fuse at their borders. The collision neoplasms in our compilation consist of granular cell tumors in conjunction with either astrocytomas or oligodendrogliomas. Furthermore, neural tumors may appear as multiple or multicentric masses (i.e., two or more foci in distant locations, such as bicentric astrocytoma in the brain and spinal cord).
CNS Tumors
Glial Tumors
Astrocytoma
Location
In rats, astrocytomas in the CNS occur predominantly in the rostral half of the cerebrum and mainly affect regions adjacent to the ventricles. In our RccHan:WIST rats, predilection sites included the limbic system and basal ganglia followed by the periventricular regions. Two tumors were located in the spinal cord.
Primary Diagnostic Features
Astrocytomas do not have pathognomonic structural characteristics. The neoplastic cells are arranged in clusters, whorls, cell strands, and, occasionally, pseudopapillary structures. In the periphery, satellitosis of tumor cells around neurons is a common feature (Figure 2a). Pseudopalisading is very rare, and it usually develops at the margins of necrotic areas (Figure 2c). Regressive changes are not common except for neuronophagia in aggressive tumors, but these changes include cysts, hemorrhage, mineralization, necrosis, pyknosis, and Rosenthal fiber formation. Although rat astrocytomas are far less well differentiated than those in humans, an attempt has been made to classify the tumors in RccHan:WIST rats according to the known types reported in humans (i.e., fibrillary, gemistocytic, pilocytic, or protoplasmic). The following astrocytoma types were recorded in the brain: fibrillary, 22.2%; pilocytic, 37.8%; pilocytic-fibrillary, 15.6%; protoplasmic, 13.3%; protoplasmic-pilocytic, 2.2%; and protoplasmic-fibrillary (Weber 1994), 4.4%; astrocytomas in the spinal cord (4.4%) were all of the fibrillary type (Weber 1994). Gemistocytic astrocytes were seen focally in protoplasmic astrocytomas, but not in other kinds. Therefore, subclassification of astrocytomas as previously proposed (Koestner et al. 1999) appears to be a useless exercise.
Ancillary Diagnostic Features
Neoplastic astroglia in rat brain tumors stained positively with antibodies against ED1 (a macrophage marker; Figure 2e) and RCA1 (a microglial marker). The positive reaction for ED1 was consistently present within all investigated tumors, suggesting that astrocytes are capable of expressing macrophage antigens. A positive reaction of rat astrocytomas to RCA1 has also been reported by Krinke and Germer (1993), suggesting that the surface of neoplastic cells could have glycosylation patterns similar to that observed in microglial cells.
In general, neoplastic astrocytes were negative for glial fibrillary acidic protein (GFAP), Fc receptor (FC), fibronectin (FN), major histocompatibility complex type II (MHCII, another microglial marker), and S100. However, occasional cells are labeled by some markers. For example, in spontaneous astrocytomas in rats, antibodies against GFAP show staining only of reactive astrocytes at the tumor periphery, but the tumor cells themselves do not stain. However, astrocytomas induced by avian sarcoma virus (ASV) or ethylnitrosourea (ENU) contain tumor cells with positive reactions against GFAP, as described by Solleveld et al. (1990). Also, Zook, Simmen, and Jones (2000) observed GFAP positivity in ENU-induced gliomas in rats, although only a portion of the neoplastic cells was reactive. Similarly, recent IHC investigations have revealed MHCII expression in scattered cells within previously diagnosed acrylonitrile-induced astrocytomas (Kolenda-Roberts and Hardisty 2010), and the diffuse expression of leukocyte antigens in tumors diagnosed as malignant reticulosis (Halm 2010) that are in many aspects similar to astrocytomas. Therefore, it is possible that some neoplasms previously classified as astrocytomas may actually represent microglial neoplasms.
Astrocytoma Variants
Low-grade astrocytomas ranged from 0.4 up to 14.4 mm in diameter and were in most cases composed of small, fairly uniform cells (5–25 µm in diameter). These cells typically had indistinct cell borders. Neoplastic cell nuclei were between 5 and 15 µm in diameter, rich in chromatin, and usually contained two nucleoli. Up to four mitotic figures were evident per high-power field (hpf). All tumors infiltrated the surrounding parenchyma but, in single cases, infiltrated into the meninges, choroid plexus, or ventricles. In most cases, vascularization was not distinct.
A high-grade astrocytoma variant was identified with binucleate giant cells (Figures 2d, 2e) of unknown origin. Relative to low-grade astrocytomas, these high-grade neoplasms were larger (3.2 × 2.8 to 13.8 × 6.8 mm in diameter) and had higher mitotic rates. Regressive changes were much more common in the high-grade tumors and included fibrosis, hemorrhage, hemosiderin deposition, mineralization, necrosis, and neuronophagia. Increased vascularization was a distinct feature. The giant (15–45 µm in diameter) binucleated cells contained eosinophilic cytoplasmic granules (1–2 µm in diameter). The cytoplasm of the neoplastic astrocytes, but not that of the binucleated giant cells, was stained with periodic acid-Schiff (PAS), but the giant cell granules stained diffusely with PAS. The cell membranes and cytoplasmic granules of the neoplastic astrocytes prominently expressed RCA1, but in the giant cells, RCA1 labeled only the peripheral cytoplasmic granules. The binucleate cells did not express ED1.
The presence of granular cells in a rat astrocytoma was first described three decades ago (Dagle et al. 1979), but the first detailed investigation of this variant was not provided until twenty-five years later (Pruimboom-Brees et al. 2004). An astrocytoma in a two-year-old SD rat contained two populations, a small one that was positive for lysozyme (LYS), phosphtungstic acid-hematoxylin (PTAH), and vimentin (VIM) and a binucleated cohort that stained with PAS and PTAH, but not LYS. The presence of phagolysosomes containing myelin figures and glycoprotein or glycolipid was demonstrated in the binucleated cells. The authors concluded that neoplastic astrocytes were the origin of the binucleated cells in this variant because of the absence of GFAP, and they further hypothesized that accumulation of autophagocytic vacuoles developed via a lysosomal enzyme defect. Furthermore, the absence of proliferative cells in this tumor was considered to result from an additional mitotic defect. In humans, an astrocytoma variant has been described in which granular cell changes derive from the accumulation of autophagocytic vacuoles or excess production of a substance normally processed in lysosomes (Dickson et al. 1986; Geddes et al. 1996; Kornfeld 1986). However, the cells in our material resemble the “granular metrial gland cells” of deciduomas in rats, which have been identified as natural killer (NK) cells (Linnemeyer and Pollack 1991). Therefore, a possible NK (i.e., lymphocytic) origin of the neoplastic cells recorded in this subtype of astrocytoma cannot be definitively excluded.
Another rat astrocytoma variant—high-grade astrocytoma without binucleated giant cells (also referred to as “astrocytic gliomatosis”)—was characterized by diffuse infiltration of tumor cells throughout the brain (Figures 2b, 2c). In this neoplasm, mitotic figures were frequently encountered, just as in high-grade astrocytomas with giant cells.
Differential Diagnoses
The differential diagnoses for astrocytoma include induced glioblastoma multiforme (GBM), anaplastic glioma, mixed glioma, high-grade oligodendroglioma, pituicytoma, and neuromyoblastoma (Triton tumor).
Oligodendroglioma
Location
In RccHan:WIST rats, all oligodendroglial tumors occured in the diencephalon and telencephalon, with the majority of tumors affecting the middle part of the cerebrum adjacent to the ventricles. Predilection sites included the limbic system, basal ganglia, cerebral cortex, and walls of the ventricular system.
Primary Diagnostic Features
Honeycomb structures and the arrangement of cells in short rows within an eosinophilic ground substance along with vascular (glomerular-like) convolutions are the most striking characteristics of oligodendroglial tumors (Figures 2f, 3a). The tumors in our material were well vascularized, especially at the periphery (Figure 3d). Regressive changes consisted of cysts, hemorrhage, and necrosis. Mineralization, hemosiderin deposition, and occasional cholesterol crystals were encountered. However, mineralization of oligodendrogliomas in rats does not possess diagnostic importance comparable to that in human tumors.
Low-grade oligodendrogliomas in our series infiltrated the surrounding parenchyma, compressed the surrounding parenchyma, and infiltrated the ventricular system. However, in contrast with astrocytomas (which diffusely invade at the periphery), oligodendrogliomas were rather sharply delineated. These tumors reached sizes of 1.0 to 12.0 × 5.0 mm in diameter and were composed primarily of neoplastic cells with diameters of 10 to 25 µm. The tumor cells had chromatin-rich nuclei that contained up to three nucleoli. Anisocytosis (or even pleomorphism) with nuclear membrane invagination was common. Between zero and five mitotic figures per hpf were present.
In high-grade oligodendrogliomas (oligoblastomas), tumor cell clusters were embedded in broad bands of an eosinophilic, PAS-positive ground substance (Figure 3b). Perivascular rosettes and clefts mimicking ependymal cell tubes were common. The neoplastic cells were slightly smaller than in low-grade tumors (7.5–20 µm in diameter) and contained chromatin-rich nuclei without nucleoli. One to five mitotic figures per hpf were present. Several oligodendrogliomas in male rats had areas with both low-grade and high-grade features.
Ancillary Diagnostic Features
The cytoplasm of neoplastic oligodendrocytes reacted positively with antibodies against carbonic anhydrase C (CAC), and often against myelin basic protein (MBP), but never with RCA1 or VIM (Figure 3e).
Differential Diagnoses
Oligodendrogliomas, in contrast to astrocytomas, do not express RCA1. Other tumors in the differential list for oligodendroglioma include ependymomas and induced GBM.
Pituicytoma
Pituicytoma is generally rare in all species, including monkeys (HogenEsch et al. 1992) and humans (Shah et al. 2005).
Location
A single case of pituicytoma was observed in an aged female RccHan:WIST rat. The tumor replaced the pituitary gland almost completely and infiltrated along the blood vessels into the meninges and adjacent diencephalon (Figures 3f, 4a).
Primary Diagnostic Features
The neoplastic cells of the pituicytoma resembled those in an astroglioma. Tumor cells were spindle-shaped and had abundant, pale eosinophilic cytoplasm; variably sized nuclei; and indistinct cell borders. Cells were arranged in solid sheets.
Ancilliary Diagnostic Features
Two cases similar to the pituicytoma in an RccHan:WIST rat were identified in aged, female F344 rats (Satoh et al. 2000). These authors concluded that the tumors were related to astrocytomas based on their morphological traits and the GFAP reactivity of tumor cell foot processes. However, in a human case (Takei et al. 2005), positive IHC reactions to GFAP, S100, and VIM, as well as focal reactivity for epithelial membrane antigen, were considered crucial for distinguishing the pituicytoma from other intrasellar tumors, such as granular cell tumor and pilocytic astrocytoma.
Differential Diagnoses
Granular cell glioblastoma, a malignant granular cell neoplasm of astrocytic origin, has also been reported in the pituitary, as have granular cell tumors, pilocytic astrocytomas, and spindle cell tumors such as fibroblastic meningiomas, schwannomas, and solitary fibrous tumors of the posterior pituitary gland (Figarella-Branger et al. 2002; Hurley et al. 1994; Kowalski et al. 2004; Schultz et al. 2001). The unique pattern of antigenic expression—strongly immunoreactive to neural cell adhesion molecule (NCAM), neuron-specific enolase (NSE), S100, VIM, and very late antigen α2 (VLAα2), but negative for epithelial and specific neuronal markers (chromogranin, neurofilament, synaptophysin) and VLAα5—as well as the characteristic ultrastructural features (i.e., presence of tumor/blood vessel basal lamina and cytoplasmic intermediate filaments, but absence of desmosomes or pericellular basal lamina) distinguish this rare tumor (Figarella-Branger et al. 2002). Depending on their state of differentiation, some pituicytomas do not express GFAP. However, ultrastructural features comparable to those described in human tumors (Nakasu et al. 2006) have been previously described in pituicytoma cells positive for GFAP (Cenacchi et al. 2001). These cells had features consistent with both pituicytoma and pituitary adenoma differentiation (i.e., aggregates of intermediate filaments and secretory granules), suggesting an origin of this tumor from stromal folliculostellate cells of the adenohypophysis.
Ependymoma
Location
Tumors of ependymal origin are uncommon in rodents (Solleveld et al. 1991). To our knowledge, all reported cases have been experimentally induced by carcinogen exposure or were misdiagnosed oligodendrogliomas. These growths occur in the wall and/or the lumen of the ventricles, and occasionally in the spinal cord central canal.
Primary Diagnostic Features
Ependymomas are characterized by a dense growth pattern of primarily uniform cells with mostly indistinct cell margins. The typical architecture in humans and domestic animals is that of ependymal rosettes (ependymal tubules, Figure 4c), perivascular pseudorosette formation, and nuclear-free zones (Figure 4d).
Ancillary Diagnostic Features
In rats, pathognomonic features are typically absent so that these tumors should be diagnosed based on their positive IHC reactions for VIM and/or the ultrastructural presence of blepharoblasts (basal bodies) and microvilli. Regressive changes such as hemorrhage and necrosis are common. Tumor cells are negative for CAC and MBP.
Differential Diagnoses
The rare tumors recorded in our rat material as possible ependymomas were typified by ependymal tubes with short rows of cells and nucleus-free zones (Figure 4c). In some tumors, the architecture focally resembled oligodendrogliomas because of the presence of honeycomb structures. In mixed tumors, the oligodendroglial component may contain convoluted vessels. Oligodendroglioma can be excluded based on the IHC staining pattern.
Mixed Glioma
Mixed glioma is typically selected as the diagnostic term when each cell population represents at least 20% of the total neoplastic mass, but it may be diagnosed even if the second component of the tumor constitutes less than 20% of the total tumor mass. For example, the senior author has personally found very small-sized focal oligodendrogliomas within induced astrocytomas.
It is important to recall that a certain portion of any neural neoplasm may be composed of cells that differ from the primary neoplastic cell type (e.g., reactive astrocytes within an oligodendroglioma). Mixed gliomas are not often encountered as spontaneous neoplasms in rats but may exist as mixed oligoastrocytomas or as oligoependymomas (Figures 4b, 4c). Oligoependymomas are high-grade oligodendrogliomas with anaplastic cells that have large nuclei arranged in honeycomb formations, as well as ependyma-like structures such as ependymal tubes and rosettes or perivascular pseudorosettes (Figure 4c).
Tumors of the Choroid Plexus
Neoplasms of the choroid plexus are generally rare events in all species. These tumors are located intraventricularly.
Plexus Papilloma
To the authors' knowledge, plexus papillomas have not been described in laboratory animals. However, in the senior author’s case series, two plexus papillomas were diagnosed in aged Shoe:WIST rats. These tumors were characterized by papillary structures formed by the choroid plexus, and the original choroid plexus morphology was retained. The stroma supporting the papillae was formed by branched connective tissue with formation of intrapapillary threads. The tumors were glandlike in architecture and were restricted to the ventricles. Mitotic figures were not found (Figure 4e).
Plexus Carcinoma
The malignant tumor of choroid plexus epithelium is a rare lesion in all species, including humans and domestic animals. Individual cases have been documented in aged rats (Pace 1998; Shimomoto et al. 2004), and a murine case has been reported (Krinke and Kaufmann 1996). In humans, a greater percentage of plexus carcinomas occur in children than in adults (Newbould et al. 1995; Rickert and Paulus 2001).
Primary Diagnostic Features
Plexus carcinomas are usually solid epithelial tumors with dense growth patterns that arise within the ventricles. The tumor pattern is a result of multilayered cell cords that transition to solid sheets of cells that have no discernable pattern. Necrosis is a common regressive change, and invasive growth into the periventricular parenchyma is a consistent feature (Figure 4f).
Ancillary Diagnostic Features
In rats, plexus tumors exhibit positive IHC reactivity for Lu5 (a cytokeratin; Pace 1998) and for pancytokeratin but are negative for GFAP, S100, and VIM (Shimomoto et al. 2004). Cytokeratin positivity has been reported in domestic animals, as well as positivity for VIM in a goat (Klopfleisch et al. 2006) and, in several cases, in dogs (Cantile et al. 2002). In dogs, the latter authors also reported positivity for carcinoembryonic antigen (CEA); however, the reactions for epithelial membrane antigen Ber-EP4 and for S100 were negative. In human pathology, pancytokeratin is used as an important marker (Barreto et al. 2004). Transthyretin (TTR) and S100 protein are also present in a high proportion of human cases (Kunishio et al. 1991; Megerian et al. 1997; Rickert and Paulus 2001).
Differential Diagnoses
Ependymoma needs to be considered in the differential diagnosis for plexus carcinoma but does not show the same degree of positivity for cytokeratin. Transthyretin can be a very useful diagnostic marker for differentiating pleux papilloma versus ependymoma (Kunishio et al. 1991). Medulloblastomas may resemble plexus carcinomas but do not express epithelial markers, TTR, or carbonic anhydrase II (Newbould et al. 1995).
Primitive Neuroectodermal or Bipotential Tumors
Tumors of Pineal Gland
Among laboratory animals, pinealoma has been described in rats but is extremely rare. Single cases were reported in aged rats of different strains including Crj:CD (SD) IGS (Furukawa et al. 1999), WIST (Yamamoto et al. 1991), F344 (Heath and Winokur 1998), and Osborne-Mendel (Dagle et al. 1979).
Location
The typical location of pinealoma is between the cerebral hemispheres in the dorsal and caudal brain, since this is the normal location of the pineal gland.
Primary Diagnostic Features
A tumor diagnosed in RccHan:WIST rats consisted of two densely packed cell types: a large (12–15 µm), pale cell with large nucleus (10–12.5 µm) containing multifocal chromatin condensations, and small, dark cells resembling lymphocytes. The latter cell type tended to cluster. The large, pale elements had morphological similarities to seminoma cells (Figures 5a, 5b).
Ancillary Diagnostic Features
By IHC, the cytoplasm and cell membranes of large cells were positive for desmoplakin 2 and cytokeratin AE1/AE3. Positive staining with synaptophysin may also support the diagnosis (Heath and Winokur 1998).
Differential Diagnoses
However, pinealomas in rats may be benign or malignant in nature. Transformation of the pineocytoma into the less differentiated pineoblastoma has not been described in rodents. Teratoma (dysgerminoma) should be excluded.
Medulloblastoma
The medulloblastoma is not often encountered in rats and is typically seen in younger animals. However, medulloblastomas may also be found in animals necropsied at greater than 100 weeks of age. In humans, medulloblastoma is the most common malignant brain tumor noted in children (Eberhart 2003).
Location
Medulloblastoma is a primitive neuroectodermal tumor (PNET) that forms in the cerebellum but often extends into adjacent regions.
Diagnostic Features
Medulloblastomas are composed of primitive cells with the potential to differentiate along either neuronal or glial lines. In rats, PNETs show a very densely cellular growth pattern, often with pleomorphic cells with indistinct cell margins and nuclei that are rich in chromatin (Figure 5c). Nuclei may exhibit a characteristic “carrot” shape (Figure 5d). Pseudopalisading and necrosis are common.
Differential Diagnoses
Similar lesions outside the posterior fossa are classified as PNETs in humans (Eberhart 2003). This entity is deemed to be malignant. Primitive neuroectodermal tumors are expected to react positively for synaptophysin.
Neuromyoblastoma
Neuromyoblastomas are very rare in humans and are usually recorded as Triton tumors in peripheral nerves. These neoplasms are composed of intermingled mature neural and striated muscle elements (Vajramani et al. 1999). This entity was reported in the trigeminal ganglion of an aged CD-1 mouse, where it consisted of well-differentiated striated muscle fibers intermingled with nerves and ganglion cells (Ernst et al. 1993). Another case was described in a control RccHan:WIST rat in the basal portion of a cerebral hemisphere. This primitive malignant tumor contained pleomorphic elements resembling GBM. By IHC, rhabdomyoblasts reacted positively with desmin and actin (Figures 5e, 5f). This hamartomatous lesion of neuroectodermal-mesenchymal nature may be considered to have arisen either when mesenchymal tissue was incorporated into the neural tube during neurulation or by aberrant differentiation of neuroectodermal components into mesenchymal elements.
Tumors of the Meninges
Location
A preferred location for meningioma could not be established in our material, and a connection to the meninges was not clearly proven in all cases.
Primary Diagnostic Features
Generally, meningiomas exhibit a dense growth pattern. The cells are often uniform with definite margins and oval nuclei. The architecture is characterized mainly by cell whorls and dense cell nests. Regressive changes consist of increased connecting tissue and, rarely, psammoma bodies. Occasionally, scattered granular cells may be detected. Morphological subtypes currently recognized in the rodent literature consist of meningotheliomatous (lobules of large uniform cells similar to arachnoid cells surrounded by thin collagenous septae), fibrous (spindle-shaped cells resembling fibroblasts; Figure 6a), anaplastic/undifferentiated, and granular cell meningiomas (Bach et al. 2010b). A rare case of psammomatous meningioma in an F344 rat has been described in which the mass contained numerous foci of hyalinized collagen and mineralization (psammoma bodies) (Bach et al. 2010b). In addition, myxoid malignant meningioma has historically been seen in mice, and these tumors have been classified as a variant of the fibrous meningioma phenotype.
Granular cell tumors are the most common meningeal tumor in rats, and it is the most frequently recorded benign neoplasm in the brain. These neoplasms are a meningioma variant (Krinke et al. 1985; Mitsumori et al. 1986; Yoshida et al. 1997). The tumor tissue is compact and often exhibits syncytial areas (Figure 6c). Other common features include connection to the meninges, compression of the underlying brain tissue, and perivascular spread. Reticulin fibers may also be present. Tumor lobulation by connective tissue is frequently observed. Vascularization is not prominent in most cases. Regressive changes consist of fibrosis, dilated venous vessels, focal hemorrhage, focal edema, cysts, and in larger tumors, pyknosis or focal necrosis. Rarely, psammomatous bodies or hemosiderin may be evident near the periphery.
One-third of all granular cell tumors in RccHan:WIST rats caused brain atrophy by compression. In contrast, only one tumor in 149 cases invaded into the ventricular system, and only one other invaded focally into the skull. The sizes of these tumors ranged from 0.1 to 17 mm in diameter. Cells reached diameters ranging from 12.5 to 50 µm. The small tumor cells containing few or no granules in the cytoplasm are called Type I cells. The larger Type II cells contained numerous intracytoplasmic granules and round nuclei (5 to 17.5 µm) that contained 2 or 3 nucleoli (Figure 6d). Approximately one-half of granular cell tumors consisted of a uniform cell population. Multinuclear cells or cells with giant nuclei were seen in only two of the invading tumors. Mitotic figures were generally rare. The characteristic feature of the granular cell tumor is the presence of intracytoplasmic granules that are positive for PAS, Luxol fast blue, and Nissl stain and stain orange to red brown by Masson Goldners Trichrome.
A malignant meningioma (meningiosarcoma; Figure 6b) was seen in our SHOE:WIST rat material and classified as an angiomatous meningioma. The appearance of the tumor cells is similar to benign meningioma; however, it contains pleomorphic cells and multiple mitotic figures and grows aggressively. This tumor also was characterized by numerous vessels present within the background of an otherwise typical meningioma. In such a case, angioma and arteriovenous malformation (an abnormal anastomosis between arteries and veins, which is usually congenital in nature) should be excluded as a differential diagnosis.
Ancillary Diagnostic Features
The granules of granular cell tumors react positively with antibodies against RCA-1 (Figure 6e), and to a lesser degree with anti-cytokeratin AE1/AE3. In addition, a positive reaction is recorded in the cytoplasm of many Type II cells with anti-desmosome (32-2B) D2 (Figure 6f) and in Type I cells for anti-VIM (Figure 7a). The combination of VIM and D2 in neoplastic cells within a single mass is very unusual, because D2 is an epithelial marker, whereas VIM is a mesenchymal marker. Except in arachnoid cells in the meninges, this combination of markers has been recorded only in myocardial cells and Purkinje fibers (Moll et al. 1986). By electron microscopy, the granules appear as “angular bodies” and are considered to represent a form of lysosomes.
Differential Diagnoses
In the differential diagnosis of meningioma, especially in mice, the hamartomatous entity “meningioangiomatosis” must be taken into consideration. This lesion is characterized by plaque-like thickening of the cerebral meninges composed of proliferating perivascular, pluripotent mesenchymal cells characterized by fibroblastic or meningothelial differentiation. Perivascular penetration of Virchow-Robin’s spaces can be confused with the invasive growth of a malignant tumor.
Other Tumors of the CNS
Craniopharyngioma
Craniopharyngiomas are epithelial neoplasms thought to arise from the remnants of Rathke’s pouch. They are usually located in the sellar and suprasellar regions. These neoplasms are rare in humans (Nakajou et al. 1994) and laboratory animals (Guzmán-Silva et al. 1988; Heider 1986; Pace et al. 1997).
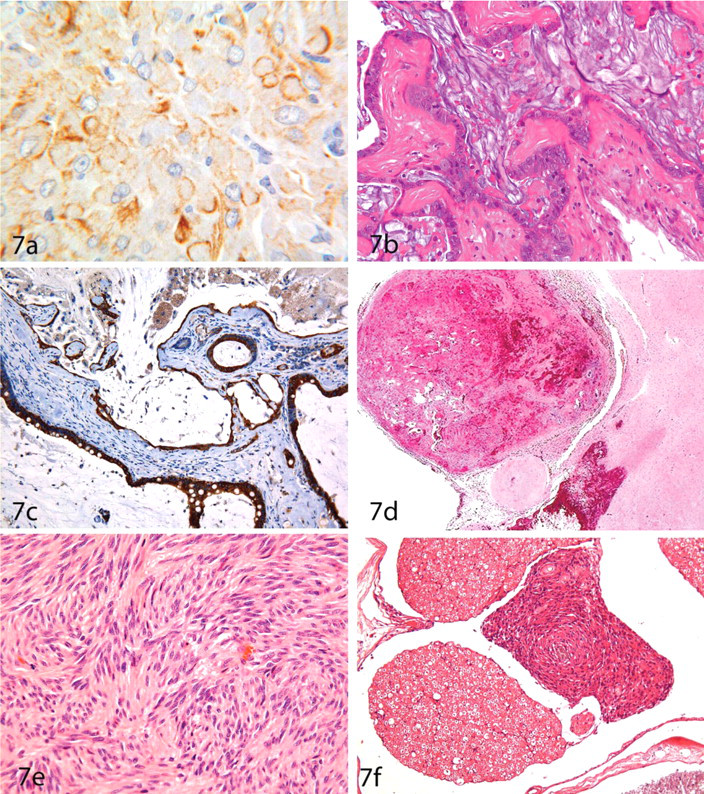
One tumor in an RccHan:WIST rat showed adenocarcinoma-like structures with tubes and cysts lined by goblet cells and ciliated cells (Figure 7b). Rathke’s cysts should be excluded as a differential diagnosis. In contrast to the present case, in human pathology, Rathke’s cysts express cytokeratins 8 and 20 (Figure 7c), whereas craniopharyngiomas do not (Xin et al. 2002). Aberrant craniopharyngeal structures in the neurohypophysis may occasionally be misdiagnosed as craniopharyngioma because they do not react with cytokeratins (Schaetti et al. 1995).
Other Mesenchymal Neoplasms
Malignant reticulosis, which histologically consists of clusters and perivascular pseudorosettes of lymphoid, histiocytic, and fibroblastic cell elements, spreads diffusely into the brain parenchyma via the Virchow-Rubin and submeningeal spaces. By histogenesis, this tumor may represent a primary neoplasm originating from microglia or from peripheral leukocytes (lymphocytes or macrophages). Accordingly, no CNS tumor should be termed “malignant reticulosis” unless more common systemic neoplasms (e.g., lymphoma, histiocytic sarcoma) have been excluded with certainty. Differentiation of these entities is possible only by the use of IHC markers. In rats, however, ED1 or lectins as markers for microglia or other cells of histiocytic origin cannot be recommended for this purpose, because neoplastic rat astrocytes also react positively with these markers.
Miscellaneous
There are other CNS tumors that are not directly related to neural cell lineages, and so they are not included in the present publication. Other such lesions recorded by the authors within the CNS of RccHan:WIST rats include one case of hemangiosarcoma that may have originated in the pituitary gland (Figure 7d), pituitary gland tumors, and a single case of chordoma affecting the lumbar spinal cord.
Peripheral Nervous System Tumors
Schwannoma
Schwannomas may be found in any organ. In the rat PNS, these tumors appear to occur in approximately 1% of aged animals (Table 5 ) (Charles River 2009; Harlan 2008; NTP 2004a). In the PNS of laboratory animals (mainly rats), schwannomas are typically found in peripheral somatic nerves. They have also been found to involve cranial and spinal cord nerves, but these latter sites are not a common location for this tumor.
Prevalence and Localization of Schwannoma in Different Rat Strains
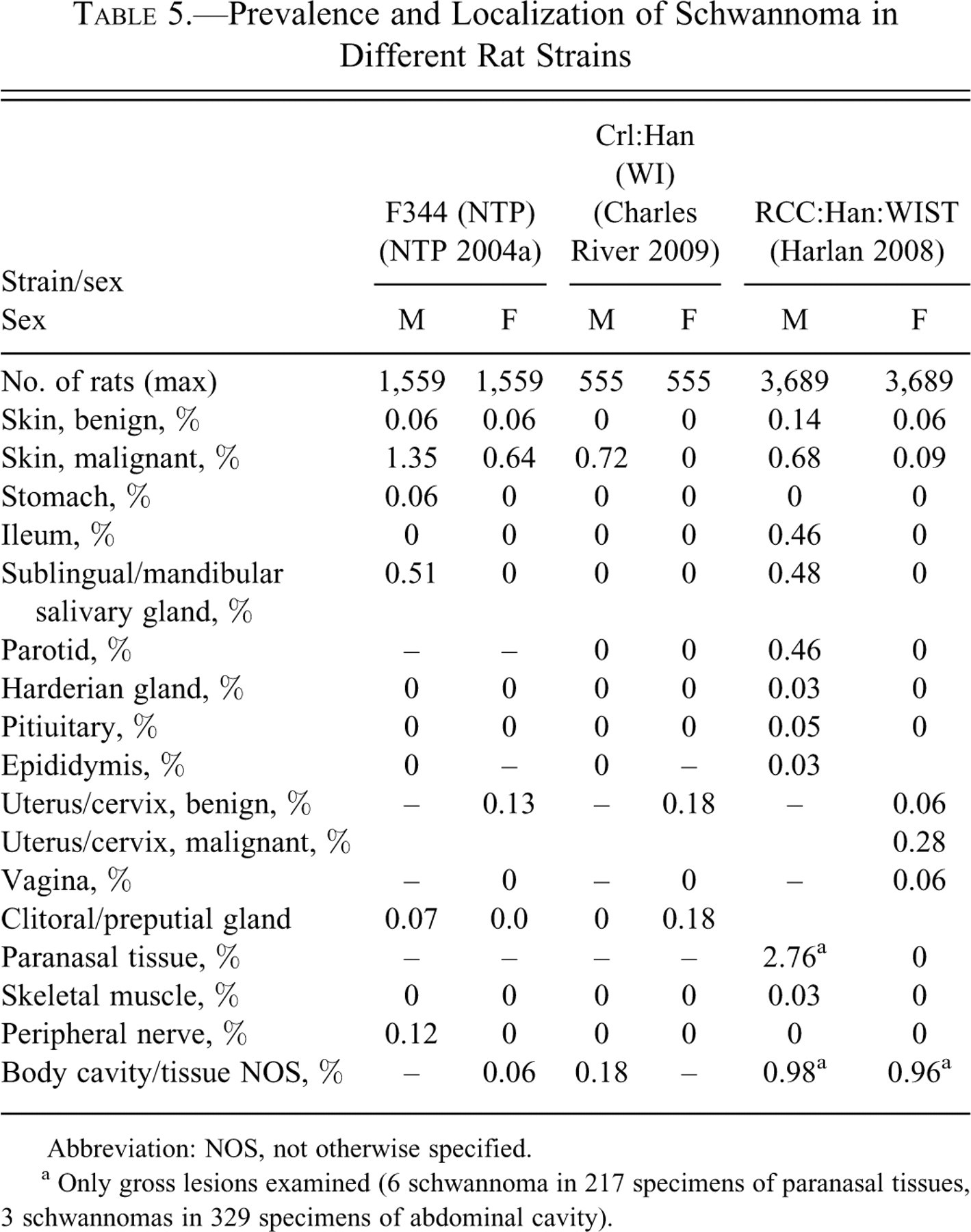
Abbreviation: NOS, not otherwise specified.
a Only gross lesions examined (6 schwannoma in 217 specimens of paranasal tissues, 3 schwannomas in 329 specimens of abdominal cavity).
In humans, schwannomas are usually considered to be benign, whereas malignant tumors arising from a peripheral nerve or showing nerve sheath differentiation (except for masses originating from the epineurium or the peripheral nerve vasculature) are instead classified as “malignant peripheral nerve sheath tumors” (MPNST) (Woodruff et al. 2000a; Woodruff et al. 2000b). The concept of benign and malignant peripheral nerve sheath tumors (BPNST, MPNST) reflects the presence of tumors that exhibit features of both schwannoma and neurofibroma or neurofibrosarcoma (see below). For practical purposes, PNS tumors in the rat exhibiting Schwann cell differentiation and that are positively labeled for S100 protein should be diagnosed as schwannomas.
Primary Diagnostic Features
Schwannomas in rodents are characterized by a dense growth pattern composed of cells that often have indefinite cell margins and that contain oval to round, hyperchromatic nuclei. The typical tumor architecture consists of cell whorls, “fish swarms,” and palisades (Figure 7e). The latter pattern is more clearly visible in mouse tumors than in those from rats. Further architectural patterns often include the Antoni Type A (densely packed cells) and/or B (loosely packed tissue) areas. The Antoni Type B appearance may be regarded as a region of regressive change characterized by formation of pseudocystic spaces and edematous foci; these zones also frequently appear to be less well differentiated and more malignant than the Antoni Type A regions. Regressive changes are rare in benign tumors.
Schwannomas in rodents are usually malignant in nature. These malignant tumors may be anaplastic, which may make differentiating this lesion from other poorly differentiated neoplasms quite difficult. An example in our series is a single case recorded in the optic nerve of an RccHan:WIST rat. The tumor mass consisted of poorly differentiated, uniform-appearing cells with indistinct margins and round nuclei and did not exhibit any particular growth pattern. However, the optic nerve, by itself, cannot be regarded as the origin for this tumor because this nerve is a direct extension of the CNS, which does not have its own Schwann cells; instead, it is myelinated by oligodendroglia. Thus, nerves of the surrounding meninges or blood vessels may have been the site of origin.
Ancilliary Diagnostic Features
By IHC, neoplastic cells in schwannomas react positively for MBP, neurofilament protein (NFP), and S100. A survey of schwannoma incidence in different organs in various rat strains is given in Table 5.
Differential Diagnoses
Depending on the PNS site at which the schwannoma is identified, neoplasms that should be considered in the differential diagnosis include fibrosarcoma, leiomyosarcoma, histiocytic sarcoma, and, in the uterus, stromal sarcoma. Non-neoplastic proliferative lesions of the PNS such as neuromas may also resemble schwannomas, but schwannomas consist entirely of Schwann cells, whereas neuromas contain a tangled mixture of Schwann cells and many axons.
Endocardial Schwannoma
Endocardial schwannoma is a neoplastic lesion recorded in many rat strains (Table 6 ), usually at incidences <1% (Brix et al. 2005; Charles River 2009; Harlan 2008; NTP 2004a). However, an incidence of 1.8% has also been reported in one compilation (Teredesai and Wöhrmann 2005). This neoplasm may be spontaneous but can also be induced, for example by N-nitroso-N-methylurea (Alison et al. 1987; Berman et al. 1980; Novilla et al. 1991; Tanaka et al. 1983). Small lesions may be recorded as hyperplasia rather than as neoplasia. A perineural or Schwann cell origin is favored for the histogenesis of this neoplasm.
Prevalence of neural tumors in the hearts of different rat strains.
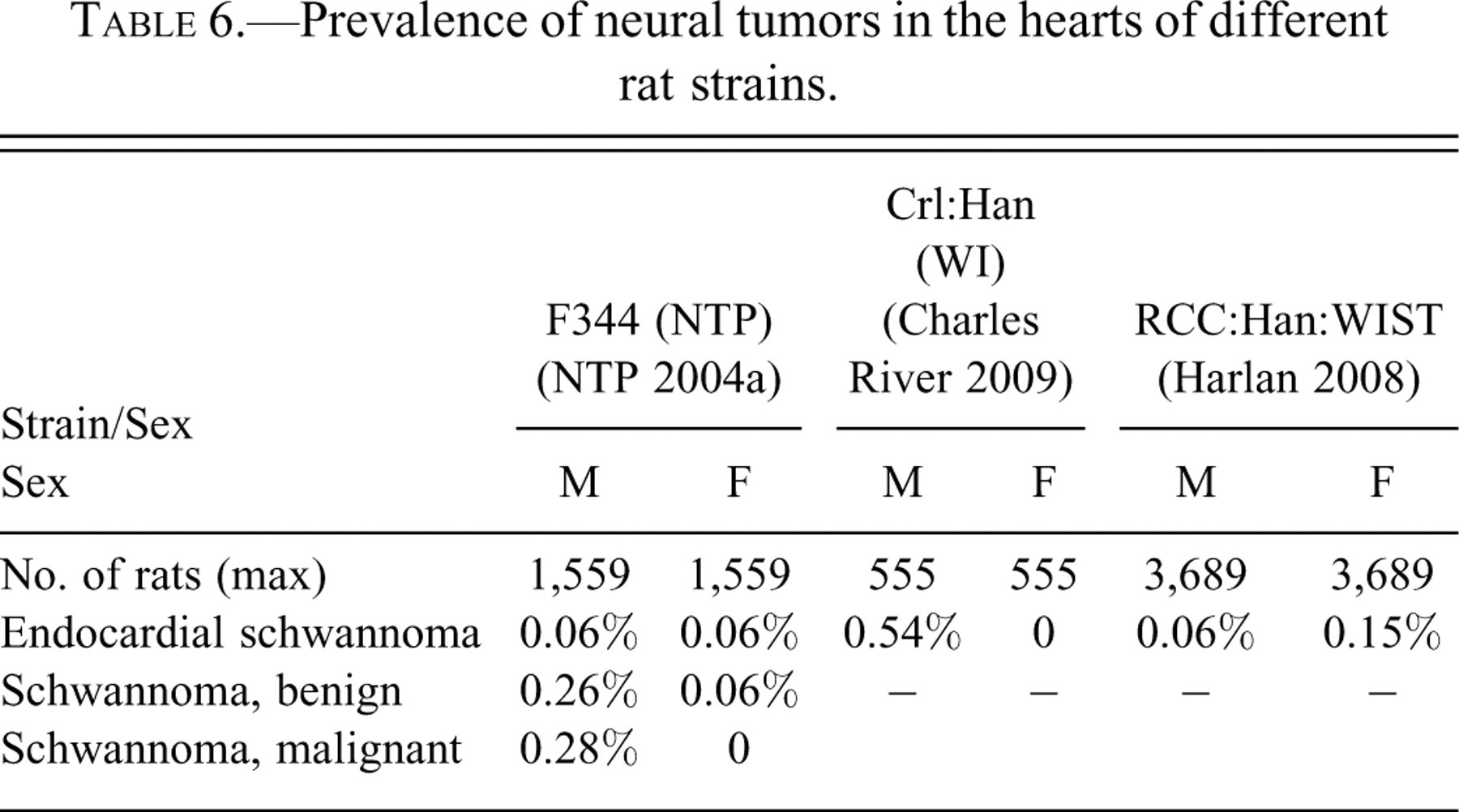
Endocardial schwannomas are limited to the endocardium and consist of round to ovoid cells interspersed with spindle-shaped cells enmeshed in a fine reticulin network (Figure 8b ). Verocay bodies (collections of fusiform cells arranged in whorls, herringbones, or palisades) are occasionally formed. Larger tumors may involve extensive areas of the myocardium and extend along the aorta into the pericardium. Malignancy as indicated by metastasis (to the lung) has been observed in one RccHan:WIST rat in our experience and has also been reported in two WIST rats (Teredesai and Wöhrmann 2005). The schwannoma character of these lesions is confirmed by their positivity for S100 protein (Landes et al. 1988).
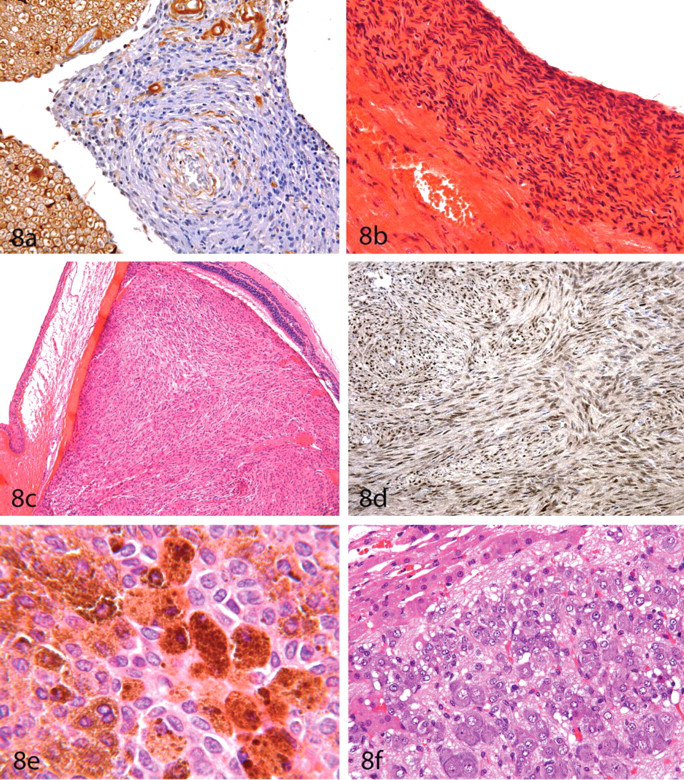
(A) Neurofibroma (same tumor as in Figure 7F). The tumor cells are S100 negative; formaldehyde-fixed, paraffin-embedded section subjected to indirect immunoperoxidase immunohistochemistry, ×400. (B) Endocardial schwannoma in the heart of an RccHan:WIST rat. The neoplasm conssts of round to oval cells interspersed with layers of spindle cells; hematoxylin and eosin (HE), ×400. (C) Amelanotic melanoma in the eye of an RccHan:WIST rat; HE, ×40. (D) Amelanotic melanocytes react with PNL2, a fixative-resistant melanocyte antigen; formaldehyde-fixed, paraffin-embedded section subjected to indirect immunoperoxidase immunohistochemistry, ×200. (E) Melanoma in a Syrian Golden (Lak:LVG(SYR)BR Outbred VAF/Plus) hamster; HE, ×1,000. (F) Ganglioneuroma in the adrenal medulla of an RccHan:WIST rat; HE, ×400.
Neurofibroma
The structure of a neurofibroma is very similar to that of the benign schwannoma. The difference relates to the increased amount of connective tissue in the neurofibroma, resulting in a positive IHC reaction against FN. In our material, this entity was recorded as a single case within the cauda equina of the spinal cord of a control RccHan:WIST rat (Figure 7f). Neoplastic cells in the neurofibroma were arranged in whorls, with thin layers of connective tissue present between layers of tumor cells. The neoplastic elements were oval to spindle shaped and had oval-shaped, invaginated nuclei of variable size. Mitotic figures were not present.
Schwannoma should be excluded as a differential diagnosis. The distribution of IHC markers varies between neurofibromas and schwannomas. In the neurofibroma, a positive reaction for FN was evident within some myelin sheaths, the cytoplasm of some tumor cells, and isolated fibers within the tumor parenchyma; these structures all have a staining intensity similar to the normal surrounding tissue. In contrast, neoplastic cells in neurofibromas are almost all negative for S100 in comparison to the positive reaction in the surrounding normal nerve (Figure 8a).
Perineuroma
Proliferative lesions of the peripheral nerves with concentrically arranged cells resembling Schwann cell–derived “onion bulbs” have been described as perineuromas. They are considered to originate from perineural cells. In contrast to Schwann cell–derived lesions, they are negative for S100 but are positive for claudin-1, epithelial membrane antigen (EMA), and glucose transporter-1 (GLUT-1). So far, perineuromas have not been described in the toxicologic pathology literature, but these tumors must be considered in the differential diagnosis of schwannomas.
Paraganglioma
These lesions are neuroendocrine tumors that originate from chromaffin-negative glomus cells that are derived from the neural crest and function as part of the sympathetic portion of the autonomic nervous system. These cells act as special chemoreceptors and are located along blood vessels, particularly in the carotid bodies (at the bifurcation of the common carotid artery in the neck), aortic bodies (near the aortic arch), and within the retroperitoneum caudal to the kidneys in the organ of Zuckerkandl. Paragangliomas are sometimes still referred to using older terms (“chemodectoma” or “glomus tumor”).
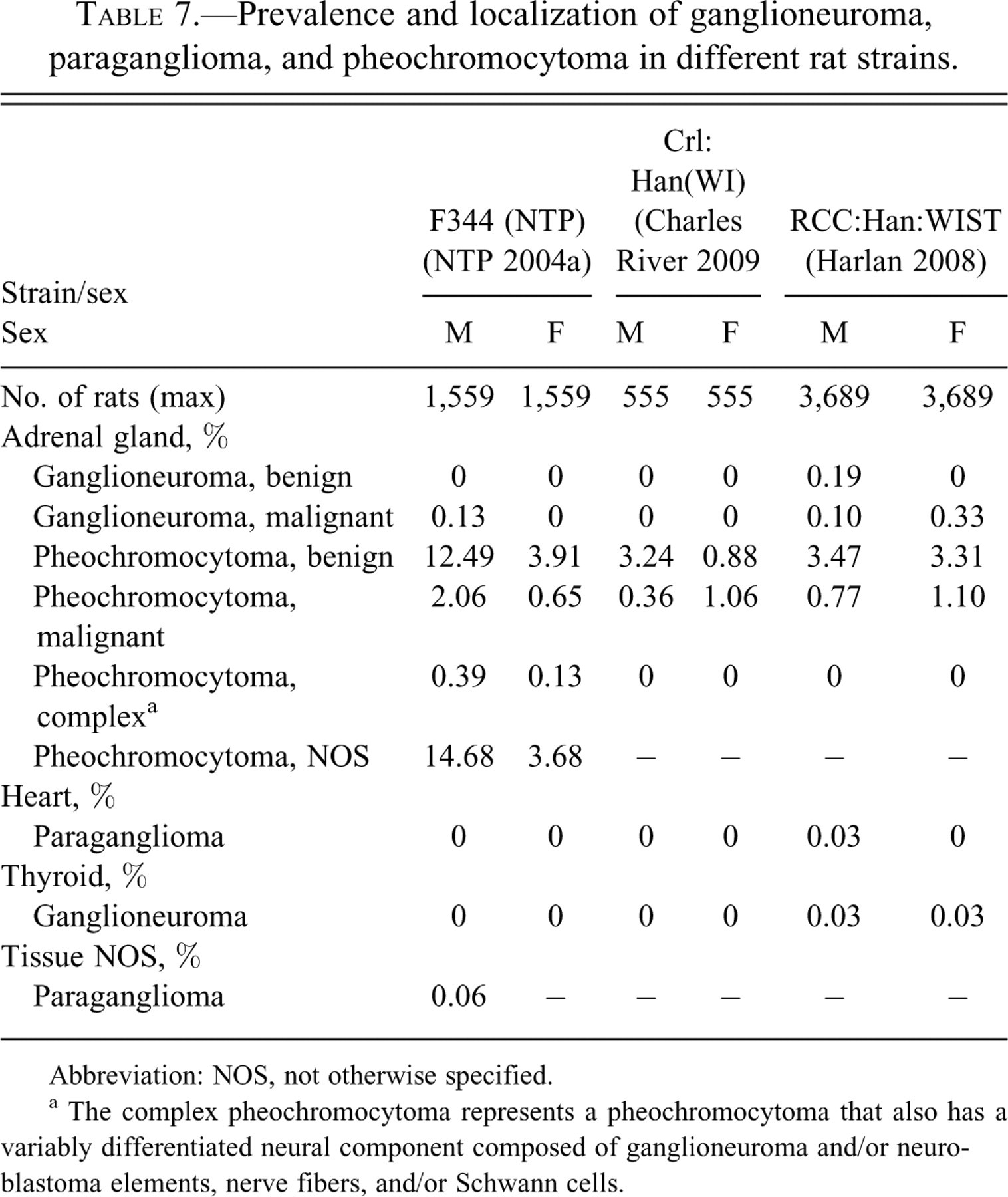
Although very rare (Charles River 2009; Harlan 2008; NTP 2004a), these tumors have been reported to occur in F344/N rats in the retroperitoneum adjacent to the vertebrae and aorta near the kidneys, as well as at the base of the heart (Table 7 ). Most paragangliomas were grossly visible (Hall et al. 1987). In the senior author’s series of RccHan:WIST rats, one very large, malignant, retroperitoneal paraganglioma (in the organ of Zuckerkandl) was observed in a treated rat and another small mass was located in the. Aortic body lesions (hyperplasia and neoplasia) have been reported in fifty-six rats, with lesions occurring more often in WAG/Rij rats than in BN/Bi rats or their hybrids (van Zwieten et al. 1979).
Prevalence and localization of ganglioneuroma, paraganglioma, and pheochromocytoma in different rat strains.
Abbreviation: NOS, not otherwise specified.
a The complex pheochromocytoma represents a pheochromocytoma that also has a variably differentiated neural component composed of ganglioneuroma and/or neuroblastoma elements, nerve fibers, and/or Schwann cells.
The main differential diagnosis for abdominal paraganglioma is pheochromocytoma.
Adrenal Medullary Tumor (Pheochromocytoma)
Pheochromocytoma is histologically similar to paraganglioma, but the tumor cells are chromaffin positive. This adrenal medullary neoplasm is among the most common tumors in rodents and also appears in domestic animal species and humans. In comparing different rat strains (Table 7), F344 rats are considered to be more affected by sponatenous adrenal medulla tumors than are CD(SD) or WistarHan rats (Charles River 2009; Harlan 2008; NTP 2004a).
Pheochromocytomas consist of polygonal, occasionally spindle-shaped cells arranged in sheets and/or multilayered cords. The neoplasms usually compress the surrounding tissue. The differentiation of benign and malignant tumors is very difficult and should include the presence of metastases and invasion rather than reliance on cytological features. Evaluation of cell proliferation using Ki67 has been regarded as helpful in supporting the differential diagnosis between benign and malignant pheochromocytomas (Pace et al. 2002).
The complex pheochromocytoma (with a <1% prevalence in RccHan:WIST rats) is rare in rats, and there are not many reports in the literature (Martinez and Mog 2001). In this tumor type, some cells undergo transformation into neural cells. Therefore, these tumors are composed of pheochromocytes and neurons, as well as occasional neuroblasts.
Carcinoids: Tumors of Neuroendocrine Cells
Carcinoids, also called apudomas (APUD denotes amine precursor uptake and decarboxylase cell), are neuroendocrine tumors. In humans, over two-thirds of carcinoids are found in the gastrointestinal tract (Modlin et al. 2003), where they arise from enterochromaffin cells. Other locations include the pancreas, lungs, and prostate. Neoplastic cells have scant cytoplasm and are grouped into small lobules separated by fine connective tissue septa, which are the characteristic feature of neuroendocrine organs. Tumor cells contain neuroendocrine vesicles by electron microscopy and react positively with neuroendocrine markers such as adrenocorticotropic hormone, glucagon, growth hormone releasing factor, insulin, and so on. Under experimental conditions, carcinoids may be induced by proton antagonists (Betton et al. 1988) but have also been reported in the stomach of an aged mouse as a spontaneous lesion (Okimoto et al. 2003).
Ganglioneuroma and Ganglioneuroblastoma
Ganglioneuromas arise from the sympathetic portion of the autonomic nervous system and are composed entirely of ganglion, satellite, and Schwann cells in a neurofibrillary matrix of nerve fibers. These tumors are rare (Brix et al. 2005; Charles River 2009; Harlan 2008; NTP 2004a) but may appear in various organs (Table 7, Figure 8f). There are several case reports of this lesion in rodents. Ganglioneuromas of the thyroid gland, unrelated to treatment or sex, occurred at an incidence of 7.2% in SD rats during a two-year carcinogenicity bioassay (Crissman et al. 1991). These tumors had coincidental C-cell proliferations within the same or in the contralateral lobe and were composed of large ganglion cells and/or neuronal precursors in a matrix of neurites and Schwann cells. The tumors were reported to react positively for calcitonin, S100 protein, and NFP (Crissman et al. 1991). Three cases of ganglioneuroma have been reported in the adrenal medulla of WIST-derived rats (Todd et al. 1970) and one case has been reported in an aged F344 rat (Goelz et al. 1998). Furthermore, there were single cases described in the pituitary gland of an SD-derived rat (Pace and Perentes 2001) and an aged F344 rat (Okazaki et al. 1997). Ganglioneuroma also may occur as a spontaneous neoplasm in hamsters (Rustia and Cardesa 1975) and has been shown to arise from the trigeminal ganglion in an aged B6C3F1 mouse (Yasui et al. 2009).
The malignant form of ganglioneuroma is the ganglioneuroblastoma, a neuroblastoma variant characterized by a small region of dark basophilic, oval, stemlike cells surrounded by ganglion cells. In humans, approximately 50% of neuroblastomas are in children (Lacayo and Davis 2010), and occur mainly in the adrenal glands, although they may also develop at other sites. In laboratory animals, neuroblastomas are recorded mainly in the adrenal glands, sometimes as a component of malignant pheochromocytomas (Maekawa et al. 1984).
Esthesioneuroblastoma (olfactory neuroblastoma) is believed to arise from the olfactory epithelium and thus is observed within or near the caudal nasal cavity. This tumor may be induced in rodents (Pino et al. 1999) but has not been described as a spontaneous entity.
Melanoma and Amelanotic Melanoma
Melanoma (Figure 8e) and amelanotic melanoma (Figure 8c) are derived from neural crest cells, which may appear as slightly pleomorphic but are mainly spindle-shaped (epitheloid) cells, often with vacuolated cytoplasm. The presence of cytoplasmic melanin granules is an important diagnostic feature in pigmented animals; melanin is absent in albino rodent strains. Melanomas assume several patterns. The usual appearance is dense, interlacing cell bundles. Another fairly common arrangement is perivascular pseudopalisading, which is a helpful diagnostic feature in amelanotic melanomas in albino laboratory animals. Another typical pattern is “phalanx” formation (i.e., cells forming parallel bands of nuclei).
Preferred locations for melanoma initiation vary with both the species and the strain. For example, a main predilection site for melanomas in rats is the pinna of the ear, but other major sites differ by strain: F344/N rat, pinna > scrotum > eyelid > perianal region; hooded rat, pinna > anogenital/tail > eyelid; and F344/duCrj rat, pinna (Yoshitomi et al. 1995). In the RccHan:WIST rat, single cases have been recorded in the pinna and in the eys (uvea). Similar cases in the uvea have been published in pigmented Long-Evans rats (Mannig et al. 2004) and in SD rats (Magnusson et al. 1978). No predilection site has been identified in the hamster (Yoshitomi et al. 1995). In humans, melanomas exhibit a sex predilection for some sites (man: ear; woman: face and shoulder; Yoshitomi et al. 1995) and also occur in the uvea (Li et al. 2003). Melanomas have also also been reported in domestic animals (Patnaik and Mooney 1988; Pfeiffer et al. 1977). These tumors are also inducible by a variety of chemicals (Albert et al. 1982; Kendrey and Roe 1969).
Melanomas have been shown to arise from dermal melanocytes in hooded rats, from perifollicular melanocytes in hooded mice, and from melanocytes of the skin and outer sheath of the hair follicle in albino guinea pigs (Nakashima et al. 1996; Yoshitomi and Boorman 1993; Yoshitomi et al. 1995). The origin of melanomas in abino rats is unknown.
In the differential diagnosis of amelanotic melanoma, schwannoma should be excluded. Amelanotic melanomas are located mainly in the pinna, eyelid, uvea (Figures 8c, 8d), scrotum, and perianal region. The spindle-shaped melanoma cells exhibit a perivascular orientation in contrast to the polygonal schwannoma cells, which are arranged in Antoni Type A and Antoni Type B patterns. Premelanosomes are present in amelanotic melanoma, whereas a basal lamina is seen in schwannomas. Therefore, a diagnosis of amelanotic melanoma should be made only after electron microscopic examination or IHC to demonstrate melanocyte markers such as HMB45 for premelanosomes (Gown et al. 1986; Kapur et al. 1992; Yaziji and Gown 2003) or PNL2 (a fixative-resistant melanocyte antigen; Rochaix et al. 2003) (Figure 8d).
Tumorlike Lesions and Artifacts in RccHan:WIST Rats
In historical data for several thousand control and treated animals, only two cases of squamous cysts (epidermoid cysts) were identified in the CNS. One 8-mm–diameter cyst was present between the cerebral hemispheres, and the other small cyst was found in the cauda equina. These keratin-containing cysts were lined by squamous epithelium and were considered to be a congenital consequence of abnormal neural tube development. Similar cysts have been described in the spinal cord, with an incidence of 2.5% in CDF rats, 1.3% in Lewis rats, and 1.3% in WIST rats (Levine 1966), and in the leptomeninges of the longitudinal cerebral fissure of young SD rats (Sugimoto et al. 1994). In mice, squamous cysts were more frequently reported (Nobel et al. 1987; Satoh and Furuhuma 2001; Stroop 1984), with incidences in the range of 0.3%–1.8% in several mouse strains (Jung et al. 2004).
Rarely, cysts are present in the brain that are not lined by squamous epithelium and, therefore, resemble porencephaly (i.e., formation of an unlined cyst, generally by parenchymal necrosis). Only one case could be established in our material.
Artifacts that may result in a diagnosis of meningioma may be induced by pressing the meninges or even the pineal gland into the neuropil while opening the skull. Furthermore, inexperienced investigators may misinterpret such normal autochthonous structures as the subfornical and subcommissural organs as neoplastic lesions.
Induced Tumors
Most investigations of induced NS tumors were published between 1965 and 1978. The inciting agents included chemicals (mainly delivered by transuterine treatment), viruses (mainly inoculated by intracerebral injection), and radionuclides. Susceptible laboratory animal species included the rat, mouse, hamster, and rabbit; the most sensitive species was the rat. Strain differences were not reported. A literature survey of experimentally induced NS tumors is not within the scope of this manuscript. Furthermore, the classification of induced NS neoplasms is difficult, because these tumors may differ in many ways from spontaneous tumors, often appearing to be more similar to neoplasms originating from non-neural tissues.