
Abstract
The continuing education course “Hemostasis” provided a comprehensive review of hemostasis and selected perturbations of the underlying processes as well as an assessment of hemostasis in animal models and preclinical testing environments. The session began with a review of the current state of understanding of hemostasis and how the waterfall or cascade of activation has transformed to the current cell-based, membrane-associated sequence of highly regulated events. The specific mechanisms of drug-induced thrombocytopenia were then presented, followed by a discussion of the relationships of coagulation and platelets in inflammation and cancer metastasis and platelet activity. Evaluation of hemostasis and platelet function in animals and especially in the environment of the contract research facility concluded the session.
Keywords
The continuing education course on “Hemostasis” presented during the 2010 Scientific Symposium sponsored jointly by the Society of Toxicologic Pathology (STP) and the International Federation of Societies of Toxicologic Pathologists (IFSTP) began with an initial discussion of the current understanding of hemostasis followed by an in-depth analysis of the complex disorder of drug-induced thrombocytopenia as well as cellular and molecular interactions in inflammation and hemostasis. Following an understanding of the molecular basis of normal hemostasis and the interactions during inflammation and drug-associated thrombocytopenia, the broad topic of evolution of species-dependent variations in hemostasis and the evaluation of hemostasis in a GLP-compliant facility were discussed. Several examples of clinical study data were then presented to highlight problems in hemostasis evaluation in laboratory animals. Presenters (given in the order in which they spoke) included Andrew Gale, PhD; Jacqueline Brassard, DVM, PhD, Diplomate ACVP; Brunhilde Felding-Habermann, PhD; Marjory Brooks, DVM, Diplomate ACVIM; Jeffery McCartney, DVM, MVSc, Diplomate ABT; ACVP; and Dale Baker, DVM, PhD, Diplomate ACVP, ABT.
Current Understanding of Hemostasis
Hemostasis can be separated into three topical divisions for an easier understanding of the overall process. These three categories or divisions are primary hemostasis, secondary hemostasis, and fibrinolysis.
Primary hemostasis is the initial platelet response to vascular injury when the endothelium is perturbed and/or the subendothelial matrix is exposed in injured vessels. It consists of platelet adhesion and activation, and involves the following platelet receptors and their corresponding ligands (Nieswandt, Varga-Szabo, and Elvers 2009; Ruggeri and Mendolicchio 2007; Rivera et al. 2009): GPIb/V/IX—von Willebrand’s factor (vWF); GPVI—collagen; αIIbβ3—fibrinogen, vWF, fibronectin, vitronectin; α2β1—collagen; αvβ3—vitronectin, thrombospondin, vWF, fibronectin, fibrinogen; and PAR1, PAR4—thrombin.
In addition to collagen and thrombin, which activate platelets, thromboxane A2 (TXA2), adenosine diphosphate (ADP), serotonin, platelet activating factor (PAF, also termed C-X-C motif ligand 4 [CXCL4]), and epinephrine stimulate platelets and further promote platelet thrombus formation. Activation of platelets causes an increase in intracellular calcium and activation of other signaling pathways. This activation results in activation of platelet integrins, especially αIIbβ3, as well as secretion of platelet granules that contain multiple agonists such as adhesive proteins, growth modulators, and coagulation factors. After activation, platelets undergo a characteristic shape change (termed “spreading”) from discoid cell to sphere with fine filopodial extensions, followed by filling of the space between the filopodia to increase the surface area, and finally to the fully spread platelet that has the appearance of a fried egg as it covers a vascular defect. Platelet drug inhibitors can block cyclo-oxygenase 1 (COX1) activation and TXA2 production or interfere with ADP receptors to dampen this platelet response (Andrews and Berndt 2004; Barrett et al. 2008; Kamath, Blann, and Lip 2001).
At one time, secondary hemostasis was thought to consist solely of thrombin generation by soluble coagulation factors and cofactors leading to the formation of fibrin through a stepwise cascade or waterfall of proteolytic reactions. This concept was based on two complementary blood-clotting models that were proposed in the early 1960s (Davie and Ratnoff 1964; Macfarlane 1964). The coagulation cascade was outlined in a Y-shaped scheme with intrinsic and extrinsic pathways initiated by factor XII and factor VIIa/tissue factor, respectively. The intrinsic pathway consisted of blood components and was measured by activated partial thromboplastin time (aPTT), and the extrinsic pathway consisted of tissue factor (factor VII) and was measured by prothrombin time (PT). These pathways converged on a “common pathway” at the factor Xa/factorVa (prothrombinase) complex, and it was generally noted that phospholipid and calcium were required for all activities. Over time, the coagulation cascade/waterfall model proved insufficient in predicting a patient’s bleeding tendency, in correlating clinical outcomes with specific laboratory screening tests such as PT and aPTT, and in explaining the role of cells in controlling coagulation based on blood flow modeling studies implicating the importance of cells in the coagulation process.
A new cell-based model of coagulation represents the current understanding of secondary hemostasis and consists of three phases: initiation, amplification, and propagation. Initiation is localized to cells expressing tissue factor (TF) leading to the formation of prothrombinase on the cell surface and the formation of a small amount of thrombin. Amplification is thrombin activation of platelets and cofactors V and VIII on the activated platelet surface. Propagation is the recruitment by activated platelets of circulating platelets to the site of vascular injury to set up a burst of thrombin generation with sufficient magnitude to form fibrin in order to stabilize the platelet “plug” (Hoffman and Monroe 2001; Hoffman 2003).
Thrombin is central to further activation of platelets, coagulation (formation of factors VIIIa and Va), as well as anticoagulation (protein C), and protease-activated receptors (PARs) (Binnette et al. 2007; Mann 2003). Two important coagulation factor complexes form at the platelet surface: the tenase (a complex of factors VIIIa, IXa, and X) and the prothrombinase complexes (factors Va, Xa, II), both of which promote thrombin formation (Mann 1987; Mann, Butenas, and Brummel 2003). Activated protein C/protein S and the serine protease inhibitors (“serpins”) are important anticoagulants to prevent excessive clot formation (Shen and Dahlbäck 1994). The serpins include antithrombin, heparin cofactor II, protein Z-dependent protease inhibitor, protein C inhibitor, plasminogen activator inhibitor-1 and 2, alpha-2-antiplasmin, and C1-inhibitor (Rau et al. 2007).
The final division of hemostasis, fibrinolysis, serves to counter the procoagulant properties of primary and secondary hemostasis. Accordingly, fibrinolysis is an important mechanism to maintain blood flow and vessel patency and involves the cleavage of circulating plasminogen to plasmin by tissue-type plasminogen activator (tPA) expressed on endothelial cells. Plasmin degrades fibrin and lyses the forming platelet and fibrin plug (Nesheim 2003; Oliver, Webb, and Newby 2005).
Overall, coagulation is a balance between the prothrombotic components (activated platelets, procoagulation factors) and the anti thrombotic components (fibrinolysis, serpins, activated protein C [APC]/protein S, anticoagulant system). Defects in any of these components (either acquired or inherited) will result in either bleeding or thrombosis (thrombophilia). In humans, thrombophilia is associated with activated Protein C (APC) resistance by a mutated Factor V (Factor V Leiden) that is unable to be inactivated, prothrombin nt20210A polymorphism (excessive prothrombin), protein C heterozygosity, protein S heterozygosity, antithrombin deficiency, and anticardiolipin antibodies. Drugs used to inhibit coagulation in these conditions include heparin, low molecular weight heparin, warfarin (coumadin), factor Xa inhibitors, and thrombin inhibitors. Statins are beneficial in preventing thrombosis in addition to their plasma lipid-altering effects. Statins upregulate nitric oxide synthesis in endothelium, which inhibits platelet aggregation and leukocyte adhesion, inhibitsTF expression (Mason 2003).
Coagulation factor deficiencies (notably factors VIII, IX) are associated with bleeding and have been treated by recombinant factors, whole plasma, and factor VIIa (Novoseven?®). Von Willebrand’s disease is a common hereditary defect of vWF, a large multimeric plasma protein that is essential for platelet adhesion to subendothelial components under high shear flow. The defect can be either qualitative or quantitative and results in a mild bleeding tendency (Zimmerman and Ruggeri 1982).
Immune-Mediated, Drug-Induced Thrombocytopenia (DITP)
DITP is an unpredictable and sometimes serious side effect that can occur in up to 5% of human patients who receive certain drugs, such as unfractionated heparin. To date, there are five mechanisms for drug induction of antibodies that promote platelet destruction.
1. Hapten-Dependent Antibodies (HDAb)
HDAbs have been associated with drug-induced hemolytic anemia (DIHA) in patients treated with massive doses of intravenous penicillin and penicillin derivatives over a period of time and were presumed to also be involved with DITP. In fact, at one time it was thought that most drugs causing immune-mediated thrombocytopenia acted as haptens. With HDAb, a drug acts as a hapten by covalently linking to a cell membrane protein and inducing a drug-specific immune response. The induced antibodies are usually specific for the linked drug. The HDAb mechanism is often suspected in DITP in laboratory and companion animals, but supportive data have been lacking. Recent investigative and mechanistic studies indicate that the HDAb mechanism may be rare in DITP (Visentin and Liu 2007; Bougie et al. 2009). As with humans, other mechanisms (e.g., formation of drug-dependent antibodies [DDAb]) may be the more likely pathway in many incidences of laboratory and companion animal DITP.
2. Drug-Dependent Antibodies (DDAb)
With DDAb, a drug binds to a platelet surface protein, creating either a compound epitope or a conformational change in the protein that can react with a preexisting (naturally occurring) antibody or can stimulate the synthesis of a specific antibody. Common targets for DDAb are platelet surface proteins (listed previously above when describing primary hemostasis). Most platelet-specific DDAbs recognize GPIIbIIIa, the platelet fibrinogen receptor, and/or GPIb/IX /GPV complex, the platelet vWF receptor.
There are two proposed models for DDAb binding to a platelet protein/antigen: The “Improved Fit” model proposes that the DDAbs are derived from a naturally occurring pool of autoantibodies that weakly bind with epitopes on platelet membrane glycoproteins. Drugs capable of causing DITP possess structural elements that improve the fit between these autoantibodies and their targets. The binding-enhancing structural elements might consist of charged (+/–) or hydrophobic domains that interact with both the glycoprotein epitope and the complementarity-determining region (CDR) of the antibody, thus increasing the binding affinity (KA) between the two proteins. The Improved Fit model has been proposed for those drugs that only weakly interact with platelet proteins, such as quinidine, quinine, nonsteroidal anti-inflammatory drugs (NSAIDs), various antibiotics, sedatives, and anticonvulsants (Bougie, Wilker, and Aster 2006). DDAbs have been identified in a dog with cephalosporin-induced ITP (Bloom et al. 1988) and in companion dogs with sulfonamide-induced ITP (LaVergne and Trepanier 2007). LaVergne and Trepanier (2007) were not able to identify the platelet target of the sulfonamide-induced antiplatelet antibodies in dogs but were able to determine that GPIIbIIIa and GPIb were not targeted as occurs in humans. The “Neoepitope” model applies to “fibans,” drugs designed to tightly bind to the arginine-glycine-aspartic acid (RGD) recognition site of platelet receptor complex GPIIbIIIa with the intent of inhibiting platelet binding to fibrin(ogen). The premise is that fiban binding induces a conformational change in the receptor, thereby creating a new target epitope elsewhere in the complex that is recognized by specific antibodies. A small subset of patients treated with fibans develops acute, severe thrombocytopenia in every clinical trial (Aster 2005b). Serologic studies in some of these patients demonstrated the presence of preexisting, naturally occurring antibodies that recognize GPIIbIIIa in complex with a fiban and may cause acute DITP (Bougie et al. 2002; Brassard et al. 2002; Peterson et al. 2008). DDAbs were also detected in one chimpanzee and one rhesus macaque with fiban-induced thrombocytopenia (Bednar et al. 1999). Preexisting, naturally occurring antibodies were also found in the pretreatment samples from these two primates. In another study with eptifibatide, transient thrombocytopenia was observed in a juvenile baboon; preexisting antibodies were suspected but not investigated. Numerous fibans have been tested in other animals through the course of drug development, including mice, rats, and dogs; however, neither fiban-ITP nor preexisting antibodies have been reported in these species, for reasons that are not entirely understood.
3. Drug-Specific Antibodies (DSAb)
The best and perhaps only current example of the DSAb mechanism is seen with abciximab, a chimeric (human-mouse) Fab fragment that recognizes an epitope adjacent to the RGD binding site in the GPIIIa (or β3) subunit of the GPIIbIIIa receptor. Abciximab inhibits fibrinogen binding through steric hindrance. Acute, severe thrombocytopenia can be seen within hours of starting treatment. Thrombocytopenia occurs in 2% of patients given abciximab for the first time, and in 10-12% after the second dose. Both naturally occurring and induced antibodies specific for the murine sequences in abciximab or for the papain cleavage product have been identified in patients, suggesting that both DDAb and DSAb mechanisms may be involved with abciximab thrombocytopenia. Abciximab binds to rhesus macaque and baboon receptors with high affinity, while binding with less affinity is observed in the dog and rat (Tam et al. 1998). While induction of abciximab-specific antibody would seem possible in nonhuman primates, it has not been reported.
4. Drug-Induced Autoantibodies (DIAAb)
Very rarely, drugs induce platelet-specific autoantibodies that lead to drug-induced autoimmune thrombocytopenia. The DIAAb mechanism has been attributed to thrombocytopenias induced by gold salts, procaine amide, sulfonamide antibiotics, interferons alpha and beta, and L-dopa (Aster et al. 2009), but supportive data is often lacking. The platelet protein(s) targeted by these autoantibodies are not well-characterized with the exception of DIAAb induced by gold salts; autoantibodies that bind platelet GPV have been demonstrated in patients with gold-associated autoimmune thrombocytopenia (Garner et al. 2002). Gold-induced immune thrombocytopenia has also been reported in companion dogs (Bloom et al. 1985). Platelet associated IgGs were identified via radioimmunoassay and electron microscopy, but were not further characterized.
5. Immune Complexes
Heparin-induced thrombocytopenia (HIT) is the single well-documented example where drug-induced immune complex formation results in thrombocytopenia (Visentin 1999; Aster 2005a). Heparin binds to platelet factor 4 (PF4), a globular homotetrameric protein and platelet α-granule constituent that neutralizes heparin. Heparin/PF4 complexes are immunogenic and can elicit an antibody response. The resulting immune complexes interact with platelet Fc receptors. Ultra-large heparin/PF4 complexes, which form with the longer heparin polymers, are capable of interacting with several Fc receptors on the platelet surface, which allows for clustering of the receptors and leads to platelet activation and fragmentation (Visentin 1999; Aster 2005a; Rauova et al. 2005). Spontaneous HIT has not been described in species other than human, and efforts to establish animal models have met with little success. The absence of HIT in mice and rats can be readily explained by the absence of platelet Fc receptors in these species (Reilly et al. 2001), but the absence of HIT in those animals that express platelet Fc receptors is not understood. Immunogenicity of heparin/PF4 complexes may play a role. In humans, HIT is caused by antibodies that recognize complexes of platelet factor 4 bound to heparin (Amiral et al. 1992; Warkentin 2008), but heparin treatment has not been associated with the induction of antibodies against PF4 in other species, including nonhuman primates, even though nonhuman primate (rhesus macaque) PF4 appears to be antigenically similar to human PF4 (Untch et al. 2002). HIT may be restricted to humans because of the unique antigenic qualities of hPF4 when complexed with heparin.
Diagnosis of DITP: Confirmation of Drug Etiology
Confirmation of DITP requires meeting certain observation criteria and demonstration of DDAbs (Table 1
). Immunofluorescence by flow cytometry is the most sensitive method available. It is performed through incubation of normal (donor) platelets with patient serum in the presence and absence of the implicated drug, followed by washing in the presence of drug and detection of platelet-bound immunoglobulin. DDAbs may be present yet not detectable because assay methods are not sensitive enough, drugs that are highly insoluble in aqueous solution may not be easy to incorporate into in vitro assays, and drug metabolites produced in vivo but not in vitro may be the sensitizing agents.
Criteria for evaluating individual patient data during diagnosis of drug-induced thrombocytopenia (DITP).

From George and Aster (2009).
Intersection of Hemostasis and Inflammation
Hemostasis and inflammation are closely interrelated processes (Smyth et al. 2009). Endothelial cells, platelets, and leukocytes can all alter inflammation, and an imbalance between procoagulant and anticoagulant factors can promote inflammation. Upon activation, endothelial cells produce and release interleukin (IL)-8, IL-6, tumor necrosis factor (TNF)-α, and upregulate CD40 expression and bind leukocytes. Platelets contain α-granules and dense granules which are released upon activation. These granules contain superoxide, soluble CD40 ligand (sCD40L), chemokine (C-C motif) ligand (CCL)-5 (formerly designated Regulated upon Activation, Normal T-cell Expressed, and Secreted [RANTES]), TXA2, adenosine diphosphate/adenosine triphosphate (ADP/ATP), serotonin, and histamine among a multitude of other bioactive factors. Acting synergistically with the stimulatory mediators contributed by platelets, leukocytes can produce or release superoxide, arachidonic acid, platelet activating factor (PAF), leukotriene, TNFα, and IL-1.
In the process of platelet-mediated recruitment of leukocytes, platelets bind to activated endothelium or to exposed areas of subendothelial matrix. During this process, platelets are themselves activated and release granules that further recruit circulating platelets and leukocytes. Platelets promote leukocyte adhesion and trans-endothelial migration, and platelet-derived factors can stimulate leukocyte survival and proliferation. Leukocyte P-selectin glycoprotein ligand (PSGL)-1 tethers to platelet/endothelial P-selectin. Activation of the leukocyte integrin αMβ2, platelet αIIbβ3, and endothelial αvβ3 and αvβ5 establishes high-affinity interactions. Leukocyte adhesion receptors αMβ2, αvβ3, CD36 (GP IV), αLβ2, and CD40 coordinate firm adhesion to the endothelium and cross-linking with platelets. Fibrinogen, fibrin, vWF, and thrombospondin are soluble mediators of adhesion that serve as ligands for receptors such as GP1bα, αIIbβ3, CD36 (GPIV), intercellular adhesion molecule (ICAM)-2, junctional adhesion molecule (JAM)-3, and CD40 to stabilize leukocytes-platelet cohesion.
Platelets are also important in the development of atherosclerosis/atheromatous plaques. Atherosclerosis is a chronic inflammatory condition with a central role for platelets in the initiation of the disease (Muhlestein 2010). The process is triggered by platelet adhesion to the endothelium of arterial vessels through GPIbα interaction with vWF. This interaction establishes an adhesive mechanism that not only withstands but is actually promoted by high shear forces generated by arterial blood flow. The importance of platelets in this process is exemplified by decreased deposition of fatty streaks and reduction in plaque size in P-selectin-deficient, apoE–/–, or LDL receptor –/– mice. This sparing effect is reversed by injection of activated wild type platelets. Further support of a role of platelets in artherosclerosis stems from the finding that disruption of CCL5 (RANTES) inhibits plaque formation in hyperlipidemic mice (Hayes and Pronczuk 1996).
Platelet granules are key in the platelet-mediated inflammatory process. There are three granule types, α-granules, dense granules, and lysosomes (Blair and Flaumenhaft 2009). The contents of α-granules are released by fusion of the granule with the platelet membrane, causing a 2 to 4-fold increase in platelet surface area and enabling changes in platelet shape and function. These granules contain hundreds of bioactive proteins and other factors. A few of the important, well-characterized mediators and their functions are given in Table 2 .
Platelet granule constituents and their functions.

Platelets are important not only for leukocyte recruitment to sites of inflammation, but they can also modulate immune cell function and responses. Platelets have a central role in regulating expression of CD154 (thrombin receptor activator peptide [TRAP]-1, a thrombin analog) and CD40L (a T- and a B-cell activating molecule) on the surface of T helper cells. Platelets are also critical for proper activation of dendritic cells and regulation of B-cell function by binding to CD40 on B-cells. Platelets are further required for T-cell-dependent B-cell immunity (B-cell mobilization, antibody production), and immunoglobulin class switching and mediation of B-cell proliferation in the absence of costimuli. Support for the role of platelets in these processes stems from the finding that adoptive transfer of normal platelets into CD154-deficient hosts transiently restores antigen-specific IgG production after adenovirus immunization. Furthermore, platelets from patients with immune thrombocytopenic purpurea prompt autologous self-reactive B-cells directly to release antibodies against platelet GPIIbIIIa in a CD154-specific manner. It is important to note in this context that several autoimmune diseases with T- and B-cell components, such as diabetes, immune thrombocytopenia, systemic necrotizing vasculitis (Kawasaki disease), systemic lupus erythematosus, and inflammatory bowel disease are linked to increased platelet activation and surface expression of CD154 (Sowa et al. 2009).
Notably, platelet activation is important for the stimulation of cytotoxic T-cell (CTL) migration into the liver of patients infected with hepatitis B virus (HBV). Evidence for this concept comes from elegant studies where experimental thrombocytopenia diminished the infiltration of adoptively transferred HBV-specific CTLs into the liver and reduced lesion severity. Through serotonin release, platelets hinder CD8+ T-cell infiltration of the liver, allowing viral replication. Platelet contributions to inflammatory processes can be modulated by complement. Complement activation at the surface of platelets enhances inflammatory cell recruitment and potentiates platelet response to ADP and thrombin (Peerschke, Yin, and Ghebrehiwet 2009).
Interestingly, platelets and their interaction with leukocytes are also intimately involved with cancer cell metastasis. Recent studies indicate that arrest of circulating tumor cells can co-opt the intricate adhesive properties of platelets and utilize interaction with platelets for arrest at the vessel wall and extravasation into target tissues of metastasis (Felding-Habermann et al. 1996, 2001). In this way, tumor cells can mimic and incorporate the adhesive and stimulatory properties of leukocytes to promote cancer progression. This highly relevant pathophysiological aspect was briefly reviewed.
Hemostasis Testing in Animal Models
The testing methods for testing hemostasis are typically classified as primary hemostasis tests, or an evaluation of platelets and vWF (Favaloro, Kippi, and Franchini 2010), or secondary hemostasis tests, or coagulation factor evaluation. Sample collection is important to both of these analyses. Laboratory methods assess platelet function and coagulation factor activities separately. Platelet activation occurs through agonists stimulating surface receptors resulting in platelet shape change, adhesion, secretion and spreading, and a change of the platelet outer membrane composition to promote coagulation factor activation (Furie and Furie 2008). This functional change of the membrane is phosphatidylserine flipping from the interior surface of the lipid bilayer, where it is exclusively located in a resting platelet, to the exterior surface of the lipid bilayer. Adhesion by surface proteins varies between species (Lentz 2003; Andrews and Berndt 2004; Zwaal, Comfurus, and Bevers 2005; Jackson 2007; Pelagalli et al. 2003).
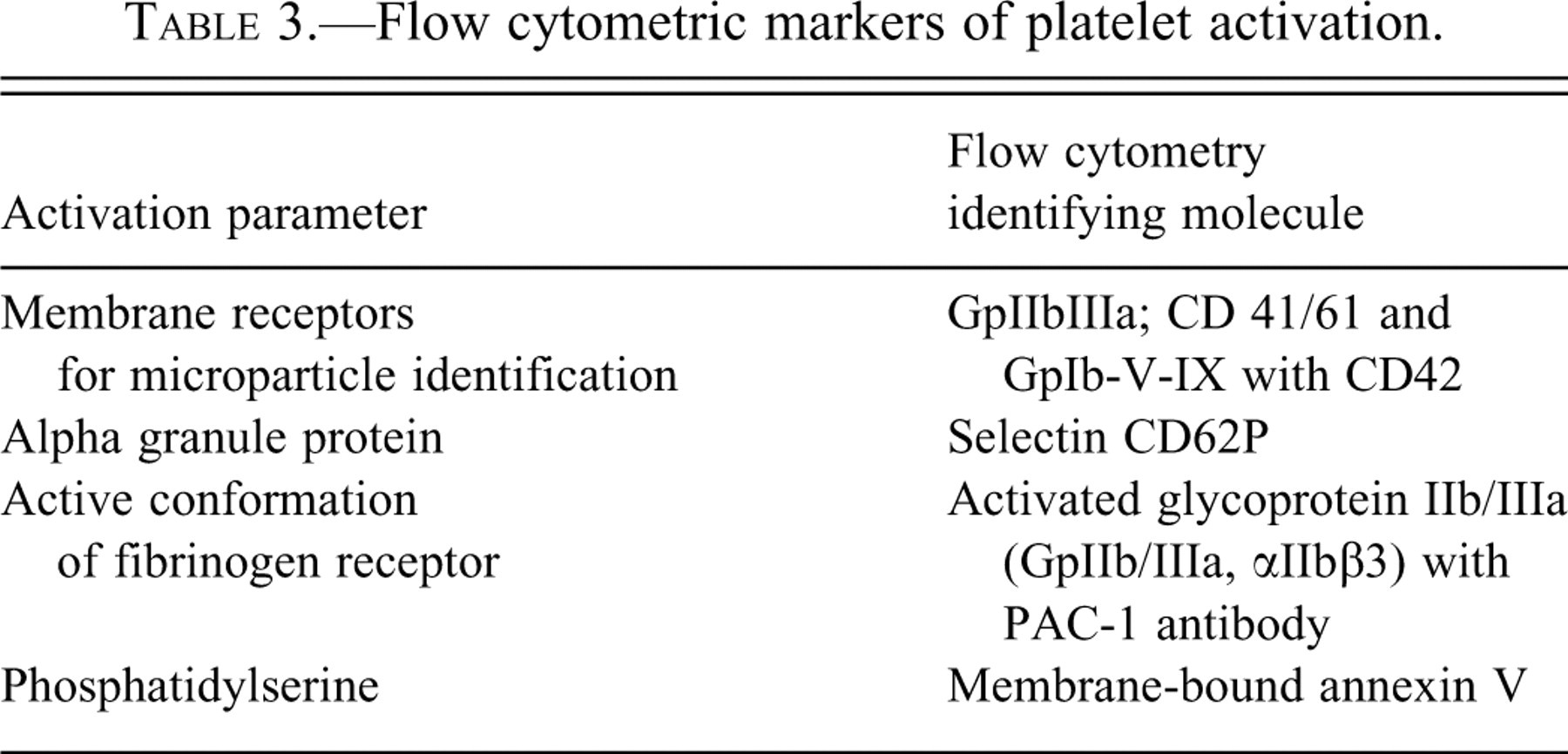
The traditional method to evaluate platelet function is light transmittance aggregometry (LTA, also known as the turbidometric method) and citrated platelet-rich plasma (PRP) (Born 1962; O’Brien 1962; Kurata et al. 1995). A change in light transmittance is detected as platelets respond to agonists by undergoing shape changes and large-order aggregate formation. Agonists for specific surface receptors on platelets include ADP, collagen (convulxin and C-reactive protein [CRP] also stimulate platelets), thrombin (TRAP), arachidonic acid, thromboxane (also U44619—a thromboxane analog), PAF, and epinephrine. The order of species reactivity by EC50 concentration reactivity to ADP is: mouse > human > rat, guinea pig > dog (Nylander, Mattsson, and Lindahl 2006). Because of the long history of use, LTA remains the optimal method for platelet testing (Cattaneo 2009); however, it is time-consuming, technically challenging, and requires large volumes of platelet-rich plasma. Whole blood platelet aggregation (WBA) was introduced in 1980 and was an improvement over LTA in that platelet function could be assessed in whole blood rather than PRP. Platelet function is also evaluated by flow cytometry (Hagberg and Lyberg 2000; Brooks et al. 2009). Smaller volume blood samples are required, and single cell activation status can be assessed. The activation markers and their respective antibodies that have been utilized are given in Table 3 .
Flow cytometric markers of platelet activation.
Flow cytometric analyses of platelet activation are limited by the availability of antibodies to all species and variable agonist reactivity (Schuberth et al. 2007). For example, the EC50 for ADP is 680 nM for humans versus 1,070 nM for dogs. Not all species react to synthetic analogs. For secondary hemostasis screening, the activated partial thromboplastin time (aPTT) evaluates the intrinsic pathway and is an endpoint assay. Thrombin generation assays (TGA) are a new method, better able to detect hypo- and hyper-coagulability based on kinetic rather than endpoint determinations (Baglin 2005). Both aPTT and TGA are influenced by the anticoagulant used and the blood collection technique; for routine in-life assessment, the preferred procedure is to use 3.2% citrate anticoagulated blood and withdraw blood from an accessible peripheral vein. TGA is influenced by centrifugation techniques (due to residual cell-derived microparticles in plasma) and the trigger reagent composition due in part to species differences in the contact pathway (Ponczek, Gailani, and Doolittle 2008). Variability for aPTT occurs in the biologic lipids used (rabbit brain cephalin, soy phosphatide) and the negatively charged surface of the activator (kaolin, celite, silica, ellagic acid) as well as the clot detection method used (optical vs. mechanical). Examples of the contact pathway variability are an aPTT in rabbits of 120 seconds for cephalin/silica and 40 seconds for cephalin/ellagic acid. Collecting blood directly into premeasured anticoagulant via atraumatic technique will minimize the release of tissue thromboplastin and contact activation. In rats at terminal necropsy, collection from the abdominal aorta rather than cardiac (or vena cava) puncture provides access to relatively large volume, nonactivated samples.
Hemostasis Application in Toxicology Studies
Hemostasis testing in toxicology studies is possible but should consider multiple variables. For platelet evaluation, consider the platelet count and evaluate the blood smear for clumping that would suggest artificially low counts. Platelet morphology is also evaluated to look for large platelets or abnormal granulation. Traumatic venipuncture resulting in platelet clumping is the most common cause of low platelet counts, even when the blood smear may indicate a normal platelet count. Increased platelet counts may reflect enhanced erythropoietin production if erythrocyte (red blood cell [RBC]) parameters are low and regeneration is taking place. Thrombopoietin and erythropoietin both stimulate platelet numbers. Platelet function can be measured by turbidimetric or impedance methods. Platelet adhesion tests to measure function include ristocetin-induced aggregation, but there is no response in the rat, rabbit, or dog. Enzyme-linked immunosorbent assay (ELISA) measurement of vWF is an important method to assess adhesion defects. Platelet release of granule contents from platelets can be measured ex vivo with a luminometer in the presence of luciferin. Core coagulation tests include PT, aPTT, and fibrinogen concentration. Blood needs to be collected in plastic or siliconized glass containers. Marked dehydration or a significant change in red cell mass can change the anticoagulant:plasma ratio (normally 9:1). Venipuncture should be clean, and blood needs to mix with anticoagulant quickly. The PT and aPTT are relatively insensitive to changes in coagulation factor concentrations. PT and aPTT prolongation may result from poor quality samples, factors <30% of their normal levels, altered lipid metabolism, low fibrinogen, disseminated intravascular coagulation (DIC, which consumes factors), or specific and nonspecific coagulation factor antagonists. Phosphorothioate oligonucleotides act as nonspecific coagulation antagonists that cause concentration-dependent increases in aPTT and, to a lesser extent, PT (Henry et al. 1999). These effects are due to inhibition of coagulation sequence at the level of the tenase complex and to direct inhibition of thrombin (Sheehan and Lan 1998). Decreased fibrinogen can be due to poor-quality samples, liver failure, and DIC, but it is not commonly observed. Thrombin time is the method used to assess fibrinogen concentration. Fibrinolysis evaluation includes d-dimer and fibrin degradation products (FDP). D-dimer is specific for fibrinolysis and requires a clean sample. Other assays that can be performed to measure concentrations of antithrombin, protein C, protein S, plasminogen, or plasmin. An example of prolonged aPTT due to the type of contact activator has been observed in plasma containing a PEGylated (polyethylene glycol [PEG]-conjugated) recombinant protein.
Case Analysis
The purpose of this section was to review a published case (De Jonghe et al. 2008) of unexpected prolongation of the PT and aPTT during rat and mouse toxicology studies. The importance of this case was to demonstrate how analysis of hemostasis can be incomplete and erroneous. There was hemorrhage and prolongation of the aPTT, so the authors postulated that vitamin K metabolism or vitamin K availability was the underlying cause. However, when coagulation factor analysis was done, both a non-vitamin K factor (XI) and vitamin K-dependent factors (II and VII) were low. While factors II and VII activity improved with vitamin K administration, factor XI activity did not normalize. Therefore, the cause of hemostatic difficulties may be complex, involving factors from multiple pathways.