
Abstract
The blood–brain barrier (BBB) is the regulated interface that mediates selective transcellular transport of nutrients and essential components from the blood into the brain parenchyma. Many neurodegenerative diseases including stroke, multiple sclerosis, rheumatoid arthritis, and AIDS dementia exhibit loss of BBB integrity. Despite the increasing body of evidence for the involvement of glia in maintaining the BBB, few studies have addressed glial/endothelial/extracellular matrix interactions. A chemically induced astrocyte lesion provides a noninvasive model to study reversible BBB dysfunction in vivo. Blood–brain barrier integrity was assessed with fluorescent dextran tracers (3-70 kDa) and magnetic resonance imaging, in parallel with confocal and electron microscopy imaging of the neurovascular unit. These studies demonstrated modified tight-junction protein expression with loss of vascular integrity. We propose that adherens junction proteins and extracellular matrix remodeling provide a temporary size-selective barrier, whereas astrocyte and microglia activation direct tight-junction proteins to paracellular domains and restore BBB integrity. Morphological comparisons were made with the area postrema, a circumventricular organ with a naturally porous BBB. Further studies into cellular mechanisms of glial/endothelial/extracellular matrix interactions may identify novel glial-based therapeutic targets and innovate therapies for modulating diseases in which gliosis and raised levels of pro-inflammatory mediators are central components.
Keywords
Introduction
The blood–brain barrier (BBB) is a dynamic interface that separates the peripheral circulating blood supply and the central nervous system (CNS) (Wolburg and Lippoldt 2002). Brain capillary endothelial cells form the structural basis of the BBB, where they restrict the passage of blood-borne proteins, many neurotoxic agents, and hydrophilic molecules while allowing diffusion of small hydrophobic molecules (O2 and CO2) and selective transcellular transport of nutrients and other essential components into the brain parenchyma (Abbott 2002; Wolburg and Lippoldt 2002). The low and selective permeability of the BBB has been attributed to a number of anatomical features: (1) vascular endothelial cells lack fenestrations; (2) adjacent endothelial cells are joined by various adherens and tight-junctional complexes located in the paracellular clefts restricting paracellular diffusion of solutes (Furuse et al. 1993; Kniesel and Wolburg 2000); (3) expression of transport proteins in both apical and basolateral endothelial cell membranes, such as P-glycoprotein (Loescher and Potschka 2005); and (4) decreased pinocytosis compared to other endothelial cells.
A schematic representation of the BBB is shown in Figure 1A . A close physical and functional interaction exists between the vascular endothelial cells and surrounding cells, including microglia, pericytes, neurons, the extracellular matrix, and astrocytes, whose processes and astrocytic end-feet ensheath the capillary endothelium covering over 99% of the vascular endothelium. Thus, increasingly the BBB is no longer viewed in isolation, but as a component of an integrated neurovascular unit (Banks 2009) (Figure 1B).
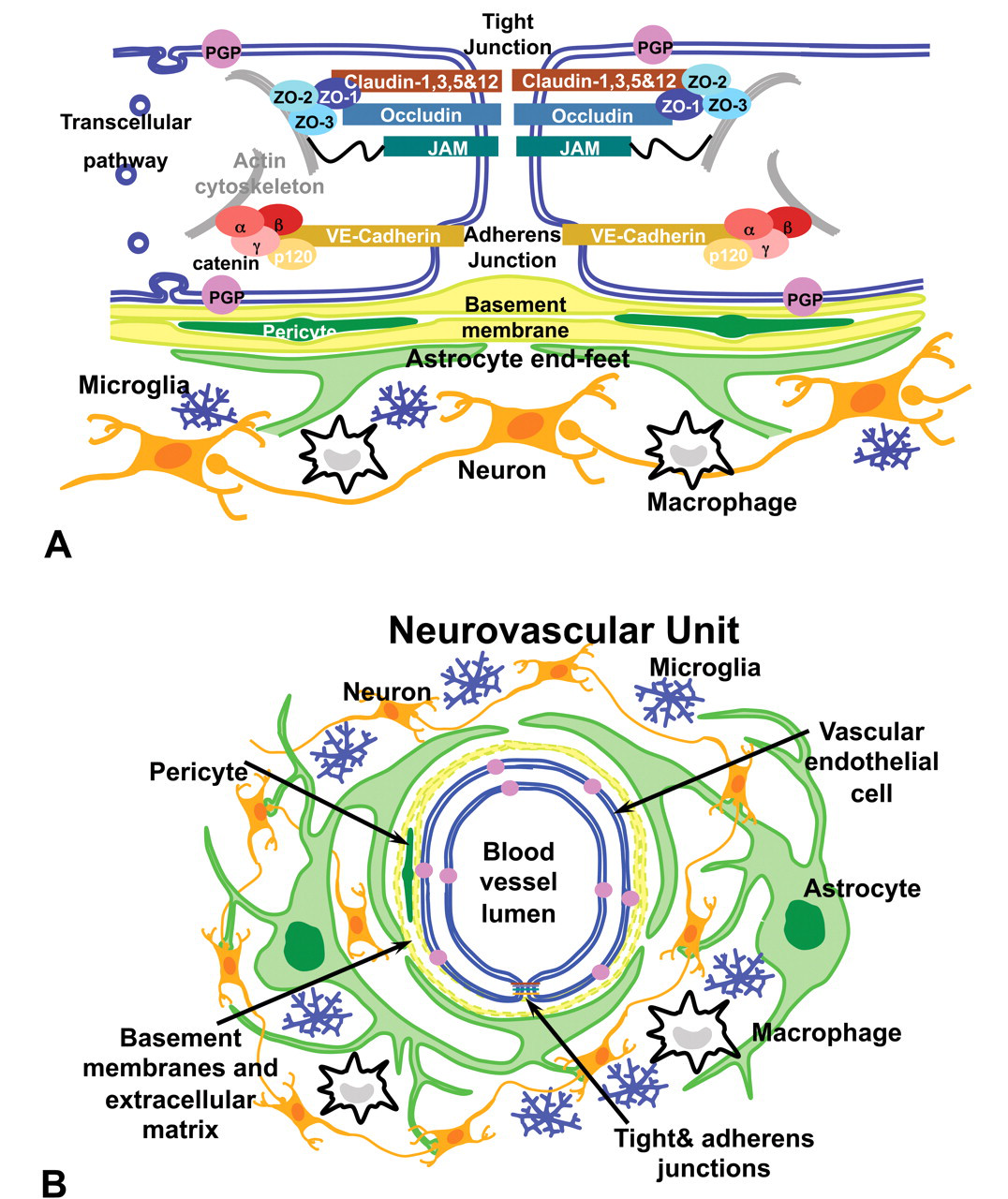
(A) Schematic representation of tight and adherens junctional proteins organization at the neurovascular unit. Tight junctional proteins, claudin, occludin, and junctional adhesion molecule and accessory proteins seal apposing membranes of the endothelial cells. Cadherin-based adherens junction proteins include vascular endothelial cadherin, which mediates a calcium-dependent, homophilic binding between cells. Cytoplasmic tail of vascular endothelial cadherin binds β-catenin, α-catenin, plakoglobin, and p-120, linking it to actin cytoskeleton. (B) Schematic representation of the neurovascular unit. Vascular endothelial cells form the blood vessel and are connected by tight and adherens junction proteins forming the structural basis of the blood–brain barrier. Basal lamina, formed by the endothelial cells and astrocyte end-feet, surround the endothelial cells. Other components of the neurovascular unit are neurons, pericytes located in the basal lamina, and microglia. Figures reprinted with permission from Bentham Science Publishers Ltd. (Willis and Davis 2008).
In the adult, although the BBB prevents the entry of many potential neurotoxic agents, this protection is not complete, since a number of neurotoxic agents are lipophilic (e.g., hexacarbons, carbon disulfide, and organophosphorus compounds) or can access an endothelial cell carrier mechanism (such as valproate, a drug used in the treatment of epilepsy, which crosses the BBB via the monocarboxylic acid transporter). In the fetus and early postnatal period, there is increased risk of neurotoxic effects. In humans, the fetal BBB develops during pregnancy and is not fully developed until the middle of the first year of life (Rodier 1995). Thus, during these pre- and postnatal periods, the BBB is leaky and some toxic agents such as cadmium, which in the adult is very unlikely to enter the brain, may pass into the developing brain (Levin and Miller 1981).
The BBB is not a static barrier but is tightly regulated, allowing changes in vascular permeability. In health, there may be a regulated temporary increase in permeability to allow increased access of nutrients and oxygen to tissues and drug accessibility. However, in disease, an uncontrolled and prolonged increase in BBB permeability results in vasogenic edema and leakage of potentially neurotoxic plasma constituents into tissue. Many acute and chronic neurodegenerative diseases including stroke, multiple sclerosis, rheumatoid arthritis, and AIDS dementia show changes in BBB integrity (Abbott et al. 2006; Plumb et al. 2002). However, the development of drugs for the treatment of these diseases is severely restricted by the BBB. Over 98% of small-molecule drugs and all large-molecule drugs are excluded from the brain (Pardridge 2007). An additional complication in many of these diseases is that changes in BBB integrity and expression of efflux transporter proteins affect drug delivery to the brain and may result in either CNS toxicity or ineffectual dosing (Wolka et al. 2003). However, therapy for Parkinson’s disease with L-3,4-dihydroxyphenylalanine takes advantage of BBB transport systems to deliver dopamine to surviving dopaminergic terminals. Dopamine does not cross the BBB, whereas L-3,4-dihydroxyphenylalanine is transported across the BBB via the L1 facilitative transporter (Hawkins et al. 2006) and then decarboxylated and converted to dopamine.
Despite the importance of the BBB to homeostasis and correct functioning of the CNS, the nature of factors responsible for the induction and maintenance of BBB properties in development or maturity remains unclear. In vitro evidence suggests that astrocytes play a key role in inducing BBB properties. Endothelial cell monocultures lose BBB characteristics (Rubin et al. 1991), which are restored when co-cultured with cells of astroglial origin that are incubated with astroglial membrane fractions or astrocyte/glioma conditioned medium (Prat et al. 2001; Sobue et al. 1999). However, in vivo, the role of astrocytes on BBB properties is less clear. One study has shown that cultured astrocytes exert barrier-inducing properties on endothelial cells when injected into the brain (Janzer and Raff 1987), whereas in another study, injection of 6-aminonicotinamide induced loss of rat spinal cord astrocytes, which resulted in local protein extravasation and horseradish peroxidase, but the expression of a number of BBB-associated proteins (e.g., γ-glutamyltranspeptidase) was unaffected (Krum 1994).
In this report, we demonstrate a critical role of astrocytes in regulating the integrity of the mature rat BBB but also show the close interaction between other components of the neurovascular unit to maintain a degree of BBB integrity in a pathological environment. Comparisons are drawn with the area postrema (AP), a circumventricular organ that lacks BBB properties. These studies not only demonstrate the importance of a greater understanding of intercellular interactions at the neurovascular unit, but also pave the way for studying intracellular signaling mechanisms at the BBB/neurovascular unit. They also suggest that a more target-directed approach to drug design and delivery is needed in the therapy of neurodegenerative diseases in which gliosis and loss of BBB integrity are features.
Astrocytes: Induction of Focal Loss with Subsequent Repopulation
To test the hypothesis that astrocytes play a critical role in maintaining BBB integrity in vivo, we have selectively induced astrocyte loss by intraperitoneal (i.p.) injection of the metabolic toxicant 3-chloropropanediol (α-chlorohydrin) into male Fisher F344 rats (180–220 g). 3-Chloropropanediol induced lesions in a number of specific nuclei including the inferior (caudal) colliculus, superior (dorsal) olivary nucleus, and red nucleus, whereas the cerebral cortex and hippocampus were unaffected (Willis 2004b). (Correct terminology for quadrupeds based on the Nomina Anatomica Veterinaria is “caudal,” but have used the classic biped term “inferior” to conform to the conventions in a standard rodent neuroanatomy atlas [Paxinos and Watson 1998]). This approach has several advantages. It is noninvasive and uncomplicated by trauma or ischemia, and it produces only mild, reversible ataxia (unlike cold injury or fluid percussion models), allowing long-term survival for studies into mechanisms of both degenerative processes and repair. The ability to study astrocytic loss and repopulation in vivo also has advantages over isolated microvessels or cultured cells, since it allows the study of interactions between astrocytes and other components of the neurovascular unit.
In undosed animals, immunohistochemical studies have shown the presence of resident glial fibrillary acidic protein (GFAP)-positive, vimentin-negative astrocytes in both the vulnerable inferior colliculus and the nonvulnerable cerebral cortex (Figure 2A ). Over the course of a twenty-eight-day study period, administration of 3-chloropropanediol (i.p.) induced a degenerative phase followed by a regenerative phase (Willis 2004a). During the degenerative phase, electron microscopy revealed acute swelling of astrocyte cell bodies at six hours post-dosing; by twelve hours, swollen astrocyte end-feet were seen adjacent to vascular endothelial cells with no apparent effect on tight junctions (Figure 2B). Confocal microscopy demonstrated a loss of the GFAP-immunoreactive astrocytes in the inferior colliculus. Two days after 3-chloropropanediol dosing, GFAP immunoreactivity appeared very fragmented (Figure 2C). By four days after treatment, most GFAP expression within the inferior colliculus had been lost (Figure 2D). Electron microscopy confirmed the loss of astrocyte-like processes in the lesioned areas. Regeneration of astrocyte morphology was seen after eight days. Glial fibrillary acidic protein and vimentin expression was up-regulated in astrocytes around the margin of the lesion, and a few GFAP-immunoreactive astrocytes were present within the lesioned areas (Figure 2E). Reactive GFAP-positive astrocytes continued to repopulate the inferior colliculus over the remaining period of study, so that by fourteen to twenty-eight days after treatment, astrogliosis was present in the center of the lesion (Figure 2F).
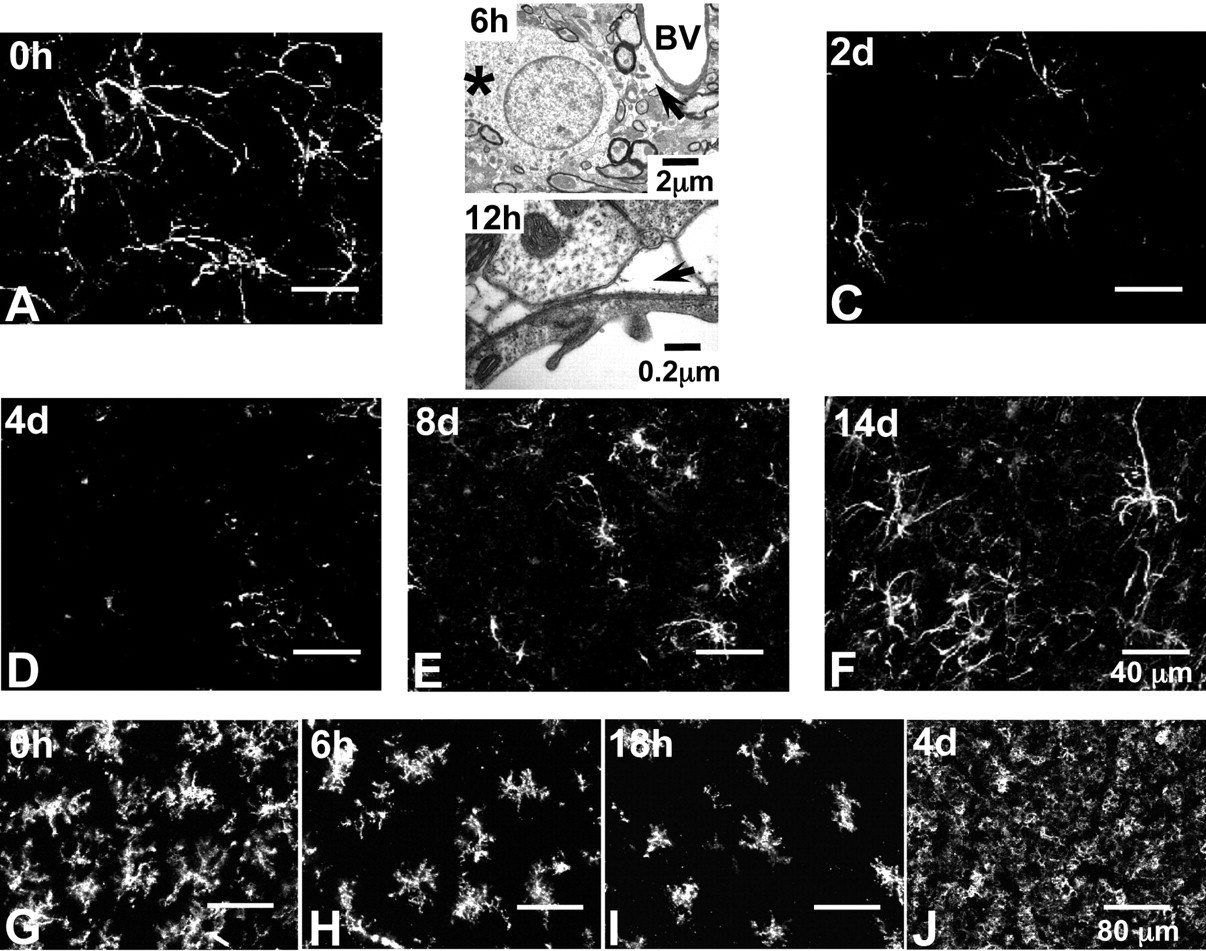
Confocal micrographs showing changes in glial fibrillary acidic protein (GFAP)-positive astrocytes and CD11b-immunoreactive microglia following 3-chloropropanediol administration. (A) At zero hours, the inferior colliculus shows GFAP-positive astrocytes throughout the tissue forming a fine network of processes. (B) At six and twelve hours, electron micrographs show early changes in astrocyte ultrastructure, including swollen astrocyte cell bodies (six hours) and swollen astrocyte end-feet (twelve hours). (C) At two days, few GFAP-positive astrocytes remain within the inferior colliculus. (D) At four days, GFAP-positive astrocyte immunoreactivity is completely lost. (E) At eight days, reappearance of GFAP-positive astrocytes is evident within the lesion. (F) At fourteen days, increased numbers of large, GFAP-positive astrocytes with many fine processes are seen within the inferior colliculus. Nonvulnerable cerebral cortical regions showed no change in GFAP-positive astrocytes. Scale bar = 40 µm (refers to A, C–F). (G) At zero hours, homogeneous distribution of CD11b-immunoreactive microglia in the inferior colliculus. (H) By six hours, a marked loss of CD11b immunoreactivity within the inferior colliculus was apparent. (I) By eighteen to twenty-four hours, loss of CD11b immunoreactivity continued. At forty-eight hours, CD11b expression increased at the margin of lesion, with cells beginning to repopulate the lesion. (J) By four days, CD11b-immunoreactive microglia are concentrated in the inferior colliculus. Scale bar = 80 µm (refers to G–J).
The question as to why astrocytes in specific brain regions are lost whereas other areas, such as the cerebral cortex, hippocampus, striatum, and cerebellar cortex, never develop any lesions remains unanswered, although we speculate that this finding reflects a differential degree of dependence on glycolysis (Willis et al 2004b).
Microglia: Rapid Loss Followed by Inflammatory Response
CD11b-positive microglia were distributed evenly throughout the inferior colliculus and adjacent cerebral cortical tissue in control tissue from nondosed rats (Figure 2G). Following 3-chloropropanediol injection, reduced CD11b expression in the inferior colliculus was seen by six hours, and by eighteen hours, there was extensive loss of CD11b-positive microglia (Figure 2H&I). Cerebral cortical tissue, which we used as control tissue, was unaffected. CD11b-positive microglia began to repopulate the inferior colliculus lesion after twenty-four hours. By four days, very intense CD11b immunoreactivity was present throughout the inferior colliculus (Figure 2J) and remained elevated until fourteen to twenty-eight days after treatment.
Together, these studies show that during the early degenerative phase, there was loss of both astrocytes and microglia. However, during the subsequent regenerative phase, there was prolonged gliosis with activated astrocytes and microglia in the inferior colliculus (Willis et al. 2004a).
BBB Integrity: Functional Assessment of Loss and Size-Selective Restoration of Vascular Integrity
To determine the effect of astrocyte loss and gliosis on vascular integrity, we used a series of variably sized molecular markers ranging from 0.5 to 70 kDa to visualize size-selective changes in BBB integrity. Magnetic resonance imaging (MRI), using a 0.5 kDa gadolinium complex of diethylenetriamine pentaacetic acid (Gd-DTPA; Magnevist, Bayer Schering Pharma AG, Berlin-Wedding, Germany) as a contrast agent, revealed some loss of vascular integrity in the inferior colliculus twelve hours post 3-chloropropanediol administration at a time when electron microscopy showed swollen astrocyte end-feet (Prior et al. 2004). Leakage of Gd-DTPA continued to increase and showed maximal loss of BBB integrity to Gd-DTPA at forty-eight hours. During the regenerative phase, in which GFAP-positive astrocytes were seen to repopulate the lesion, there was reduced leakage of Gd-DTPA from nine to thirty days post-administration, with levels of Gd-DTPA comparable to control levels at the end of the study period. This finding suggests that BBB integrity was restored. Blood–brain barrier integrity was also studied by visualizing leakage of intravenously (i.v.) administered, fluorescently tagged dextran (10 kDa) and by visualizing the presence of extravascular fibrinogen (300 kDa), as previously shown (Willis et al. 2007). In control rats with an intact BBB, the vascular endothelium was impermeable to the 10 kDa dextran and fibrinogen, which showed an intravascular localization. Following 3-chloropropanediol administration, fluorescently tagged dextran (10 kDa) could be seen leaking from the vasculature into the brain parenchyma after twenty-four hours. Maximum leakage of 10 kDa dextran occurred after forty-eight hours and continued until day 6 post-treatment. As shown previously with Gd-DTPA, some degree of restoration of BBB properties took place after this time. At eight days, no dextran (10 kDa) leakage was detected. Vascular leakage of the serum protein fibrinogen showed a similar time course to that of dextran. In control rats, fibrinogen was retained within the vasculature of the inferior colliculus and was not seen in the parenchyma (Figure 3A ). At twenty-four hours to two days after dosing, tissue sections showed sharply defined clusters of fibrinogen immunoreactivity in the inferior colliculus parenchyma, suggesting focal leakage from the vasculature (Figure 3B arrows). From eight days, fibrinogen was once again retained within the vasculature, and no new extravascular clusters of fibrinogen immunofluorescence were detected (Figure 3C).
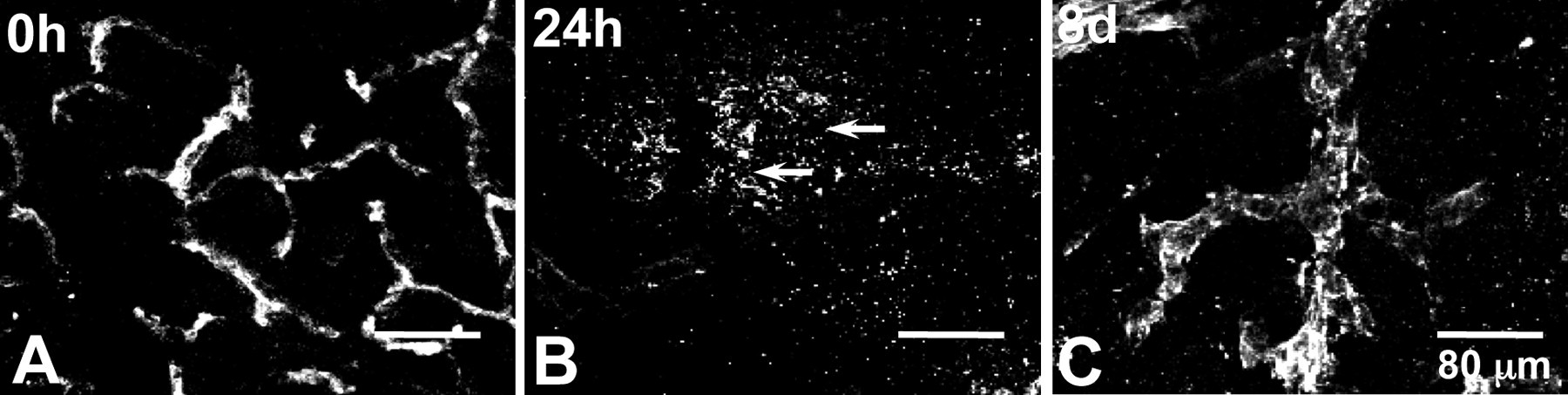
Confocal micrographs showing leakage of fibrinogen following 3-chloropropanediol-induced changes in the inferior colliculus. (A) At zero hours, fibrinogen is retained within the vasculature and is absent in the parenchyma. (B) At twenty-four hours, fibrinogen immunoreactivity shows a few focal areas of extravascular (arrow) fibrinogen. (C) At four days, some extravascular fibrinogen is still present, whereas at eight days, no further leakage occurs into the parenchyma. Scale bar = 80 µm.
The cessation of leakage to 10 kDa dextran and fibrinogen seen after six to eight days took place in the absence of GFAP-positive astrocytes in the inferior colliculus. It may be proposed that the cessation of leakage could be due to a lack of blood flow in the region. To test this hypothesis, we used hydrogen washout polarography (Ray 1982) to determine blood flow in the inferior colliculus over the thirty-day study period. Briefly, platinum/iridium (90/10) glass-coated polarographic electrodes were implanted in the inferior colliculus and in the nonvulnerable cerebellar cortex. Following recovery from surgery, rats inhaled hydrogen gas (10% in air) until an equilibrium was reached. The washout of hydrogen was then recorded, and absolute blood flow was calculated. Results showed a large, sustained increase in blood flow in the inferior colliculus at a time when 10–300 kDa tracers had stopped leaking from the vasculature (Willis et al. 2004a), thereby confirming that restoration of BBB integrity was well underway.
Tight Junction Protein Expression: Reversible Disruption Correlates with Transitory Loss of Astrocytes and BBB Integrity
A major contributing factor to the low permeability of the BBB is the presence of tight and adherens junctional protein complexes located in the paracellular cleft of adjacent vascular endothelial cells. These junctional proteins impart a high transendothelial electrical resistance (TEER) of 1500–2000 Ωcm2 (Butt et al. 1990) and restrict paracellular movement of molecules with a molecular weight of greater than 180 Da (Petty and Lo 2002). Tight junction complexes are composed of transmembrane proteins: (1) the claudin multigene family of proteins (Tsukita et al. 2001); (2) occludin (Furuse et al. 1993; Hirase et al. 1997); and (3) the junctional adhesion molecule (JAM) proteins (Martin-Padura et al. 1998). Claudin proteins are believed to form the primary seal of the tight junction by forming dimers through homophilic and heterophilic interactions with other claudin molecules in adjacent endothelial cells (Furuse et al. 1998). Occludin is proposed to play a role in electrical resistance and barrier properties of the BBB (Antonetti et al. 1998; Martin-Padura et al. 1998). Disrupted tight-junction protein expression is seen in many neurodegenerative diseases, including multiple sclerosis (Plumb et al. 2002).
The mechanism(s) that regulate the opening of the BBB at both the intracellular level and the intercellular level are, at present, not clear. We hypothesized that the loss of BBB integrity to Gd-DPTA, dextran, and fibrinogen was due to a critical role played by astrocytes in maintaining tight-junction integrity. To test this hypothesis, we visualized changes in expression and endothelial cellular localization of the tight-junction proteins claudin-5, occludin, zona occludens-1 (ZO-1) (Willis et al. 2004a). In nondosed rats, both the inferior colliculus and cerebral cortex showed claudin-5 immunoreactivity, which appeared as a continuous, sharply defined pattern of single or double lines marking the margins of adjacent platelet endothelial cell adhesion molecule-1 (PECAM-1)-immunoreactive endothelial cells (Figure 4A , main image). Claudin-5 also showed diffuse immunoreactivity within the PECAM-1–positive vascular endothelial cells (Figure 4A, inset). Occludin and ZO-1 immunoreactivity showed a pattern of expression similar to that of claudin-5 in these areas, but lacked the diffuse immunoreactivity (data not shown). At the ultrastructural level, electron microscopy of the endothelial cells showed intact paracellular junctions with “kissing points” formed by junctional proteins at the paracellular cleft between two rat brain vascular endothelial cells (Figure 4A inset, arrows).
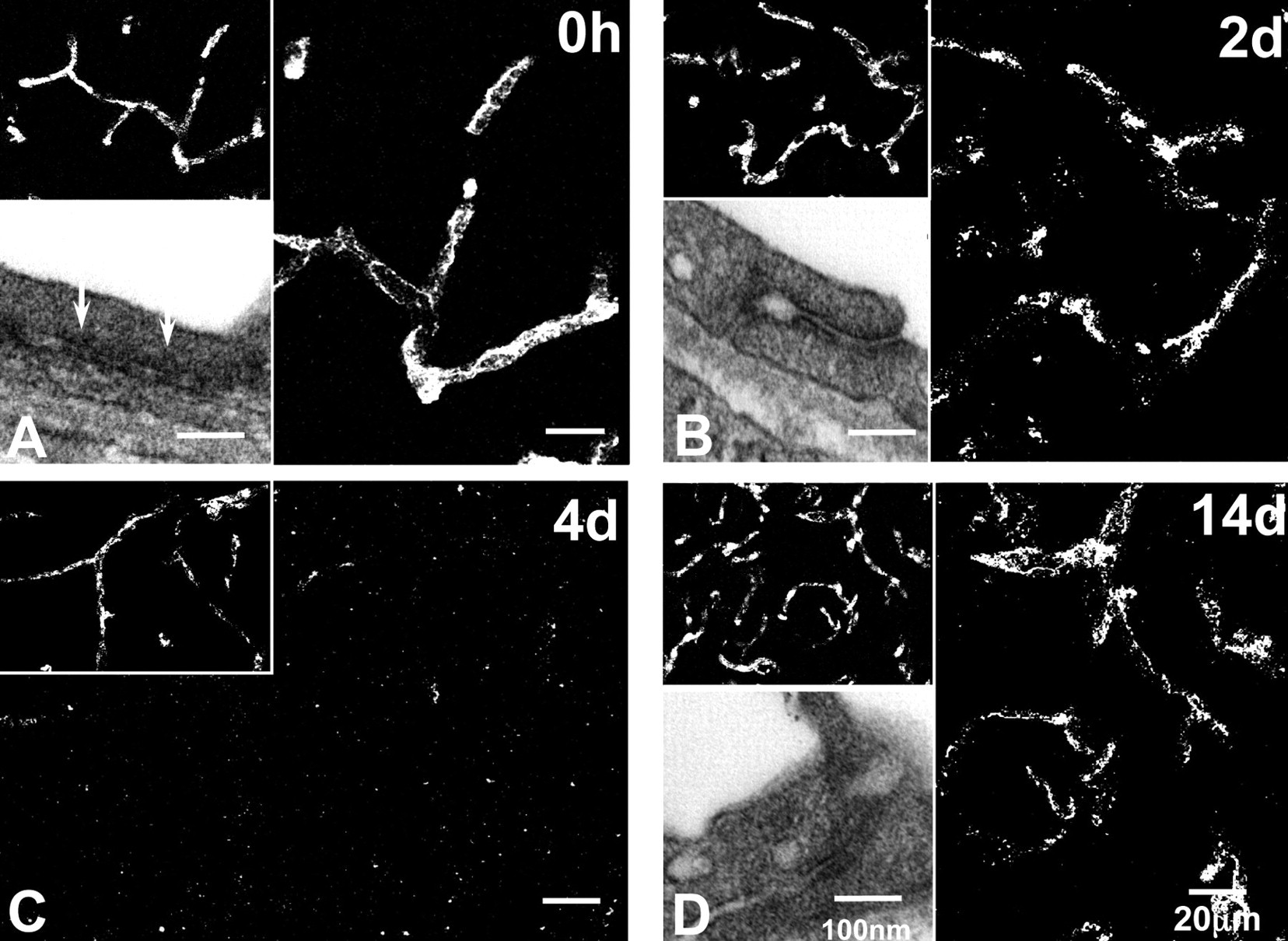
Confocal and electron micrographs showing changes in claudin-5 expression and ultrastructural changes at the tight junction. (A) At zero hours, claudin-5 expression appears as both a continuous, sharply defined network and as diffuse immunoreactivity (main images) within PECAM-1-immunopositive microvasculature (insets). Electron micrograph shows close association between adjacent endothelial cells. (B) At two days, the sharply defined paracellular claudin-5 immunoreactivity is lost, but diffuse staining is still present. Electron micrograph shows separation between endothelial cells. (C) At four days, widespread loss of claudin-5 immunoreactivity is evident, although endothelial cells still express PECAM-1 immunoreactivity. (D) At fourteen days, sharply defined paracellular cleft staining and diffuse cytoplasmic claudin-5 immunoreactivity is restored. Nonvulnerable cerebral cortical regions showed no change in claudin-5 immunoreactivity. Scale bars = 20 µm for confocal microscopy and 100 nm for electron microscopy.
The pattern of claudin-5 expression in the inferior colliculus changed markedly as astrocytes were lost. Eighteen hours post-dosing, the sharply defined claudin-5 immunoreactivity marking the paracellular cleft between endothelial cells was disrupted, although the diffuse immunoreactivity was retained. At two days, further disruption of claudin-5 immunoreactivity could be seen (Figure 4B, main image). Disruption of the close association between adjacent endothelial cells could be visualized by electron microscopy, with loss of kissing points and greater separation between adjacent endothelial cells (Figure 4B, inset), consistent with increased vascular porosity. This morphological profile corresponds with loss of vascular integrity to Gd-DTPA (0.5 kDa), dextran (10 kDa), and fibrinogen (300 kDa). Four to six days post-dosing, both the sharply defined paracellular claudin-5 immunoreactivity and most of the diffuse cytoplasmic immunoreactivity was lost within PECAM-1–immunoreactive vessels (Figure 4C). Restoration of claudin-5 immunoreactivity started eight days after dosing, and by fourteen to twenty-eight days, claudin-5 immunoreactivity was restored to sharply defined cellular domains, with diffuse immunoreactivity within the PECAM-1–positive endothelial cells (Figure 4D, main image). This pattern of immunoreactivity resembled that seen in control inferior colliculus and the cerebral cortex, which did not change over the twenty-eight-day period. Similar reversible changes in the expression of occludin and ZO-1 were seen in the inferior colliculus following toxicant-induced loss of GFAP-positive astrocytes. The close association between endothelial cells also appeared to be restored at the ultrastructural level (Figure 4D, inset). Other claudin proteins (-1,-3,-12) were also studied, since loss of one claudin isoform may result in the up-regulation of another claudin isoform. Claudin-1 showed a similar loss as that described for claudin-5, whereas claudin-3 and -12 were not detected in the rat brain.
Early Size-Selective Restoration of BBB Properties in the Absence of Astrocytes and Tight Junctions
These studies demonstrate that GFAP astrocytes play a critical role in the expression of BBB tight-junction proteins (claudin-5, occludin, and ZO-1) and vascular integrity during both the degenerative phase and subsequent regenerative phase, in which we see gliosis. However, this study also revealed a surprising anomaly regarding the mechanisms and factors regulating vascular leakage. During the early degenerative phase (zero to four days) of this study, widespread loss of GFAP astrocytes in the inferior colliculus was associated with loss of marginal tight-junction protein expression and loss of vascular integrity, as defined by the leakage of Gd-DPTA, fluorescent-tagged dextran (10 kDa), and fibrinogen. However, later, during a recovery phase beginning at six days, vascular integrity to solutes >10 kDa was restored, despite the apparent absence of claudin-5, occludin, and ZO-1 and GFAP-astrocyte expression, as determined by immunofluorescence. Only at eight days did diffuse or fragmented claudin-5, occludin, and ZO-1 immunoreactivity reappear within the vascular endothelial cells, together with some GFAP-positive astrocyte expression. To explain this anomaly of early cessation of vascular leak, we hypothesize that the partial restoration of BBB properties is a result of modifications of the neurovascular unit, such as changes in adherens junction protein and extracellular matrix expression, thereby restoring barrier function to compounds >10 kDa.
Neurovascular Unit Components: Adherens Junction Proteins and Extracellular Matrix
Each component of the neurovascular unit may contribute to the regulation of microvascular permeability and BBB function (Hawkins and Davis 2005). In our study, we proposed that loss of astrocytes and BBB integrity with leakage of blood-borne proteins into the parenchyma will induce a reaction in other neurovascular unit components. Adherens junctional complexes are found in the paracellular cleft between endothelial cells (Figure 1A) and are formed by members of the cadherin family of proteins. Endothelial cells express two major cadherins, vascular endothelial (VE) cadherin and neuronal cadherin (Bazzoni and Dejana 2004). Adherens junctions are formed in the early stages of developing endothelium, before tight-junction formation (Lampugnani and Dejana 2007). At present, it is unclear whether adherens junctions play a critical role in regulating BBB permeability in the established BBB. However, a role for β-catenin has been shown in vessels from β-catenin–null mouse embryos (embryonic day 11.5–13.5); these vessels are frequently hemorrhagic and show a different organization of adherens junctions (Cattelino et al. 2003). Another important component of the neurovascular unit is the extracellular matrix. Extracellular matrix constituents such as laminin, heparin sulphate proteoglycans, fibronectin, and collagens reside predominantly in basement membranes surrounding brain capillary endothelial cells (Colognato and Yurchenco 2000). The basement membranes are a structural component of cerebral blood vessels and appear to be involved in the regulation of cell behavior and differentiation of brain capillary endothelium, leading to the formation of a functional barrier (Hartmann et al. 2007). Arthur and coworkers (1987) reported that tight-junction formation in cultured brain capillary endothelial cells induced by astrocyte-conditioned medium also required an endothelium-derived extracellular matrix as a growth substrate. In many pathological states, disruption of the extracellular matrix is associated with increased BBB permeability (Rascher et al. 2002). Early breakdown of the BBB with perivascular infiltration and accumulation of leukocytes and changes in the basement membrane are features of multiple sclerosis pathology (van Horssen et al. 2006).
We are currently testing our hypothesis that other neurovascular components also play a critical role in restoring some degree of barrier integrity by studying changes in the adherens junction protein expression and changes in the extracellular matrix profile. This hypothesis is based on our earlier studies directed at visualizing vascular barrier properties in a circumventricular organ, the AP, which we used as a model to investigate morphological features of a neurovascular unit in a region that shows a more open BBB compared to many other CNS areas.
A Size-Selective Vascular Barrier in the Rat AP
Circumventricular organs are recognized as important sites for communication between the brain and peripheral organs via hormones and other blood-borne products. The AP is involved in emesis, neurosecretion, cardiovascular and respiratory control, blood osmoregulation, control of renal function, caloric homeostasis and autonomic immune interaction (Goehler et al. 2006). Unlike other areas of the CNS, studies have shown the presence of fenestrations in the vascular endothelial cells of the AP giving a less restrictive vascular barrier (Dermietzel and Leibstein 1978). The same limited BBB function has also been shown for other circumventricular organs.
Our investigations and others have demonstrated that the structural and molecular properties of the BBB differ between the AP and other brain regions that have a traditional BBB. Glial fibrillary acidic protein–positive astrocytes in the AP showed a different morphology to that seen in other areas of the brain (Figure 5A ), and astrocytic processes did not make close contact with endothelial cells (Figure 5B, arrows) (Willis et al. 2007). This observation is supported by an earlier electron microscopy study that demonstrated that AP astrocytes did not contact the endothelial cells (Shimizu and Ishii 1964). Expression of claudin-5 and occludin were absent in nearly all PECAM-1-immunoreactive vessel profiles within the AP (Figure 5C). In contrast, claudin-5 and occludin immunoreactivity was present in vascular endothelial cells of the surrounding nucleus of the solitary tract (NST) and other non-AP CNS areas (Figure 5C). In these non-AP regions, claudin-5 and occludin immunoreactivity appeared as continuous, sharply defined staining along margins of adjacent endothelial cells, as described earlier. However, diffuse claudin-1 immunoreactivity (data not shown) and the adherens junction protein VE-cadherin were expressed in the vascular endothelial cells of the AP (Figure 5D).
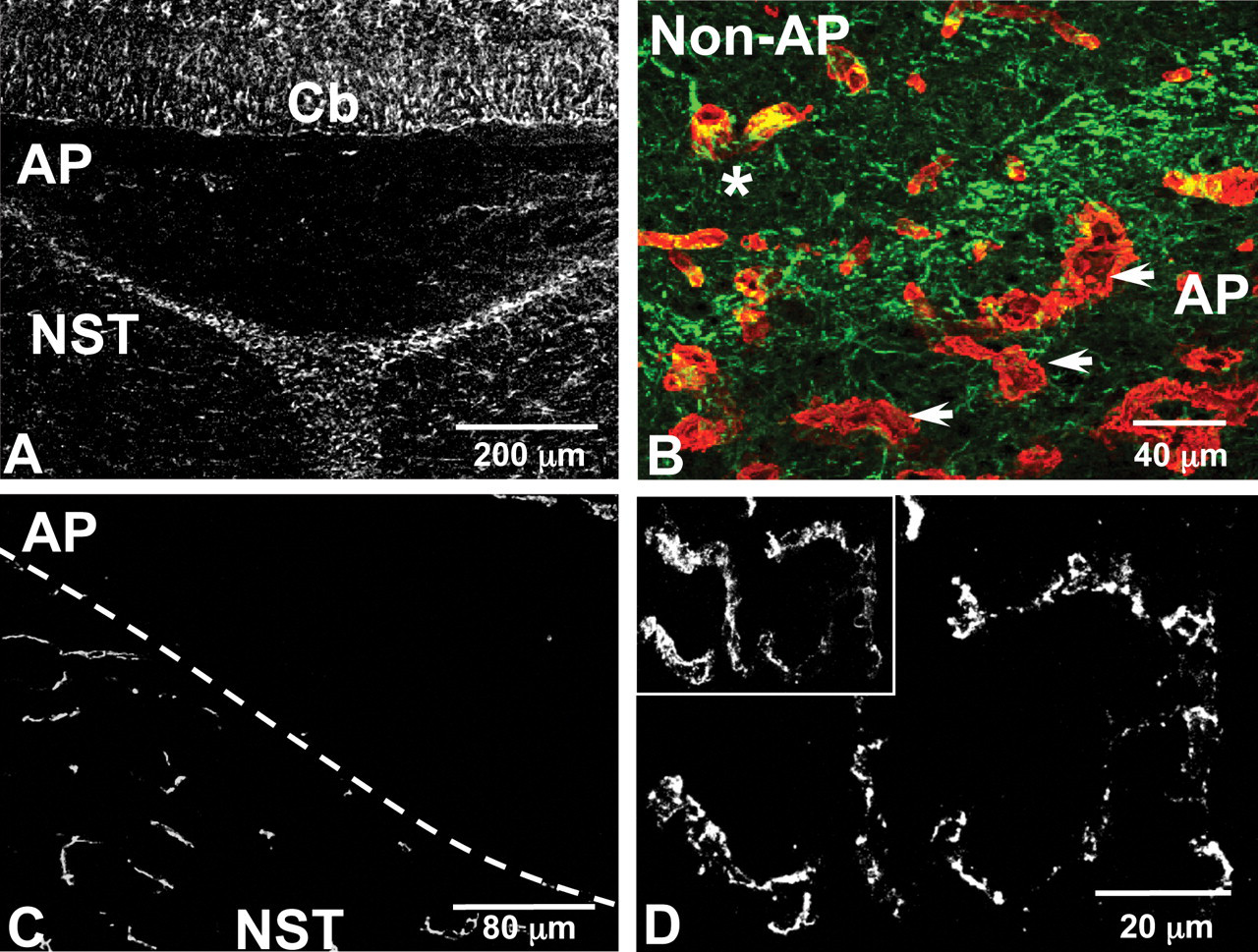
Confocal micrographs showing the distribution of GFAP, laminin, claudin-5, and VE-cadherin immunoreactivity in the area postrema (AP). (A) Glial fibrillary acidic protein (GFAP)–immunoreactive astrocytes in the AP and surrounding cerebellar (Cb) and nucleus of the solitary tract (NST) tissue. Glial fibrillary acidic protein–positive astrocytes in the AP show a different morphology to those in surrounding tissue. Scale bar = 200 µm. (B) Asterisk shows vasculature, as visualized by laminin (red) immunoreactivity, is almost completely enveloped by GFAP-immunoreactive astrocytes (green) in the NST, whereas arrows highlight few GFAP-immunoreactive astrocytes associated with the laminin-positive vasculature in the AP. Scale bar = 40 µm. (C) Claudin-5 immunoreactivity can be seen in surrounding NST tissue, but it is absent from the AP. Scale bar = 80 µm. (D) Vascular endothelial cadherin expression is present in PECAM-1–immunoreactive vessels in the AP. Scale bar = 20 µm.
The extracellular matrix is considered to be an important part of the BBB and neurovascular unit and may have a physiological function in BBB formation and maintenance of barrier properties. Within the AP, the extracellular matrix as visualized by laminin immunoreactivity appeared ruffled and made only loose association with the underlying endothelial cells, resulting in the apparent formation of lacunae (Figure 6A ) (Willis et al. 2007). This finding contrasts to non-AP brain regions, which showed that the laminin component of the extracellular matrix tightly enveloped the vascular endothelial cells with no lacunae (Figure 6B). The difference in extracellular matrix morphology between the AP and non-AP regions was also seen at the ultrastructure level (Willis et al. 2007). Endothelial cells of the AP were seen to be surrounded by two distinct, widely separated layers of basement membranes. Within the space between these two basement membrane layers were perivascular macrophages with processes that almost completely encircled the fenestrated endothelial cells. Blood vessels in the NST and other regions showed fused layers of basement membrane with no lacunae.
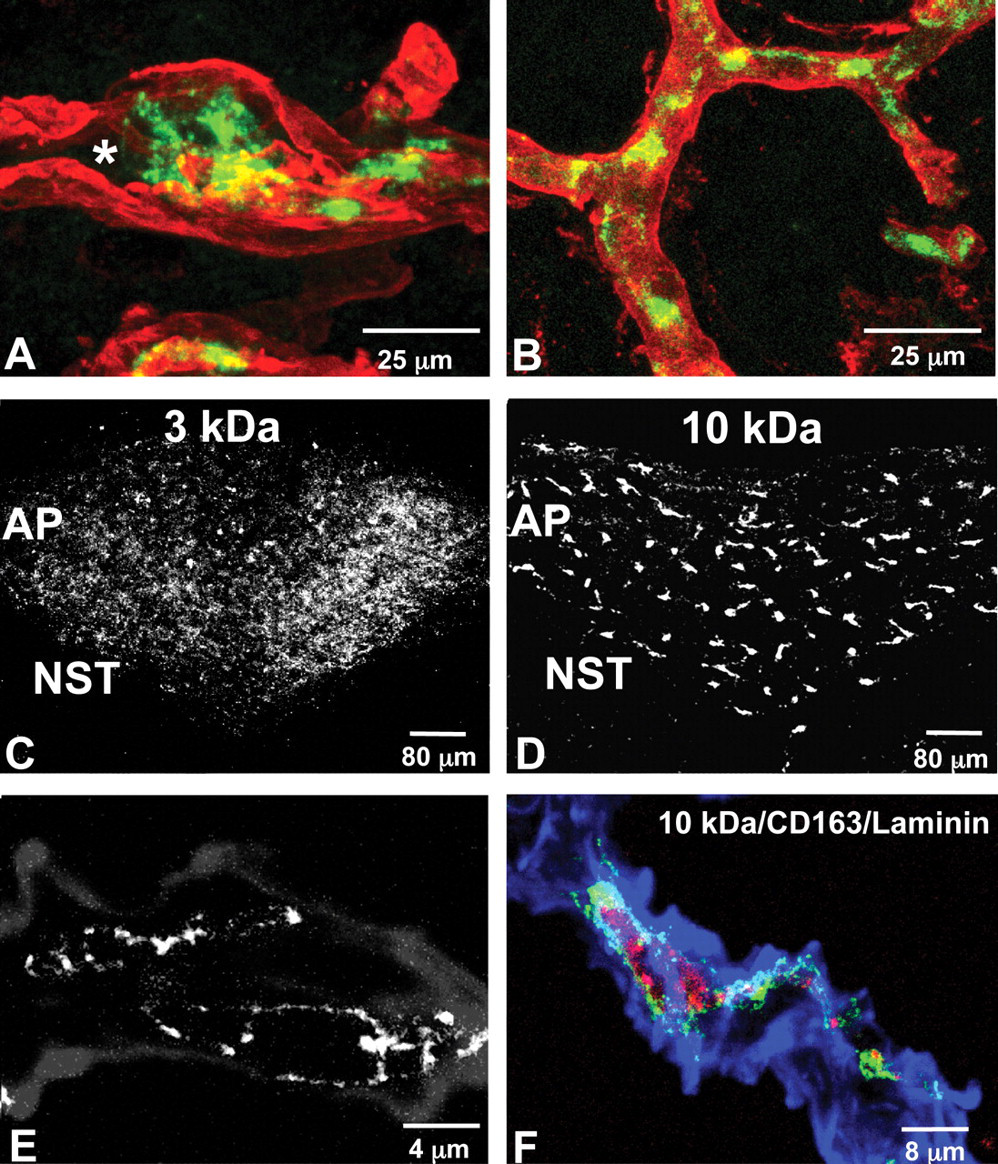
Confocal micrographs of laminin immunoreactivity in the area postrema (AP) and non-AP regions. (A) Laminin immunoreactivity (red) completely surrounds the PECAM-1 (green) immunoreactive endothelial cell, although there is a loose association between the laminin-immunoreactive basement membrane and the PECAM-1–immunoreactive endothelial cells forming lacunae (*). (B) Close association between the laminin-containing basement membrane and endothelial cells is seen in the nucleus of the solitary tract and other non-AP regions without formation of lacunae. Scale bar = 25 µm (refers to A and B). Injection of fluorescent dextran tracers. (C) Diffuse distribution of 3-kDa dextran is confined within the AP and absent in surrounding tissue, thirty minutes after administration. (D) Cellular distribution of 10 kDa dextran thirty minutes after administration. Scale bars = 80 µm (refers to C and D). (E and F) Merged image showing 10-kDa dextran (red) accumulated within CD163-positive macrophage (green) located within laminin-immunoreactive basement membranes (blue). Scale bar = 8 µm.
The nonfused basement membranes in the AP resembled basement membranes during vascular development and wound healing following CNS injury. Neonatal rat brains show large, perivascular spaces between the nonfused basement membranes, which later fuse into one common membrane with no perivascular space as tight gliovascular connections develop (Caley and Maxwell 1970). In a stab wound injury model, vessels exhibited increased laminin immunoreactivity, which was later reduced during a healing phase coinciding with the formation of gliovascular connections (Szabo and Kalman 2004). Our study and others suggest that the AP presents a morphological profile similar to an arrested stage in developmental or a regenerative phase following injury.
Using the same approach to assess vascular integrity in the inferior colliculus, we used a range of molecular-sized tracers (0.3–70 kDa) to visualize the vascular permeability of the AP. We found that the AP exhibited some size-selective, barrier-like properties, despite lacking many characteristics seen in other areas of the CNS (Willis et al. 2007). Administration of sodium fluorescein (0.3 kDa i.v.) rapidly diffused through the AP parenchyma and appeared as a zone of increased fluorescence in the parenchyma of the surrounding nucleus of the solitary tract. Dextran (3 kDa i.v.) was seen to have crossed the vascular endothelium and was distributed throughout the entire AP parenchyma, but in contrast to sodium fluorescein, 3 kDa dextran was contained within the AP and did not diffuse further (Figure 6C). Larger dextrans (10 kDa or 70 kDa) were also seen to have crossed the vascular endothelium, but in contrast to the 3 kDa dextran, they were concentrated in cells adjacent to the vascular endothelium (Figure 6D). No diffuse parenchymal fluorescence was seen in the AP or non-AP tissue with the 10- to 70 kDa dextrans. Fluorescent dextrans (3–70 kDa) were not observed in other areas of the brain beyond the AP, indicating that non-AP domains possessed an intact BBB. These results suggest that despite showing a fenestrated endothelium the AP shows some size-selective barrier properties.
Immunofluorescence revealed that these dextran-containing cells were CD163- and CD169-immunoreactive macrophages that were evenly distributed throughout the AP, closely associated with the vasculature, and located within the laminin-immunoreactive extracellular matrix (Figure 6E) (Willis et al. 2007). Following injection of dextran (10 or 70 kDa), CD163- or CD169-macrophages rapidly sequestered the dextran tracers (Figure 6F). The 3 kDa dextran was not sequestered, as it could not be seen within any cellular structure. Other studies of the AP have described phagocytic cells (macrophages) with long cellular processes encircling fenestrated capillaries that sequestered horseradish peroxidase (Murabe et al. 1981; Sano and Murabe 1980). These macrophages are well placed to sequester any solutes that pass through the vascular endothelium and the inner laminin barrier but are detained by the outer laminin barrier. The phagocytic function of these macrophages may indicate that this cell type may play a role in selective barrier functions.
Thus, we propose that a modified neurovascular unit exists in the AP and that it shows some GFAP-positive astrocytes, VE cadherin expression in the endothelial cells, a modified extracellular matrix, and the presence of resident macrophages within the extracellular matrix, which may impart a degree of barrier function to the vasculature of the AP. A size-selective barrier at the AP would play an important role in the function of the AP as a sensory organ. It would allow small molecules such as catecholamines, amino acids, steroid hormones, and neuropeptides to pass through the vasculature and enter the AP parenchyma, whereas larger proteins such as albumin (68 kDa), albumin-bound complexes (such as the thyroid hormone triiodothyronine), precursor proteins, and blood enzymes could be predicted to be retained within the limit of the outer basement membrane and sequestered by resident macrophages. We are currently investigating whether similar changes occur in the inferior colliculus under pathological conditions to explain the early cessation of leakage. An early cessation of leakage would offer a degree of neuroprotection to the lesioned area during astrogliosis and while tight-junction proteins are restored to paracellular domains.
Pharmacological Modulation of Glia for the Therapy of Disease
The studies reviewed demonstrate the critical role that astrocytes play in regulating BBB integrity. We have shown that selective loss of astrocytes induced changes in tight-junction protein expression and BBB permeability and triggered a cascade of events involving an inflammatory response with microglia activation and infiltration. In addition, during the regenerative phase of our study, we saw the return of tight-junction proteins and a reactive gliosis with increased expression of GFAP-positive astrocytes and reactive microglia. Gliosis is a characteristic feature of CNS inflammation that has been demonstrated in many neurodegenerative diseases, including vascular dementia (Ellison et al. 2003), and in conditions with chronic inflammatory pain, such as rheumatoid arthritis and active multiple sclerosis. Reactive astrogliosis is a defensive brain reaction aimed at (1) isolating the damaged area from the rest of the CNS tissue, (2) reconstruction of the BBB, and (3) facilitation of the remodeling of brain circuits in areas surrounding the lesioned region (Pekny and Nilsson 2005). Activated astrocytes and microglia show increased production of pro-inflammatory mediators including cytokines (IL-1, IL-6, TNF-α and interferon-γ), growth factors (TGF-β and VEGF), chemokines, reactive oxygen species (including O2*), and nitric oxide (Abbott et al. 2006; Dohgu et al. 2004; Dong and Benveniste 2001; Willis and Davis 2008), all of which may modulate BBB integrity and lead to a decreased expression or altered localization of tight-junction proteins at endothelial paracellular domains (Didier et al. 2003; Hansson and Ronnback 2003; Ibuki et al. 2003). In addition, astrocytes, leukocytes, and macrophages express activated matrix metalloproteinases, which leads to amplification of extracellular matrix and BBB damage (Dzwonek et al. 2004). Therefore, one therapeutic approach is to direct pharmacological interventions at reducing the release of pro-inflammatory mediators in the neural parenchyma. Modulating glial activation is thought to improve CNS repair/regeneration and represents a novel approach for controlling these diseases. Some of these glial based strategies are shown in Figure 7 and are discussed below.
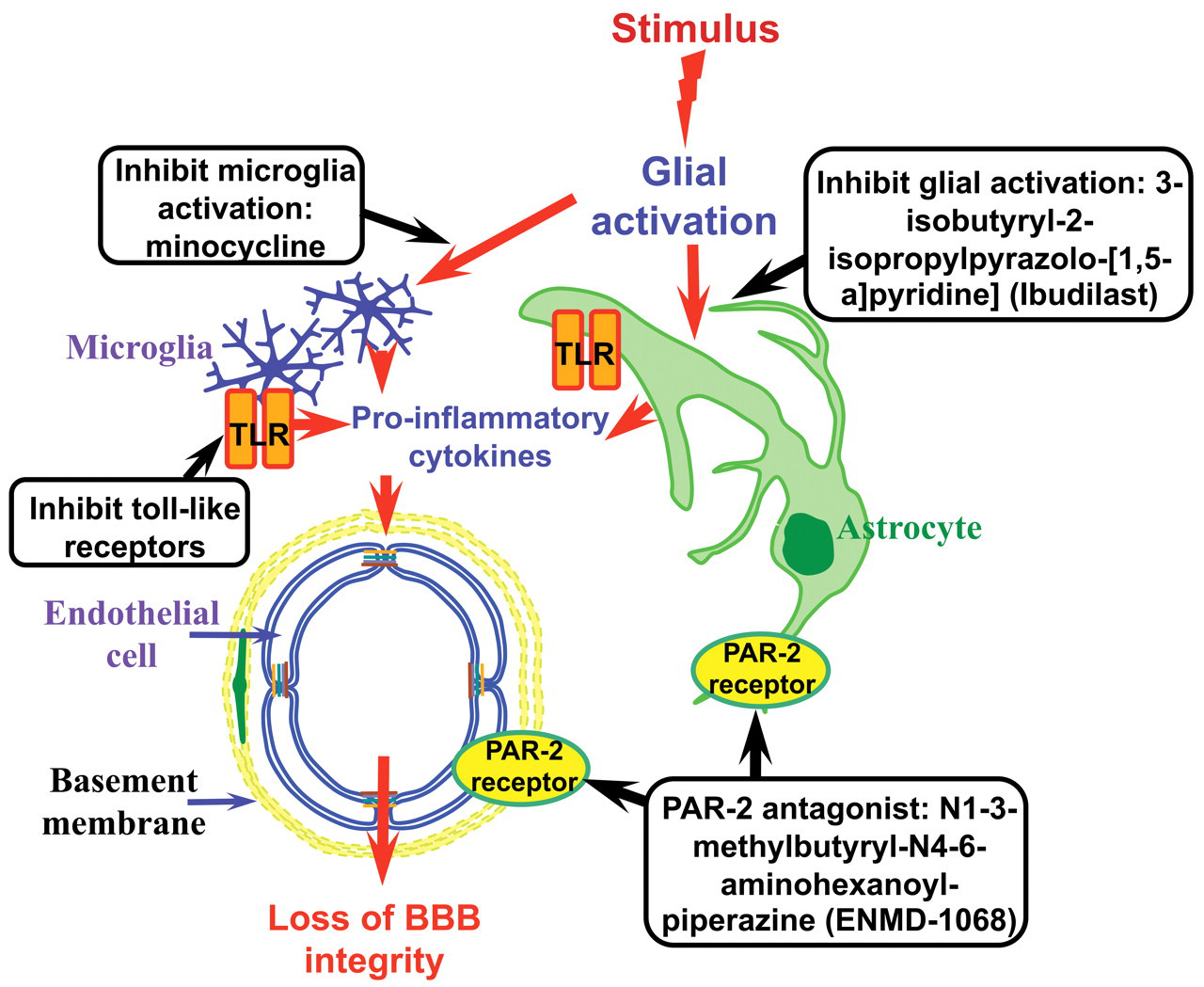
Schematic representation of potential pharmacological modulation of glia. Gliosis is a characteristic feature of central nervous system inflammation that shows increased production of pro-inflammatory mediators, which may lead to modified tight-junction protein expression and changes in blood–brain barrier integrity. Glia are now emerging as potential drug targets in the treatment of conditions in which chronic inflammation is a feature. Inhibition of glial activation with ibudilast results in decreased pro-inflammatory cytokine levels in multiple sclerosis patients. Minocycline inhibition of microglia activation reduces blood–brain barrier disruption after experimental stroke. Inhibition of proteinase-activated receptors with N1-3-methylbutyryl-N4-6-aminohexanoyl-piperazine has been shown to attenuate joint inflammation. Toll-like receptors on astrocytes and microglia are considered profound contributors to chronic pain. Figure modified from original (Willis and Davis 2008). Reprinted with permission from Bentham Science Publishers Ltd.
Ibudilast (AV411 [3-isobutyryl-2-isopropylpyrazolo-[1,5-a]pyridine]) has been found to result in decreased production of pro-inflammatory cytokines (IL-1β, TNF-α, IL-6), increased production of the anti-inflammatory cytokine IL-10, and up-regulation of the release of neurotrophic factors (Mizuno et al. 2004; Suzumura et al. 1999). Ibudilast may be effective as a neuroprotective and anti-dementia agent (Mizuno et al. 2004) and as a potential therapy for multiple sclerosis (Feng et al. 2004). In multiple sclerosis, patients show increased pro-inflammatory cytokine levels with reduced anti-inflammatory cytokine values. Clinical studies have shown the anti-inflammatory actions of ibudilast to reduce pro-inflammatory cytokine levels in multiple sclerosis patients (Feng et al. 2004; Kishi et al. 2001). Although no human clinical trials have yet been performed, one may speculate that the loss of BBB integrity seen under inflammatory conditions may be attenuated by ibudilast through its glial modulation properties and altered cytokine levels.
Minocycline is a tetracycline with anti-inflammatory properties that inhibits microglial activation. Microglia are among the first cells to respond to brain injury (Stoll and Jander 1999). Blocking microglial activation may limit BBB disruption and reduce edema and loss of BBB integrity. Minocycline has been shown to protect cultured neurons from excitotoxic insults by preventing microglial generation of glutamate, IL-1β and NO (Tikka et al. 2001). In vivo, minocycline has been shown to reduce BBB disruption after experimental stroke (Yenari et al. 2006). In vitro, inhibition of microglial activation by minocycline limited ischemia-like damage and reduced the release of superoxide after oxygen glucose deprivation, but it did not prevent tumor necrosis factor-α release by microglia (Yenari et al. 2006). Thus, it appears that activated microglia potentiate BBB damage under ischemic and inflammatory pain conditions and that this damage can be attenuated by inhibiting microglial activation and generation of superoxide. Therefore, targeting microglia with minocycline may represent a novel treatment of diseases in which CNS inflammation is a feature (Figure 7).
Proteinase-activated receptor (PAR) antagonists may quench some pro-inflammatory activities of CNS glia. Astrocytes, microglia, and neurons have been shown to express PAR-2 (Bushell et al. 2006; Noorbakhsh et al. 2006). Proteinase-activated receptor-2 has been implicated in inflammatory responses, and actions include increased vascular permeability, leukocyte infiltration, and smooth muscle relaxation (Kawabata et al. 1998; Vergnolle et al. 1999). A critical pro-inflammatory role for PAR-2 has been shown in animal models of rheumatoid arthritis. Administration of specific PAR-2 agonists induced joint swelling and hyperemia, whereas in other studies, chronic joint inflammation was attenuated in PAR-2 knockout mice (Ferrell et al. 2003). Intra-articular carrageenan/kaolin injection in mice resulted in joint swelling that was associated with synovial PAR-2 up-regulation. A novel small-molecule PAR-2 antagonist, N1-3-methylbutyryl-N4-6-aminohexanoyl-piperazine (ENMD-1068), has been shown to dose-dependently attenuate joint inflammation (Kelso et al. 2006). Therefore, targeting the PAR-2 receptor on glial cells may represent an exciting new target to prevent changes in the integrity of the BBB in patients suffering chronic inflammatory pain from diseases such as multiple sclerosis and rheumatoid arthritis (Figure 7).
Toll-like receptors (TLR) are widely expressed in the human CNS, particularly by microglial cells and astrocytes (Kielian 2006). Targeting these receptors has emerged as a promising goal for therapeutic control of chronic pain. Toll-like receptor ligands induce thermal and/or mechanical hyperalgesia in rats and elicit microglial activation and pro-inflammatory cytokine production (Guo and Schluesener 2006; Raghavendra et al. 2004). Therefore, microglial cells and TLR are considered profound contributors to chronic pain and present a target in the development of novel therapeutic strategies in the prevention and reduction of persistent pain (Figure 7) (reviewed by Guo and Schluesener 2007).
Conclusions and Future Directions
The studies presented in this review demonstrate a critical role for glia in regulating the integrity of the BBB. Loss of astrocytes results in loss of tight-junction protein expression at paracellular domains and increased vascular permeability to markers ranging from 0.5 to 300 kDa. However, we propose that modification of the neurovascular unit may impart some degree of barrier integrity to solutes >10 kDa, as we demonstrated in the AP, before the return of tight-junction proteins. We have also shown a reactive gliosis with activated astrocytes and microglia, which repopulate the inferior colliculus and reestablish tight-junction proteins at paracellular domains and restore BBB integrity.
Activated astrocytes and microglia have been shown to increase production of pro-inflammatory cytokines. Thus, we are now in a position to study the mechanism(s) of astrocyte regulation of tight-junction protein expression and BBB integrity in vivo during both loss of astrocytes and under pro-inflammatory conditions. In fact, preliminary studies have shown increased expression of the pro-inflammatory cytokine tumor necrosis factor-α. Pharmacological modulation of astrogliosis and microglia activation, as suggested earlier (Figure 7) in this in vivo model, will not only allow us to study intercellular mechanisms regulating BBB integrity but will enable us to investigate intracellular signaling mechanisms mediating post-translational modification of tight-junction proteins and assess the resulting effect on BBB integrity. In a recent study, we suggested a role for protein kinase C isozymes in modifying the phosphorylation state of tight-junction proteins, thus affecting the BBB integrity (Willis et al. 2010). These studies will provide greater understanding of the mechanisms of BBB regulation and lead to the discovery and development of innovative therapies for modulating the severity of various neurodegenerative diseases in which gliosis and inflammation are central components.
Footnotes
Acknowledgments
The work was supported by a Medical Research Council (UK) Program grant and the American Heart Association (SDG2170105). The author would like to thank Dr. David E. Ray for his support and mentorship and Dr. Beth Mahler (NIEHS/NIH) for her assistance in preparation of the figures.