
Abstract
Exposure to moderately selective p38α mitogen-activated protein kinase (MAPK) inhibitors in the Beagle dog results in an acute toxicity consisting of mild clinical signs (decreased activity, diarrhea, and fever), lymphoid necrosis and depletion in the gut-associated lymphoid tissue (GALT), mesenteric lymph nodes and spleen, and linear colonic and cecal mucosal hemorrhages. Lymphocyte apoptosis and necrosis in the GALT is the earliest and most prominent histopathologic change observed, followed temporally by neutrophilic infiltration and acute inflammation of the lymph nodes and spleen and multifocal mucosal epithelial necrosis and linear hemorrhages in the colon and cecum. These effects are not observed in the mouse, rat, or cynomolgus monkey. To further characterize the acute toxicity in the dog, a series of in vivo, in vitro, and immunohistochemical studies were conducted to determine the relationship between the lymphoid and gastrointestinal (GI) toxicity and p38 MAPK inhibition. Results of these studies demonstrate a direct correlation between p38α MAPK inhibition and the acute lymphoid and gastrointestinal toxicity in the dog. Similar effects were observed following exposure to inhibitors of MAPK-activated protein kinase-2 (MK2), further implicating the role of p38α MAPK signaling pathway inhibition in these effects. Based on these findings, the authors conclude that p38α MAPK inhibition results in acute lymphoid and GI toxicity in the dog and is unique among the species evaluated in these studies.
Introduction
p38 MAP kinases are a subfamily of the mitogen-activated protein kinases (MAPKs) and consist of multiple isoforms that differ in cell and tissue distribution and likely in substrate specificity (Obata, Brown, and Yaffe 2000; Kyriakis and Avruch 1996a, 1996b). Four p38 isoenzymes (α, β, γ, δ) are expressed in rodents and humans. p38α and β are broadly expressed while γ is expressed mainly in skeletal muscle and δ in endocrine/exocrine tissues (Wang et al. 1997; Hu et al. 1999). p38α is the predominant isoform expressed in monocytes, macrophages, and fibroblasts in which it regulates the expression of tumor necrosis factor-alpha (TNFα) and interleukin-1 (IL-1) in response to inflammatory and stress stimuli (Badger et al. 2000; Hale et al. 1999) via phosphorylation and activation of one of its immediate downstream effectors, MAPK-activated protein kinase-2 (MK2; Gaestel 2006). Inhibitors of p38α kinase are currently being investigated as potential new therapies for arthritis and chronic inflammatory diseases involving TNFα and IL-1 as mediators (Henry et al. 1998; Wadsworth et al. 1999; Lee et al. 2000; Hill et al. 2008).
During exploratory toxicology studies with moderately selective p38α MAPK inhibitors (30- to 40-fold and ≥100-fold selective against p38β MAPK and a broad panel of serine/threonine and tyrosine kinases, respectively), an acute toxicity was observed in the Beagle dog characterized by mild clinical signs (decreased activity, diarrhea, and fever), lymphoid necrosis and depletion with neutrophilic infiltration in the large intestine, mesenteric lymph nodes and spleen, linear colonic and cecal mucosal hemorrhages, and mild changes in hematologic parameters (neutrophilia, lymphopenia, eosinopenia, thrombocytopenia). These effects occurred at exposures equal to or lower than exposures necessary to produce a full efficacious response in a rodent model of rheumatoid arthritis, the mouse collagen-induced arthritis model (mCIA). In a similar series of exploratory acute and repeat-dose studies conducted in the mouse, rat, and cynomolgus monkey, in which large multiples of an efficacious plasma exposure in the mCIA model were achieved, the acute lymphoid and gastrointestinal (GI) toxicity was not observed. In addition, exposure to selective MK2 inhibitors in the dog also resulted in an acute toxicity similar to that observed for p38α MAPK inhibitors, further implicating the role of p38α MAPK signaling pathway inhibition in the acute canine toxicity.
This report describes a series of studies that were conducted to characterize the relationship between p38α MAPK inhibition and the acute lymphoid and GI toxicity in the dog and the species specificity for these effects.
Materials and Methods
Test Compounds
The p38 MAPK and MK2 inhibitor compounds used in these studies are proprietary Pfizer compounds. Arbitrary identifiers have been used to denote specific compounds used in the various in vitro and in vivo studies. IC50 values for p38α, p38β, and MK2 enzyme inhibition are provided in Tables 1 and Figure 3 for the p38 and MK2 inhibitors, respectively. Formulations and compound administrations are described in the in vivo and in vitro study sections that follow.
In Vitro and In Vivo Effects of Moderately Selective p38α MAPK Inhibitors on Dog and Rat B-Lymphocyte Proliferation
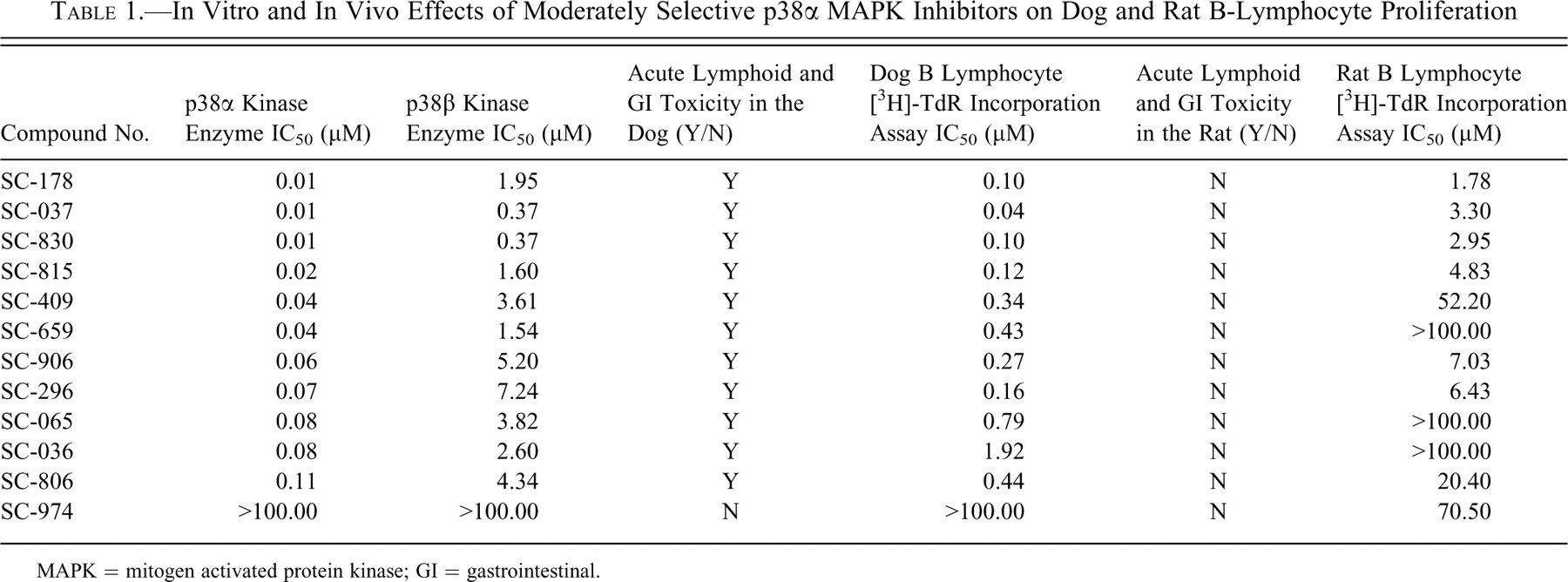
MAPK = mitogen activated protein kinase; GI = gastrointestinal.
In Vivo Toxicology Studies
The Pfizer Institutional Animal Care and Use Committee reviewed and approved the animal use in these studies. The Association for Assessment and Accreditation of Laboratory Animal Care, International, fully accredits the Pfizer animal care and use program.
Acute and 7-Day Repeat-Dose Exploratory Toxicology Studies in the Beagle Dog
Male or female Beagle dogs (Marshall Farms, North Rose, NY, USA), approximately 6 to 12 months of age and weighing between 6 and 12 kg, were randomly assigned to dose groups based on acceptable age and body weight. Animals were housed individually in stainless-steel cages. The temperature was maintained at 72 ± 5°F (22 ± 3°C) and was monitored. The humidity was set to maintain at 30% to 70% and was monitored. An approximate 12-hour light and 12-hour dark cycle was maintained. At least 400 g of Purina Certified Dog Chow 5004 was available for at least 4 hours per day. Water from the municipal source was available ad libitum. The in-life, pharmacokinetic, and pathology portions of these studies were performed at Pfizer Inc., St. Louis, Missouri, and Skokie, Illinois, facilities. Test articles were prepared daily and administered to dogs orally as neat chemical in gelatin capsules. For acute toxicity studies, q.d. doses of between 30 and 300 mg/kg (75 mg/kg was most commonly used) were administered to dogs (n = 1/dose group) at approximately 0700 hours for 1 day. For the 7-day repeat-dose toxicity study, q.d. doses of 0, 2, and 5 mg/kg were administered to dogs (n = 3/dose group) for 7 consecutive days at approximately 0700 hours each day. Animals were evaluated through 4 hours postdose, hourly from 4 to 8 hours postdose, and at 24 hours postdose for clinical observations. Rectal body temperature was monitored in conjunction with each daily dose and at approximately 8 hours postdose. For repeat-dose toxicity studies, body weights were measured pretreatment, on study days 3 and 6, and prior to necropsy. Blood samples for pharmacokinetic analysis were collected at 0.5, 1, 3, 8, and 24 hours postdose on day 1 and day 7 for repeat-dose studies. Blood was also collected for hematologic (acute and repeat-dose studies) and clinical chemistry (repeat-dose studies only) evaluation at the time of scheduled necropsy on day 2 or day 8 for acute and repeat-dose toxicity studies, respectively. In acute toxicity studies, stomach, duodenum, jejunum, ileum, cecum, colon, mesenteric lymph nodes, kidneys, liver, spleen, thymus, and any tissues with grossly observed lesions were collected and processed for histopathologic evaluation. For 7-day repeat-dose studies, adrenal glands, bone (sternum), heart, small and large intestine, kidneys, liver, lung, mesenteric lymph nodes, pancreas, skeletal muscle, spleen, stomach, testes, thymus, thyroid glands, and any tissues with grossly observed lesions were collected for histopathologic evaluation. In some studies, a section of the spleen, mesenteric lymph node, and colon was also collected for immunohistochemical evaluation. Plasma compound concentrations were determined using LC-MS/MS analysis and kinetic parameters calculated using Watson v6.4.0.04. A 24-hour time course, onset and progression, study was conducted in the dog following an acute single-dose administration of a p38 MAPK inhibitor (SC-806), where all parameters noted above for acute toxicity studies were evaluated at 6, 12, and 24 hours postdose. This study was intended to determine the temporal relationship between the lymphoid and GI effects observed in this species. A 2-week reversal study was also conducted following an acute single high dose of a p38 MAPK inhibitor to determine the reversibility of the lymphoid and GI toxicities. At the end of the 2-week observation period, all parameters noted above for acute toxicity studies were evaluated.
Acute and 7-Day Repeat-Dose Toxicology Studies in Male Sprague-Dawley (SD) Rats and C57BL6 Mice
Male rats (Charles River Laboratories, Raleigh, NC, USA), approximately 6 to 8 weeks of age and weighing between 175 and 250 g, and male mice, approximately 6 to 8 weeks of age and weighing between 20 and 40 g, were randomly assigned to dose groups based on acceptable body weights. Rats were housed individually in suspended, stainless-steel, wire-mesh cages. Mice were housed individually in polycarbonate, solid-bottom cages. The temperature in the animal room was set to maintain 72 ± 5°F(22 ± 3°C) and was monitored. The humidity was set to maintain 30% to 70% and was monitored. An approximate 12-hour light and 12-hour dark cycle was maintained. Purina Certified Rodent Chow 5002 (pellets) was available ad libitum while on study. Water from the municipal source was available ad libitum. For acute toxicity studies, animals were fasted overnight prior to dosing. Animals were fasted overnight prior to necropsy on day 8 for repeat-dose toxicity studies. The in-life, pharmacokinetic, and pathology portions of these studies were performed at Pfizer Inc., St. Louis, Missouri, and Skokie, Illinois, facilities. Suspensions of the test article in 0.5% methylcellulose (w/v) with 0.1% polysorbate 80 (v/v) were prepared on the day of dosing at concentrations ranging from 10 to 100 mg/mL. All doses were administered by oral gavage at a volume of 10 mL/kg of body weight. For acute range-finding toxicity studies, q.d. doses of 100, 300, or 1000 mg/kg were administered (n = 3/dose group) at approximately 0700 hours for 1 day. For repeat-dose toxicity studies, q.d. doses of 30, 100, or 300 mg/kg were administered (n = 5/dose group) for 7 consecutive days at approximately 0700 hours each day. Animals were evaluated hourly through 8 hours postdose and at 24 hours postdose for clinical observations and body temperature on acute toxicity studies. For repeat-dose toxicity studies, clinical observations and body temperature (rats only) were performed twice daily. Body temperature was monitored using a Biomedic transponder (Biomedic Data Systems, Seaford, DE, USA) implanted subcutaneously in the subcapsular region of the back. Blood samples for pharmacokinetic analysis were collected at 0.5, 1, 3, 8, and 24 hours postdose on day 1 or day 7 for acute and repeat-dose studies, respectively. Urine was collected (rats only) over wet ice from the time of last dose through the 24-hour blood collection time point. For acute toxicity studies, the stomach, ileum, cecum, colon, mesenteric lymph node, spleen, thymus, and any tissues with grossly observed lesions were collected for histopathologic evaluation. Blood was collected at the time of necropsy for clinical pathology determinations. For repeat-dose toxicity studies, a comprehensive list of tissues was collected and processed for histopathology evaluation. Plasma compound concentrations were determined using LC-MS/MS analysis, and kinetic parameters were calculated using Watson v6.4.0.04.
Acute Escalating Dose and 3-Day Repeat-Dose Exploratory Toxicology Studies in the Cynomolgus Monkey
Acute escalating dose and 3-day repeat-dose toxicology studies were conducted in the cynomolgus monkey. Test articles in 0.5% methylcellulose (w/v) with 0.1% polysorbate 80 (v/v) were prepared daily and administered via oral gavage to nasogastric intubated animals. For the acute escalating dose toxicity study, q.d. doses of 30, 60, 120, and 240 mg/kg were administered to a single male and a single female monkey at approximately 0700 hours on each day of dosing. Each dose was separated by a 3- to 4-day recovery period. For the 3-day repeat-dose toxicity study, b.i.d. doses of 120 mg/kg were administered to 2 male monkeys for 3 consecutive days at approximately 0700 and 1900 hours each day. The in-life, pharmacokinetic, and pathology portions of these studies were performed at Pfizer Inc., St. Louis, Missouri, and Skokie, Illinois, facilities. Animals were evaluated pretreatment, twice daily postdosing, and prior to necropsy for clinical observations. Rectal body temperature was monitored in conjunction with each daily dose. Body weights were measured pretreatment on days 1 and 3 and at the time of necropsy, day 4. Blood samples for pharmacokinetic analysis were collected. For the acute toxicity study, blood was collected 1, 3, 8, and 12 hours postdosing. For the repeat-dose study, blood was collected on day 3 at 0 (predose), 1, 3, 8, and 12 hours following the first dose and at 1, 3, and 12 hours following the second dose. Blood was also collected for hematology and clinical chemistry evaluation in both the acute and repeat-dose toxicity studies. For the acute toxicity study, blood was collected pretreatment on day 1 and at 24 hours postdosing on day 2. For the repeat-dose toxicity study, blood was collected pretreatment on day 1 and prior to scheduled necropsy on day 4. Coagulation parameters were assessed only in the repeat-dose study. Urine for pharmacokinetic analysis was also collected from repeat-dose study animals at the time of scheduled necropsy. Urine was collected directly from the urinary bladder via syringe and needle. In the acute toxicity study, animals were not sacrificed and only clinical observations and clinical pathology were assessed. For the 3-day repeat-dose study, all major organ systems were evaluated histologically, including adrenal glands, bone (sternum), heart, small and large intestine, kidneys, liver, lung, mesenteric lymph nodes, pancreas, skeletal muscle, spleen, stomach, testes, thymus, and thyroid glands. Any tissues with grossly observed lesions were also collected and evaluated. Plasma compound concentrations were determined using LC-MS/MS analysis, and kinetic parameters were calculated using Watson v6.4.0.04.
Immunohistochemistry and Hematoxylin/Eosin Staining
General Methods
Ten percent buffered formalin-fixed tissue sections (4-μm thick) were placed onto glass slides, deparaffinized in xylene, and rehydrated through graded alcohol solutions. The tissues were incubated with 3% hydrogen peroxide to quench endogenous peroxidase activity. With the exception of the p38α immunohistochemistry assay, antigen retrieval was accomplished for all tissues in citrate buffer solution (Dako, Carpinteria, CA, USA) using a heat-induced epitope retrieval method. The tissues were labeled using established immunohistochemical methods at room temperature (Warnke and Levy 1980; Whiteland et al. 1995).
B Lymphocyte (CD79a) Immunohistochemistry
B cells of rat, dog, monkey, and human were identified in formalin-fixed lymphoid tissue using a monoclonal (mouse) anti-hCD79a antibody at a 1:50 dilution (no. M7051; Dako). Negative control slides were incubated with equal concentrations of isotype-matched mIgG (no. X0931; Dako). Primary antibody was detected using a biotinylated secondary antibody and the Dako LSAB (labeled streptavidin-biotin) kit and diaminobenzidine (DAB) chromogen substrate.
T Lymphocyte (CD3) Immunohistochemistry
T cells of rat, dog, monkey, and human were identified in formalin-fixed lymphoid tissue using a polyclonal (rabbit) anti-hCD3 antibody (no. A0452; Dako) at a 1:50 dilution. Negative control slides were incubated with equal concentrations of rabbit polyclonal immunoglobulin (no. X0903; Dako). Primary antibody was detected using a goat antirabbit biotinylated secondary antibody and avidin-biotin complex labeling reagent (Vector Labs, Burlingame, CA, USA) and DAB chromogen substrate.
Macrophage (Lysozyme) Immunohistochemistry
Tissue macrophages of rat, dog, monkey, and human were detected using a polyclonal (rabbit) antihuman lysozyme antibody (no. A0099; Dako) at a 1:150 dilution. Negative control slides were incubated with equal concentrations of rabbit polyclonal immunoglobulin (no. X0903; Dako). Primary antibody was detected using a goat antirabbit biotinylated secondary antibody and avidin-biotin complex labeling reagent (Vector Labs) and DAB chromogen substrate.
p38α MAPK Immunohistochemistry
p38α MAPK of rat, dog, monkey, and human was detected using a polyclonal goat anti-p38α antibody raised against a peptide mapping at the carboxy terminus of p38α of mouse origin at a 1:40 dilution (Santa Cruz Biotechnology, Inc., Santa Cruz, CA, USA). The carboxy terminus is a highly conserved region of the p38α kinase across species and is identical to the corresponding human sequence. Specificity of the antibody for p38α was confirmed by Western immunoblotting analysis using p38α, β, γ, and δ kinases as substrate. The ability of the antibody to detect dog, rat, monkey, and human p38α MAPK by immunohistochemistry was verified by staining of macrophages in lymphoid tissues from each species. Negative control slides were incubated with equal concentrations of normal goat immunoglobulin. Prior to incubation with primary antibody, tissue sections were subjected to high-heat epitope retrieval (Bull’s Eye Decloaker; Biocare Medical LLC, Concord, CA, USA) in a pressure cooker followed by Fc receptor blocking (Innovex Biosciences, Richmond, CA, USA). Bound antibody was detected using a biotinylated rabbit antigoat secondary antibody, avidin-biotin complex labeling reagent (Vector Labs), and DAB chromogen substrate. Counterstaining with hematoxylin was carried out using standard methodologies.
Immunohistochemistry Competition Studies
Specificity of the immunohistochemical staining for p38α MAPK was determined in competition studies using a 50X excess (by molecular weight) of the purified p38α MAPK peptide used to generate the primary anti-p38α kinase polyclonal antibody. For competition assays, antibody and peptide were mixed overnight at 4°C before being applied to the tissue sections. All other parameters of the immunohistochemistry reaction were performed as described above.
Rat and Dog B-Lymphocyte Isolations
Splenic tissue from 14- to 16-week-old SD rats was dissociated, and suspensions were filtered through 70-μm mesh caps prior to red blood cell lysis with 0.16 M ammonium chloride. Cell pellets were washed in HBSS + 2% FBS/HEPES 2 times followed by density gradient centrifugation with Histopaque 1077 to remove dead and residual red cells. Mononuclear cells were harvested from the interface and washed as above prior to resuspending in serum supplemented RPMI for cell counts. Cell concentration was adjusted to 1 × 106/mL with 107 cells added per Petri dish. Dishes were incubated at 37°C, 5% CO2 for overnight adherence of macrophages. Nonadherent cells were harvested the following morning, centrifuged, and counted. Splenic B cells were isolated via negative selection using the Miltenyi AutoMACS system with biotinylated antidog or antirat CD3 antibodies and antibiotin MicroBeads. B cells were washed 2 times in phosphate-buffered saline without Ca or Mg and flash frozen on dry ice for Western blots. Lymphocyte purity was >98% as determined by Giemsa stained cytocentrifuge slides.
Western Blot Analysis of p38α MAPK Expression in Isolated Rat and Dog B Lymphocytes
Purified rat and dog B-lymphocyte pellets (2 × 105 cells) were homogenized in ice-cold RIPA lysis buffer containing a cocktail of protease inhibitors (Santa Cruz) and then centrifuged through a Qia Shredder (Qiagen). Denatured lysates were run on sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) gels and transferred to nitrocellulose membranes. The membranes were blocked for 2 hours at room temperature with Tris-buffered saline and 0.05% Tween 20. The blocked membranes were then incubated overnight at 4°C with our internally generated p38α-specific antibody (1:500 dilution) and the loading control antibody (α-tubulin, Abcam no. ab4074, 1:1,000). After four 5-minute washes, membranes were incubated for 1 hour at room temperature with a peroxidase-labeled secondary antibody (KPL no. 474-1516, 1:10,000). Immunodetection was performed using ECL+ (Amersham Biosciences, UK) for densitometry analysis of protein band intensities. The blots were then scanned on a Typhoon Trio Variable Mode Imager, and the intensity of each band was quantified using ImageQuant software (G.E. Healthcare). Band intensities were normalized against the α-tubulin (loading control), and the result was expressed as a percentage of the loading control. A duplicate blot using alkaline phosphatase detection was prepared for visual presentation of protein bands in Figure 5. Following overnight incubation with p38 and α-tubulin antibodies and four 5-minute washes, blots were incubated with a phosphatase-labeled antirabbit secondary antibody (KPL no. 4751-1506, 1:10,000 dilution). Immunodetection was performed using BCIP/NBT phosphatase substrate.
Mitogen-Stimulated [3H]-Thymidine Deoxyribose ([3H]-TdR) Incorporation Assay
Test Articles and Vehicles
Test articles were stored desiccated in well-closed, light-resistant containers at room temperature. Stock solutions of the compounds were dissolved in 100% ethanol or water at a concentration of 2 mM. Further dilutions of each test article were made in ethanol, water, or media to yield final test article concentrations of 0.1 to 10 μM. The final concentration of ethanol in all assays in which ethanol was used as a solvent did not exceed 0.25%. Vehicle controls included the same final concentration of solvent used in dissolving each test compound.
Preparation of Splenic Leukocytes
Spleens from normal male SD rats and C57BL6 mice (Charles River, Raleigh, NC, USA), approximately 8 to 12 weeks of age, and Beagle dogs (Marshall Farms, North Rose, NY, USA), approximately 6 to 12 months of age, were the source of rat, mouse, and canine splenic tissue. Nonhuman primate (cynomolgus and rhesus) splenic tissue was procured from Clonetics®/Biowhittaker (Walkersville, MD, USA) and Sierra Biomedical, Inc. (Sparks, NV, USA). Human splenic tissue was procured through the National Disease Research Interchange (Philadelphia, PA, USA). Human and nonhuman primate spleens were collected fresh into ViaSpan Cold Storage Solution (DuPont Pharma, Wilmington, DE, USA), placed in a sealed container on wet ice, and shipped by overnight courier. Single-cell suspensions of splenic leukocytes were prepared from rat, mouse, dog, human, and nonhuman primate spleens by physical disruption (mashing) of small pieces of tissue into Hank’s Buffered Salt Solution (HBSS; Gibco BRL, Grand Island, NY, USA). Leukocytes were isolated from the cell suspensions by centrifugation through Histopaque 1077 or Ficoll-Paque 1.077 (Sigma Chemicals, St. Louis, MO, USA; Pharmacia Biotech, Piscataway, NJ, USA), washed with HBSS, and resuspended at a concentration of 1 × 106 cells/mL in RPMI 1640 media supplemented with 5% fetal bovine serum, 2 mM L-glutamine, 10 mM HEPES, 100 U/mL penicillin/streptomycin (Gibco BRL, Grand Island, NY, USA), and 50 μM 2-mercaptoethanol (Sigma Chemicals).
Mitogen-Stimulated [3H]-Thymidine ([3H]-TdR) Incorporation Assay
Splenic leukocytes were cultured for 48 hours in 96-well microtiter plates in the presence of vehicle or 0.1 to 10 μM of each test article and either 10 μg/mL of lipopolysaccharide (LPS; K-235 strain; Sigma Chemicals) or 0.0005% Staphylococcus aureus, Cowan I strain (SAC; Boehringer Mannheim, Indianapolis, IN, USA) as the B-cell mitogen or 0.5 mg/mL Concanavalin-A (Con-A; Sigma Chemicals) as the T-cell mitogen. The strain of LPS was selected empirically based on the maximum stimulatory response of lymphocytes across species. Cells were pulsed with 0.5 μCi [3H]-thymidine (6.7 Ci/mmol; New England Nuclear, Boston, MA, USA) for the last 18 hours of the 48-hour culture period. Percentage inhibition of [3H]-TdR incorporation relative to vehicle control was calculated. IC50 values were calculated from linear regression analysis of semilogarithmic plots of test article concentration versus percentage inhibition of [3H]-TdR incorporation (DeltaGraph Software; DeltaPoint, Monterey, CA, USA). For canine lymphocyte assays in which multiple compounds were compared, IC50 values were normalized to the IC50 value of a positive control compound, SC-806. SC-806 was included in each assay run as a control for interassay variability. IC50 values for mouse, rat, human, and nonhuman primate were not normalized to an internal control compound response.
Results
Acute and Repeat-Dose Exploratory Toxicology Studies in the Dog
Acute, single-dose, oral toxicology studies performed in the Beagle dog with a number of p38 MAPK inhibitors, representing a diversity in physicochemical and pharmacological properties (n = 12; including pyrimidine, pyridine, and pyrazole scaffolds; basic and neutral compounds; selective and nonselective inhibitors; and compounds with a range of potencies), defined an acute toxicity that occurred with 100% incidence when efficacious exposures of potent, active inhibitors were achieved (Table 1). The toxicity observed at 24 hours postdose included mild clinical signs (decreased activity, diarrhea, and fever), linear colonic and cecal mucosal hemorrhages, lymphoid necrosis with neutrophilic infiltration in the large intestine, mesenteric lymph nodes and spleen, lymphoid depletion in the distal small intestine (Peyer patches), and mild changes in hematologic parameters (neutrophilia, lymphopenia, eosinopenia, thrombocytopenia; Figure 1). The neutrophilic infiltration and acute inflammatory response resulted in a grossly observed enlargement of the lymph nodes in these animals. It was further noted that lymphoid necrosis in the gut-associated lymphoid tissue (GALT) was not necessarily regionally associated with local injury to the mucosal epithelium and hemorrhage.
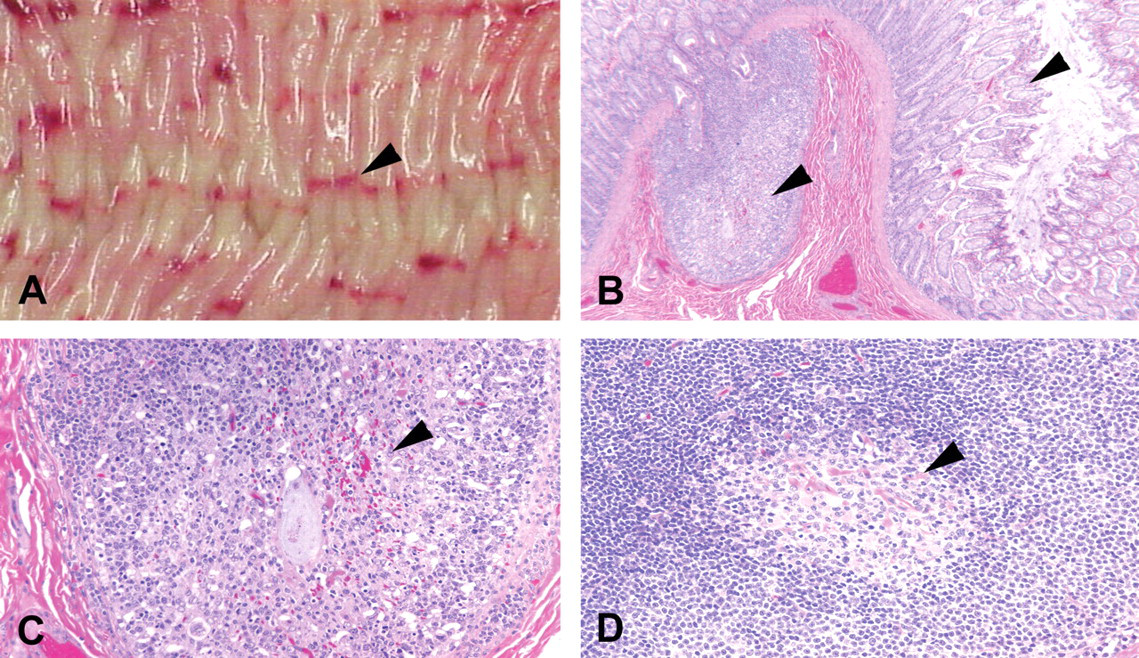
Linear colonic hemorrhages (A), lymphoid necrosis, and depletion in a lymphoid follicle of the colon and superficial hemorrhage in the lamina propria (B, C) and lymphoid depletion in the germinal center of a mesenteric lymph node (D) 24 hours following a single dose of a p38α mitogen-activated protein kinase inhibitor. Arrows indicate the linear hemorrhage observed grossly in the dog colon (A), epithelial necrosis and superficial mucosal hemorrhage and lymphoid depletion in the colon (B, C), and lymphoid depletion in the germinal center of a mesenteric lymph node (D). (B) Hematoxylin and eosin (H&E), 50× magnification. (C, D) H&E, 200× magnification.
Although several animals had clinical signs of toxicity, no deaths occurred with any of the compounds tested. A weakly active compound (SC-974), which was not efficacious in the mCIA model, did not produce the acute toxicity (Table 1).
In a 24-hour time course, onset and progression, study with a prototype p38α MAPK inhibitor, SC-806, microscopic changes were observed as early as 6 hours postdose and consisted of an increase in apoptotic lymphocytes (determined microscopically by single cell death with DNA fragmentation and chromatin condensation) within the lymphoid follicles of the GALT and mesenteric lymph nodes. By 12 hours, lymphoid necrosis was present in the germinal centers of GALT and mesenteric lymph nodes and increased apoptosis was present in peripheral lymphoid tissues (i.e., spleen). Also observed at the 12-hour time point were neutrophils marginating in the surrounding vasculature of the mesenteric lymph nodes and mild hematologic changes, including neutrophilia with a left shift (increased circulating immature neutrophils), lymphopenia, eosinopenia, and thrombocytopenia. At 24 hours postdose, all manifestations of the acute toxicity were present, including lymphoid depletion in gut-associated lymphoid follicles (large and small intestine), mesenteric lymph nodes and spleen, necrosis of the mucosal epithelium, and linear hemorrhage in the colon and cecum (Figure 1A-D). Other findings in these animals included mild, multifocal centrilobular hepatocellular necrosis and depletion of immature myeloid cells and hypertrophy of reticular stromal cells in the bone marrow. The liver and bone marrow effects were considered secondary to the acute inflammatory response observed in these animals. Based on clinical pathology assessments and GI histopathology, the acute thrombocytopenia was interpreted as being a consumptive thrombocytopenia secondary to the colonic hemorrhage in these animals.
Using immunohistochemistry, the location of the depleted lymphocyte population was found to align with B lymphocytes in the lymphoid follicles of mesenteric lymph nodes (Figure 2). This finding suggests that B lymphocytes are a primary acute toxicity target cell population in the dog. Mild fever that developed in these animals 24 hours postdose was considered secondary to a compromised mucosal barrier, which would allow entry of enteric bacteria and systemic exposure to endotoxin.
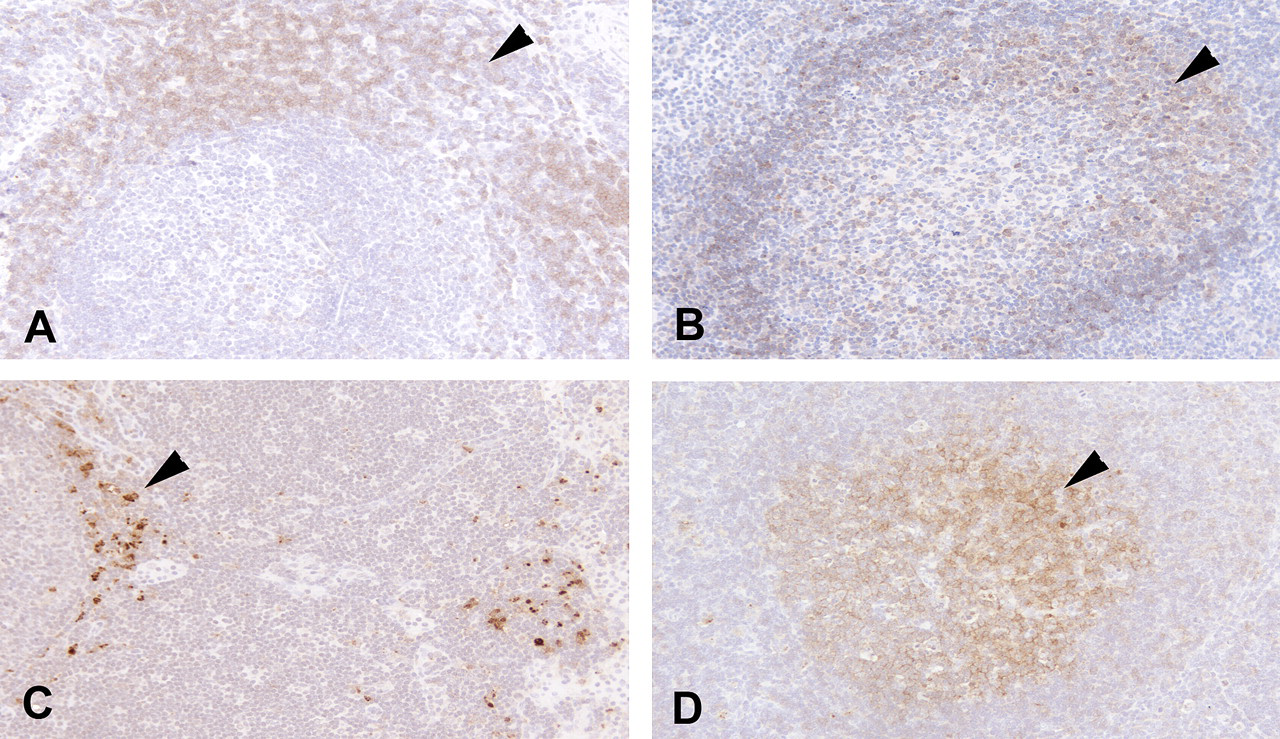
Immunohistochemical localization of (A) T lymphocytes, (B) B lymphocytes, (C) macrophages, and (D) p38α mitogen-activated protein kinase (MAPK) in a mesenteric lymph node from a normal, untreated, Beagle dog. Arrows indicate (A) anti-CD3 (T lymphocyte), (B) anti-CD79A (B lymphocyte), (C) anti-lysozyme (macrophages), and (D) anti-p38α MAPK expression. Magnification = 200×.
A 7-day exploratory repeat-dose toxicity study was also conducted with SC-036, at doses of 2 and 5 mg/kg/day, given orally (3 dogs/dose group). The purpose of this study was to define a no-observed-effect-level (NOEL) or low-observed-effect-level (LOEL) as well as an exposure multiple for the acute toxicity relative to an efficacious exposure for this compound in the mCIA model (ED80 AUC0-24 = 17 mg*h/mL). There were no clinical or pathological (gross or histopathology) findings observed in any animals at the 2-mg/kg dose level or in 2 of 3 animals at the 5-mg/kg dose level, where exposures ranged from 1- to 2-fold the ED80 AUC in the mCIA model. In 1 of 3 animals at the 5-mg/kg dose level, the acute toxicity was observed following a single dose. Results of this study demonstrated that the NOEL or LOEL for the acute toxicity with this compound is approximately equal to a fully efficacious exposure in the mCIA model. This finding further suggests a mechanism-based effect resulting from the inhibition of the primary pharmacological target enzyme, p38α MAPK. The full spectrum of the acute toxic effects of p38 MAPK inhibitors in the dog was found to be fully reversible within 2 weeks following an acute single-dose exposure to this compound.
Acute and Repeat-Dose Exploratory Toxicity Studies in the Rat and Mouse
Both acute and 7-day repeat-dose toxicity studies in rats were conducted with many of the same p38α MAPK inhibitors that were tested in the dog (Table 1). In these studies, plasma exposures representing large multiples (>30-fold) of an ED80 AUC exposure in the mCIA model were achieved. No clinical or histopathologic evidence of the canine acute lymphoid or GI toxicity was observed in the rat. The same result was observed in the mouse.
Acute and Repeat-Dose Exploratory Toxicity Studies in the Cynomolgus Monkey
An acute exploratory toxicity study was conducted in 1 male and 1 female cynomolgus monkey with SC-036. Monkeys were dosed orally with escalating doses of compound ranging from 30 to 240 mg/kg. Animals were monitored for 3 days prior to administration of the next escalating dose. In this study, plasma exposures representing an approximate 13-fold multiple of an ED80 AUC exposure in the mCIA model were achieved. No clinical or hematological evidence of the canine acute lymphoid or GI toxicity was observed in the cynomolgus monkey.
Following the acute single-dose study, a 3-day repeat-dose oral toxicity study was conducted with SC-036 in 2 male cynomolgus monkeys at a single-dose level of 120 mg/kg, b.i.d., by nasogastric gavage. In this study, plasma exposures representing an approximate 30-fold multiple of an ED80 AUC exposure in the mCIA model were achieved. No clinical, hematological, or histopathological evidence of the canine acute lymphoid or GI toxicity was observed in the cynomolgus monkey.
Expression of p38α MAPK in Lymphoid Tissues
Immunohistochemical staining of formalin-fixed, paraffin-embedded tissue, using a polyclonal anti-p38α MAPK antibody, provided qualitative evidence that p38α MAPK is more highly expressed in the germinal centers of secondary follicles in mesenteric lymph nodes of the Beagle dog than in rat, nonhuman primate, or human (Figure 3). Primary follicles were not found to have appreciable immunoreactivity in any of the species evaluated. In contrast, p38α MAPK expression in macrophages in the paracortex and in subcapsular and medullary sinuses of lymph nodes was of equal intensity across all species. A similar pattern of p38α MAPK labeling was observed in the spleen of these species (data not shown). In the rat, staining of megakaryocytes in the spleen was also observed, although no effects on megakaryocytes or platelets were observed in this species following the 7-day repeat-dose exposure to a number of p38α MAPK inhibitors. This pattern of staining suggests that while p38α MAPK is expressed at appreciable levels in macrophages of the species evaluated in this study, it is expressed at considerably higher levels in B lymphocytes of the dog.
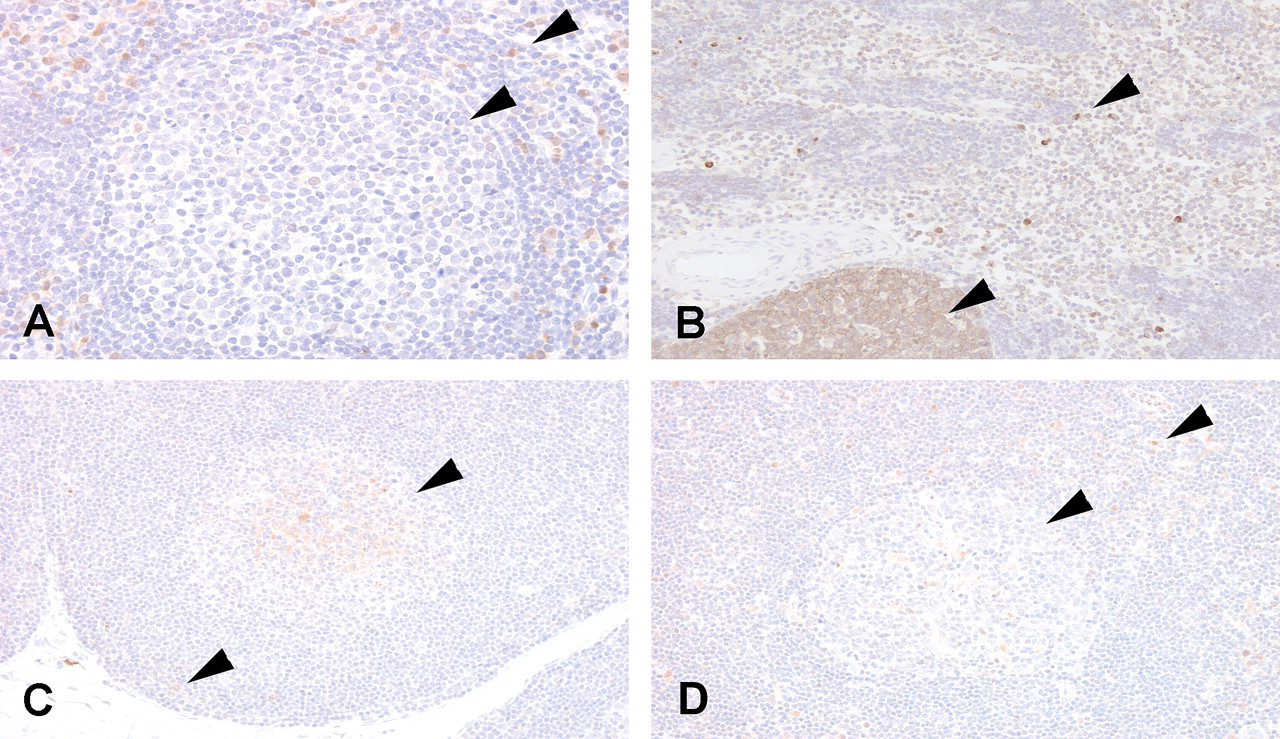
Immunohistochemical localization of p38α mitogen-activated protein kinase (MAPK) in mesenteric lymph nodes from (A) rat, (B) dog, (C) cynomolgus monkey, and (D) human. Arrows indicate anti-p38α MAPK labeling of both macrophages (paracortex and subcapsular and medullary sinuses) and germinal center B lymphocytes in the dog (B), but only macrophage labeling in the rat (A), monkey (C), and human (D). (A) 400× magnification; (B–D) 200× magnification.
Peptide Competition Experiments
To confirm the specificity of the anti-p38α MAPK antibody in tissues, peptide competition immunohistochemistry assays were conducted on lymphoid tissues (Figure 4 ). Preincubation of antibody with the p38α MAPK peptide used as immunogen to generate the antibody completely abrogated the reactivity that had previously been observed in lymphoid organs from rat, human, and monkey. In contrast, while modest residual nonspecific reactivity remained in medullary and paracortical macrophages, anti-p38α MAPK antibody reactivity was completely eliminated in the germinal centers of canine follicles after incubation with p38α peptide, confirming that the germinal center reactivity was specific for p38α.
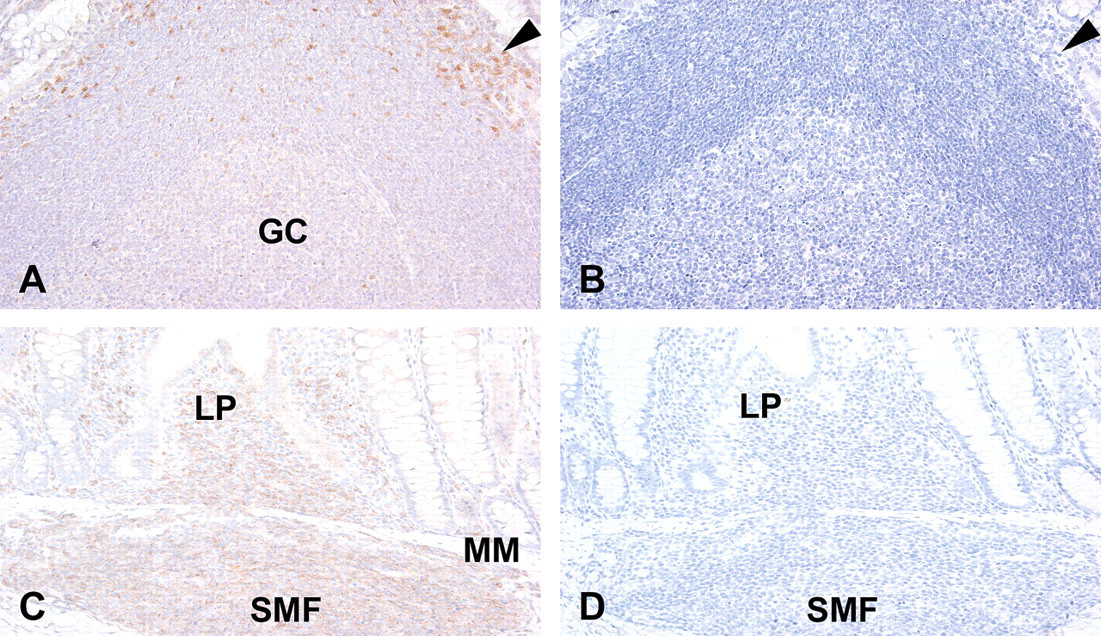
Immunohistochemical localization of p38α mitogen-activated protein kinase (MAPK) in gut-associated lymphoid tissue (GALT) from rat (A, B) and dog (C, D). p38α MAPK expression is observed in macrophages of rat GALT (A) and effectively competed using the p38α MAPK specific peptide (B) (arrows). p38α MAPK expression is observed in macrophages and lymphocytes of dog GALT (C) and effectively competed using the p38α MAPK-specific peptide (D). GC = germinal center, LP = lamina propria, MM = muscularis mucosa, SMF = submucosal lymphoid follicle. Magnification = 200×.
To further confirm the species difference in p38α kinase expression, purified splenic B lymphocytes (2 × 105 cells) from rat and dog were analyzed using SDS-PAGE and Western immunoblotting to determine the relative amount of p38α kinase expression when normalized to an internal housekeeping (control) protein, α-tubulin. As shown in Figure 5 , there is an overall 30% increase in expression of p38α kinase in the dog as compared to the rat. A higher degree of differential expression between rat and dog would not be expected in mixed B-lymphocyte populations given that the enhanced expression is observed in only secondary lymphoid follicles by immunohistochemistry in the dog and total p38α expression represents the average expression in B lymphocytes from primary and secondary follicles.
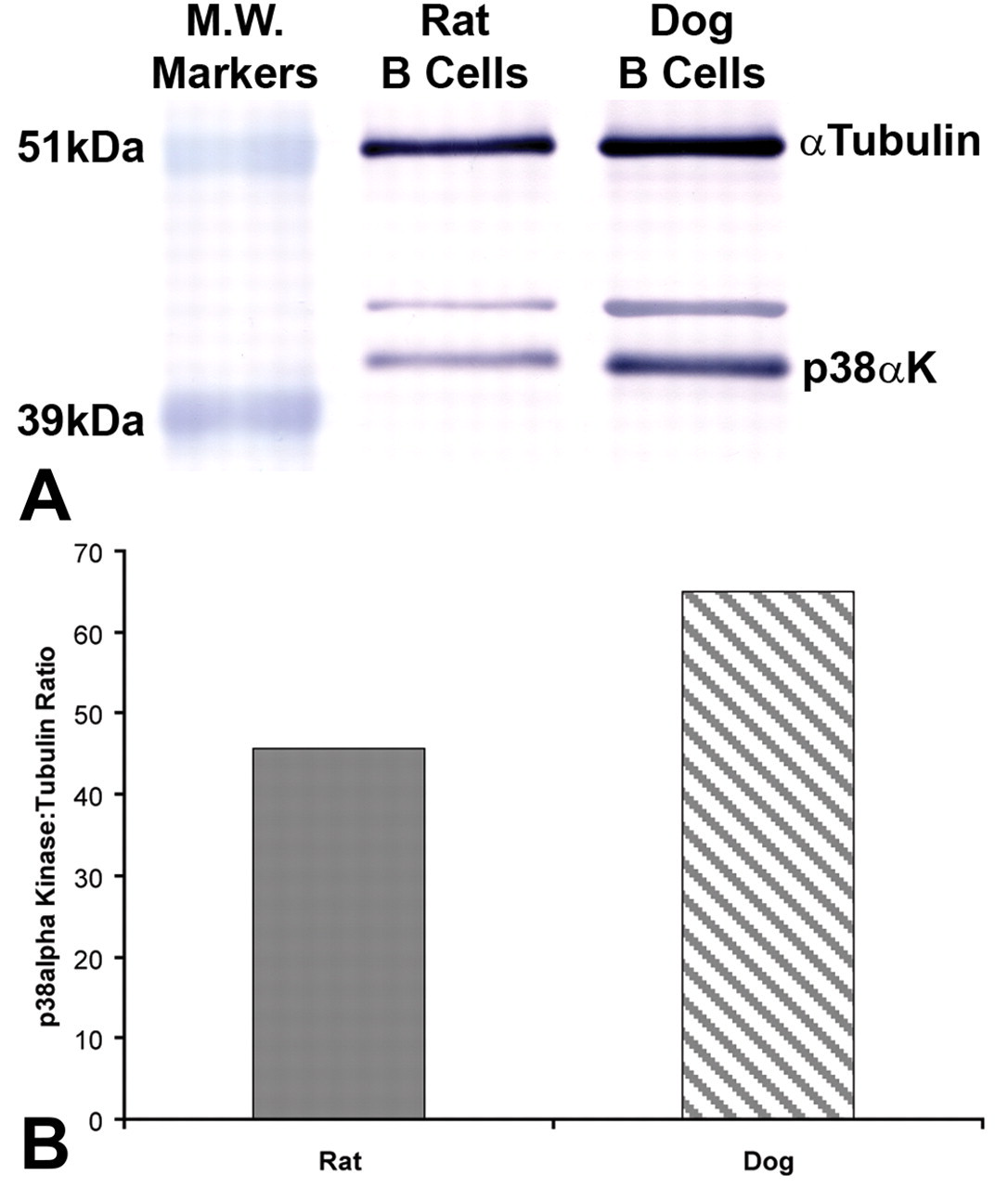
Expression of p38α mitogen-activated protein kinase in purified splenic B lymphocytes from rat and dog. Protein lysates from isolated rat and dog B lymphocytes were separated by sodium dodecyl sulfate polyacrylamide gel electrophoresis, and p38α kinase and α-tubulin were detected using p38α kinase and α-tubulin–specific antibodies and an alkaline phosphatase-labeled secondary antibody (A). Immunodetection was performed using alkaline phosphatase substrate. A duplicate blot using a peroxidase-labeled secondary antibody and ECL+ detection was used for quantitative image analysis and to determine band intensities. Data are presented as the ratio of p38α kinase to α-tublin band intensities for p38α expression in rat and dog B lymphocytes (B).
In Vitro Modeling Studies Using Primary Lymphocytes
The observation of a qualitative difference in expression of p38α MAPK in canine B lymphocytes detected by immunohistochemistry compared with human, rat, and nonhuman primate (relative to macrophage staining across all species), together with the occurrence of the lesion in dogs at plasma concentrations that are equal to a fully efficacious exposure in the mCIA model, suggests that the acute lymphoid toxicity in the dog is a consequence of p38α MAPK inhibition. To confirm this association and to further assess the relevance of the acute lymphocyte toxicity for humans, in vitro studies were conducted to determine the comparative pharmacology and relative species sensitivity for the effects of p38α MAPK inhibitors on B lymphocytes in vitro.
Initial studies were conducted to model the acute lymphoid toxicity in vitro to determine the relative species sensitivity for this effect. Primary canine splenic leukocytes were isolated to determine if mitogen-stimulated proliferation ([3H]-TdR incorporation) could be used as a surrogate endpoint for B-lymphocyte toxicity in the dog. Figure 6 illustrates that a selective p38α MAPK inhibitor, SC-036, inhibits the [3H]-TdR incorporation of LPS- and SAC-stimulated canine B lymphocytes, but not Con-A–stimulated T lymphocytes, in a concentration-dependent manner between 0.1 and 5 μM. Results of these in vitro studies demonstrated that inhibition of mitogen-stimulated [3H]-TdR incorporation (or cellular proliferation) could be used as a surrogate endpoint for modeling the selective effect of p38 MAPK inhibitors on canine B lymphocytes observed in vivo.
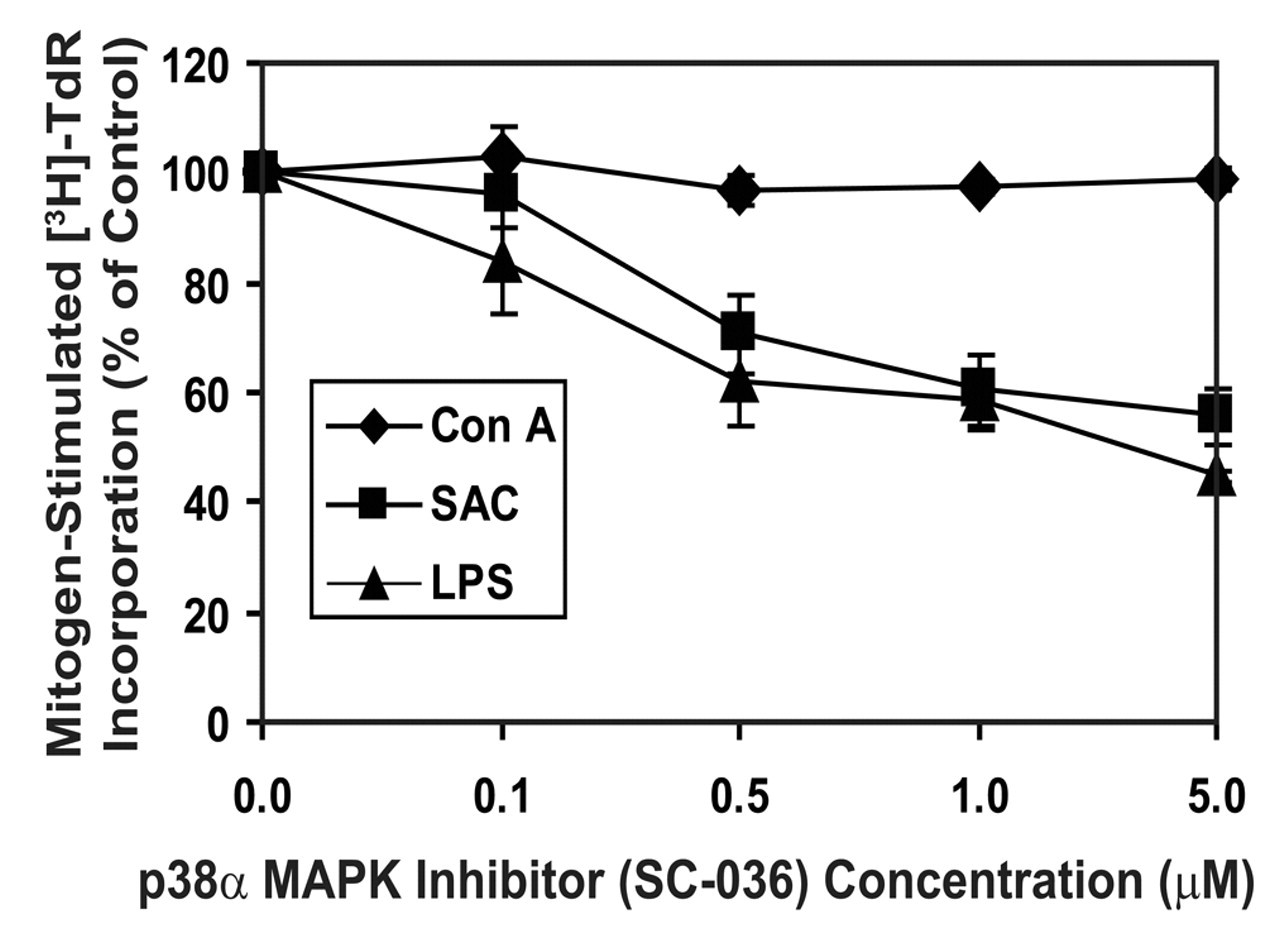
The effect of a selective p38α mitogen-activated protein kinase (MAPK) inhibitor (SC-036) on lipopolysaccharide (LPS)–, Staphylococcus aureus Cowan strain (SAC)–, and Con A–stimulated dog lymphocyte proliferation ([3H]-TdR incorporation). Primary dog splenic leukocytes were stimulated with either LPS, Con A, or SAC for 48 hours and pulsed with [3H]-TdR during the last 16 hours of culture. Results are presented as the percentage ± standard deviation of the vehicle control response and represent 1 of 3 separate experiments giving similar results. [3H]-TdR incorporation for the vehicle control, Con A, LPS, and SAC stimulation, in the absence of the inhibitor, were 806 ± 121, 58,255 ± 1791, 23,754 ± 1165, and 19,990 ± 1949 cpms, respectively.
Additional studies were conducted using the mitogen-stimulated [3H]-TdR incorporation assay to determine the relative species sensitivity and human relevance of the acute B-lymphocyte toxicity observed in the dog. In these studies, 157 p38 MAPK inhibitors, representing a diversity of physicochemical and pharmacological properties, were tested (Table 1; Figures 7 and 8). A subset of these compounds was then tested for effects on B lymphocytes from other species, including humans. A strong correlation coefficient of approximately 0.74 was obtained when IC50 values for inhibition of mitogen-stimulated dog B-lymphocyte [3H]-TdR incorporation were compared with IC50 values for inhibition of p38α MAPK activity (Table 1; Figure 7). In contrast, the correlation coefficient when compared with the IC50 values for inhibition of p38β MAPK activity was only 0.42 (Table 1; Figure 8). In addition, the weakly active p38α MAPK inhibitor, SC-974, which did not cause the acute toxicity in the dog, also did not inhibit canine B lymphocyte [3H]-TdR incorporation in vitro (Table 1).
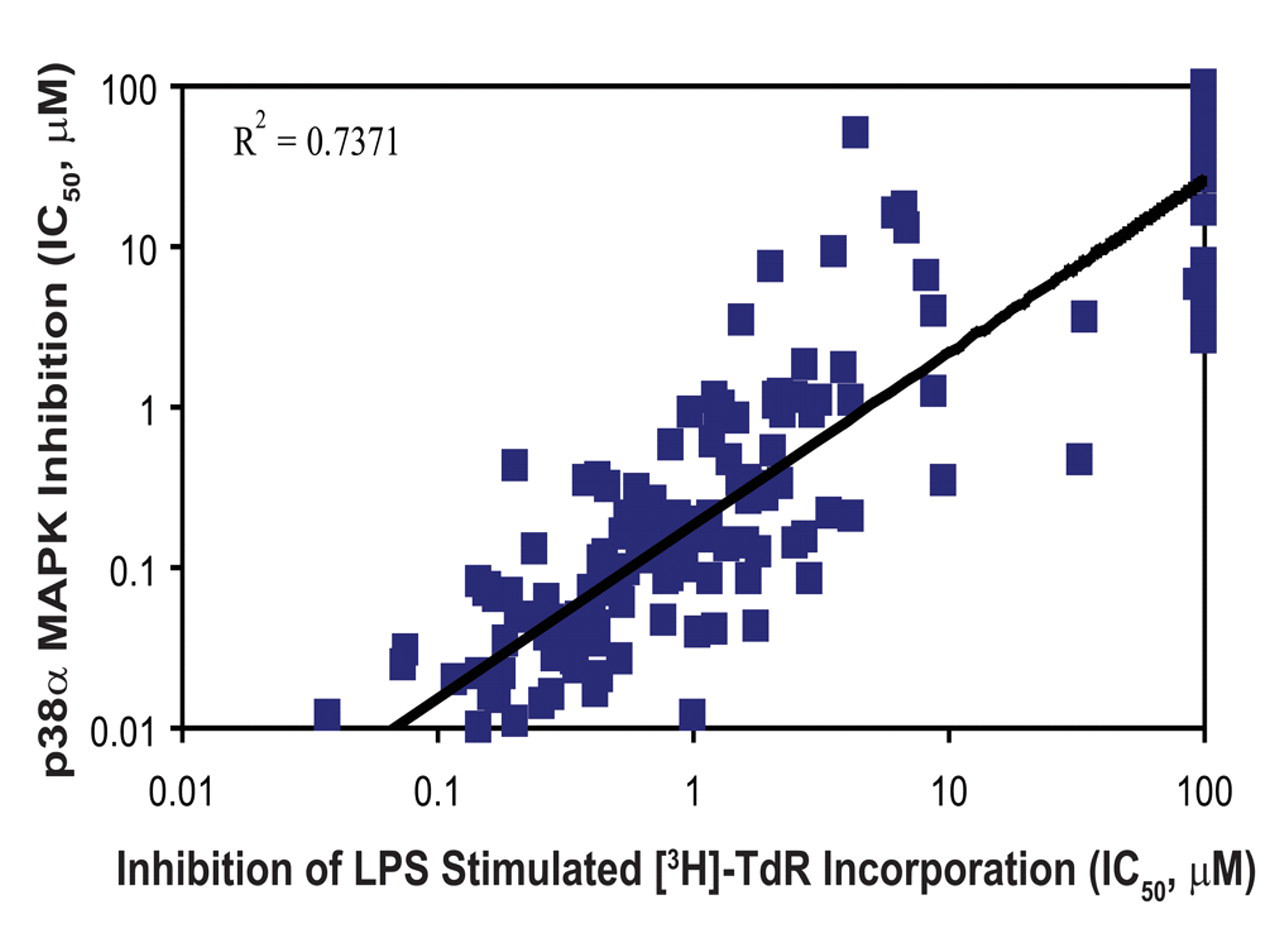
Inhibition of LPS-stimulated B-lymphocyte proliferation compared with inhibition of p38α mitogen-activated protein kinase (MAPK). The correlation coefficient between p38α MAPK inhibition and inhibition of dog B-lymphocyte [3H]-TdR incorporation for the 157 p38 MAPK inhibitors evaluated in the assay was 0.7371.
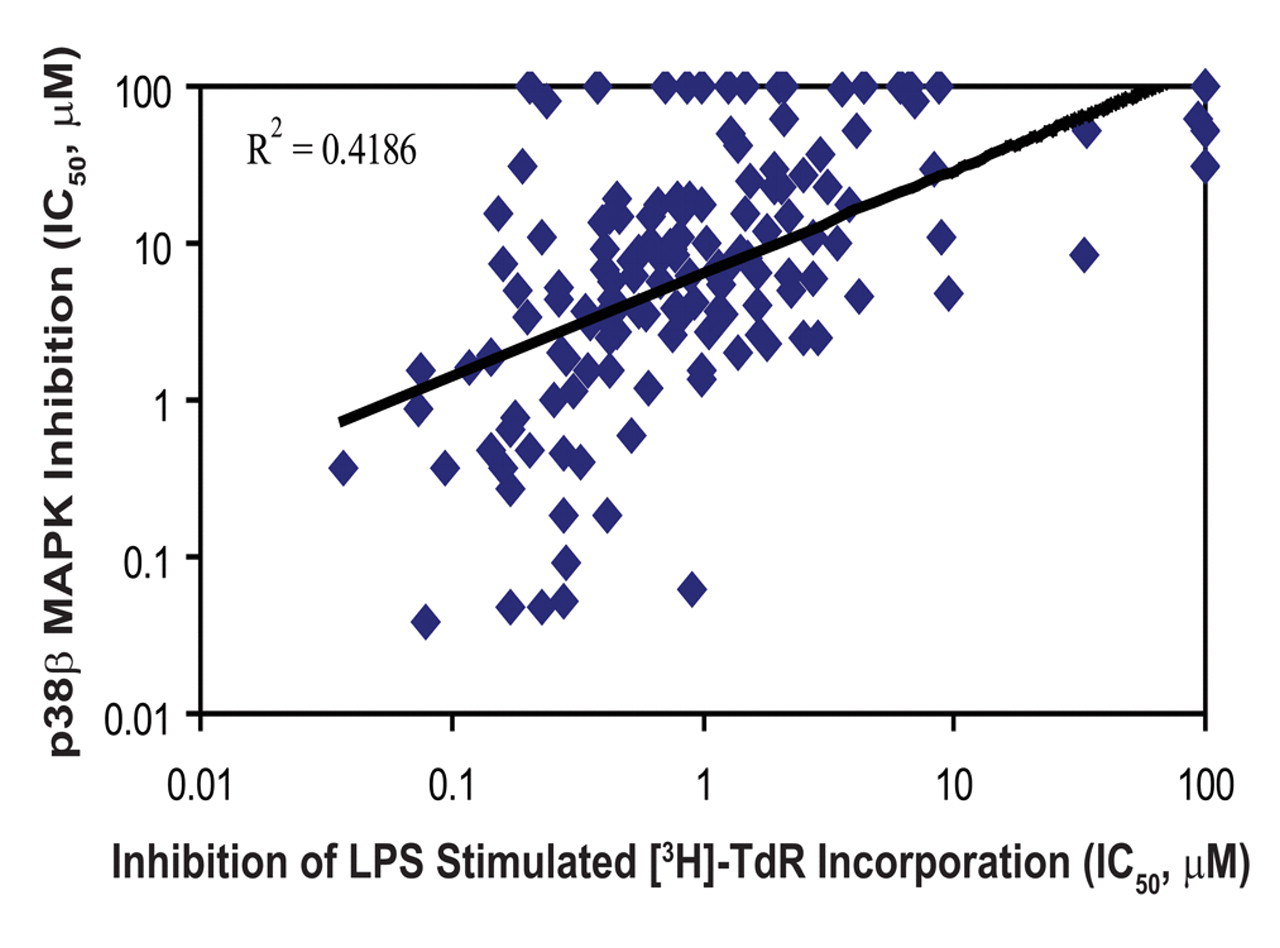
Inhibition of LPS-stimulated B-lymphocyte proliferation compared with inhibition of p38β mitogen-activated protein kinase (MAPK) activity. The correlation coefficient between p38β MAPK inhibition and inhibition of dog B-lymphocyte [3H]-TdR incorporation for the 157 p38 MAPK inhibitors in the assay was 0.4186.
In vitro studies using freshly isolated splenic leukocytes from rats and dogs demonstrated that IC50 values for inhibition of mitogen-stimulated B-lymphocyte [3H]-TdR incorporation in the rat are 10- to 100-fold higher than the IC50 values using canine B lymphocytes (Table 1). Similar results were observed using freshly isolated splenic leukocytes from mouse, cynomolgus monkey, and human for a subset of compounds shown in Table 1. This observation suggests, as in the in vivo studies, that the dog has a unique susceptibility to B-lymphocyte toxicity. Table 2 summarizes the results of a selective p38α MAPK inhibitor, SC-036, on mitogen-stimulated B-lymphocyte [3H]-TdR incorporation in different species, demonstrating the decreased sensitivity of other species, including humans, for effects of p38α MAPK inhibitors on canine B-lymphocyte proliferation. In this study, 2 different B-lymphocyte mitogens, LPS and SAC, were used, and similar results were obtained. In addition, the IC50 values for the in vitro inhibitory effects on canine B-lymphocyte proliferation with this compound (SC-036) were found to correspond to the inhibition of p38α MAPK in the human whole-blood assay and with the ED80 Cavg plasma concentration in the mCIA model.
Species Comparison for Inhibition of Mitogen-Stimulated B-Lymphocyte [3H]-TdR Incorporation by SC-036 a

a SC-036 IC50 values: p38α mitogen-activated protein kinase IC50 (enzyme assay) = 0.084 μM; human whole-blood tumor necrosis factor–α and p38α inhibition assay = 1.79 μM.
Evidence of Acute Lymphoid and GI Toxicity in Dogs Exposed to MK2 Inhibitors
In Vitro Assessment
As part of a greater effort to understand the role of downstream effectors of p38 MAPK on B-lymphocyte proliferation, MK2 inhibitors were evaluated for their ability to inhibit mitogen-stimulated B-lymphocyte [3H]-TdR incorporation in vitro. Mononuclear cells were isolated from canine spleens, as described in the Materials and Methods section, and B-lymphocyte proliferation was induced using SAC. Table 3 summarizes the effects of MK2 inhibitors on canine B-lymphocyte proliferation. In general, as the potency of MK2 inhibition increased, as determined by their ability to inhibit LPS-stimulated TNFα release in a human whole-blood cell assay, so did their ability to suppress SAC-induced [3H]-TdR incorporation. PF-318 was the most potent MK2 inhibitor tested and was, likewise, the most potent inhibitor of mitogen-stimulated B-lymphocyte proliferation. All MK2 inhibitors tested were inactive against p38 MAPKs (data not shown).
In Vitro and In Vivo Effects of Selective MK2 Inhibitors

GI = gastrointestinal; ND = not determined.
a Determined by inhibition of lipopolysaccharide-stimulated tumor necrosis factor–α production in human whole blood.
b Low-observed-effect level and no-observed-effect level were determined by comparing area under the plasma drug-concentration curves (AUCs) achieved after a single dose in preclinical species with the projected human efficacious AUC.
In Vivo Assessment
Acute oral toxicity studies in male SD rats, Beagle dogs, and cynomolgus monkeys with a selective MK2 inhibitor, PF-318, were performed to determine whether acute GI and lymphoid toxicity would be observed (Table 3). Studies were conducted following the same protocol used for the p38 MAPK compounds, as described in the Materials and Methods section.
After a single dose of up to 1,000 mg/kg to male SD rats, no gross pathologic evidence of GI or lymphoid toxicity was observed at exposures in excess of 14-fold the projected efficacious exposure. Furthermore, in repeat-dose studies in rats, exposure levels of up to 12-fold the efficacious AUC were achieved without overt, hematologic, gross, or microscopic evidence of GI or lymphoid toxicity.
The selective MK2 inhibitor, PF-318, was administered to male Beagle dogs at doses of 50, 300, and 750 mg/kg orally. Exposures at these doses were 1-, 7-, and 17-fold the projected efficacious AUC. Dogs dosed with 750 mg/kg had soft, watery feces, with red streaking. Dogs at the mid and low doses, as well as vehicle control animals, were normal. Furthermore, all dogs in the mid and high doses had gross pathologic evidence of acute GI toxicity that was described as dark red discoloration of the colonic mucosa. Histologically, the inflammation of the GALT in these dogs was also evident, although frank lymphoid necrosis could not be determined at this time point.
Finally, PF-318 was also administered to male cynomolgus monkeys. PF-318 exposure achieved systemic exposures of approximately 7-fold the efficacious AUC at the highest dose tested (1,000 mg/kg). At this dose and exposure level, no clinical signs of overt toxicity were observed, including those that would indicate GI effects. Given the lack of overt clinical signs, this study was a nonterminal study.
Taken together, these in vitro and in vivo data suggest that MK2 inhibitors cause a similar acute GI and lymphoid toxicity as selective p38α MAPK inhibitors and that the dog is uniquely sensitive to this effect.
Discussion
Data have been presented that demonstrate that the Beagle dog is uniquely susceptible, among the species evaluated in these studies, to an acute toxicity mediated by exposure to moderately selective p38α MAPK inhibitors. The clinical and pathologic manifestations of this acute toxic effect are hallmarked by mild clinical signs (decreased activity, diarrhea, and fever), lymphoid depletion in the GALT, mesenteric lymph nodes and spleen, colonic and cecal hemorrhage, and mild changes in hematologic parameters. The toxicity is fully manifested within 24 hours of an acute exposure and occurs at plasma exposures equal or similar to those achieved at a fully efficacious dose in the mCIA arthritis model. These effects were not seen in either the rat or monkey and are fully reversible within 2 weeks following an acute administration.
The original hypothesis for these acute toxic effects in the dog was a lack of selectivity of the compounds against p38β MAPK or other MAPK. However, several observations argue against that hypothesis. The GI lesions in the dog are seen at plasma exposures equal to or similar to those achieved at fully efficacious dose levels in the mCIA model, regardless of the potency with which the compounds inhibited p38β MAPK. The selectivity against p38β MAPK ranged from 30- to 40-fold over p38α inhibition in these studies. Selectivity of compounds against a broad panel of other serine/threonine and tyrosine kinases was consistently greater than 100-fold. The extent of inhibition of mitogen-stimulated [3H]-TdR incorporation in B lymphocytes in vitro was well correlated with potency of the compounds against p38α MAPK but not with their potency against p38β kinase (or other closely related MAPKs). Moreover, immunohistochemistry of lymphoid follicles detected a qualitative difference in the expression of p38α MAPK in dog B lymphocytes as compared with B lymphocytes of rats, monkeys, or humans. In these same tissues, p38α MAPK was highly expressed in macrophages of all species, indicating the ability of the antibody to detect p38α MAPK across species and a differential expression pattern in the dog. Western blot analysis of purified B lymphocytes confirmed a higher overall expression of p38α kinase (~30%) in the dog as compared with the rat. This result confirms the enhanced localized expression of p38α kinase in B lymphocytes of the dog that was detected in our immunohistochemistry studies. Finally, the dog GI lesions and lymphoid necrosis and depletion in the GALT were observed with a structurally distinct MK2 inhibitor that does not inhibit p38 MAPKs but inhibits downstream signaling in the p38α MAPK signaling pathway.
The GI injury and hemorrhage in the colon and cecum were found to be histologically consistent with ischemia reperfusion injury, but it is unclear whether these effects are a cause or a consequence of the mucosal epithelial cell injury. A number of reports have described the role of p38 MAPK in epithelial cell biology, suggesting that the toxic effects on the mucosal epithelium may be a direct consequence of p38α MAPK inhibition (Dieckgraefe et al. 1997; Bhowmick et al. 2001; Bakin et al. 2002; Sharma, He, and Bazan 2003; Stoll, Kansra, and Elder 2003). It was also noted in our studies that lymphoid necrosis and depletion in the GALT was not consistent with the linear pattern of mucosal injury and hemorrhage in the colon, and lymphoid depletion observed in Peyer’s patches was not associated with regionally localized mucosal injury or hemorrhage. However, the structure-toxicity relationship data presented in this report do support the conclusion that the mucosal injury, either directly or indirectly, is a consequence of p38α MAPK inhibition.
A role for p38α MAPK in dog B-lymphocyte proliferation and survival is further indicated by in vitro studies in which p38α MAPK inhibitors blocked LPS- and SAC-induced proliferation ([3H]-TdR incorporation) of B lymphocytes from dog but not from species (rat, monkey, human) that did not have a qualitatively higher level of expression of the enzyme in B lymphocytes compared to macrophages. Moreover, the inhibition of [3H]-TdR incorporation induced by both LPS (signaling through toll-like receptors) and SAC (signaling through the B-cell antigen receptor) suggests a distal and converging point role for p38α MAPK and MK2 in the signaling cascades leading to B-lymphocyte activation and proliferation, and it is not unique to a single signaling pathway for B-lymphocyte activation (Ogata et al. 2000; Yazawa et al. 2003; Schuurman, Gelfand, and Dosch 1980; Romagnani et al. 1981; Krakowka and Ringler 1986). It was noted in our immunohistochemistry studies that the pattern of enhanced p38α kinase expression in canine B lymphocytes was primarily observed in the light zones of secondary lymphoid follicles within the spleen, lymph nodes, and GALT, suggesting that the elevated expression is associated with a subset of B lymphocytes that are undergoing differentiation within the germinal centers. Although we are at present unclear as to the exact mechanism for the B-lymphocyte toxicity, these studies suggest that the p38α MAPK signaling pathway is important in the regulation of B-lymphocyte activation and survival in the dog following antigen or mitogen stimulation and may involve the activation or repression of cellular caspases (Graves, Craxton, and Clark 2004; Rathmell 2004).
The data presented from these studies provide a compelling correlation between p38α MAPK pathway inhibition and acute lymphoid and GI toxicity in the dog. These data collectively support the conclusion that these acute toxic effects in the dog are mechanism based but unique among the species evaluated in this report.
Footnotes
Acknowledgments
The authors would like to thank Tracy Young-Stacy and Gregory Frierdich for their expert assistance in the preparation of this article.
The work presented in this manuscript describing the acute toxicity of p38 kinase inhibitor in preclinical species was fully funded by Pfizer Inc. All contributors to this manuscript were financially supported by Pfizer Inc. at the time that the laboratory work was conducted.