
Abstract
Background:
Many inflammatory responses including chemotaxis, production of nitric oxide, and modulation of pro-inflammatory cytokines in immunological cells are mediated by p38MAPK. Due to its pivotal role, p38MAPK has been extensively explored as a molecular target for inhibition of chronic inflammation; however, it has not been successful so far due to serious toxicity issues. Among several downstream substrates of p38, mitogen-activated protein kinase-activated protein kinase 2 (MK2) has been reported to be a direct and essential downstream component in regulation of innate immune and inflammatory responses. Thus, in this study, we aimed to understand relative molecular differences between p38 and MK2 kinase inhibition in terms of a comparative anti-inflammatory potential along with molecular regulation of toxicity biomarkers such as Phospho c-Jun N-Terminal Kinase (pJNK), caspase-3, and hepatic enzyme levels in relevant human cells in vitro.
Results:
Both p38 and MK2 inhibitors attenuated lipopolysaccharide-induced pro-inflammatory biomarkers expression. In addition, both these kinase inhibitors inhibited release of Th1 and Th17 cytokines in phytohemagglutinin-induced cells with MK2 inhibitor showing a better potency for inhibition of Th1 cytokine release, interferon-γ. In the mechanistic differentiation studies, p38 inhibitors displayed an increase in pJNK and caspase-3 activity in U937 cells and elevation in aspartate transaminase enzyme in HepG2 cells, whereas MK2 inhibitor did not show such adverse toxic effects.
Conclusion:
Taken together, inhibition of MK2 kinase can be a relatively preferred strategy as an anti-inflammatory therapy over direct inhibition of p38 kinase in p38MAPK pathway.
Introduction
The mitogen-activated protein kinases (MAPKs) are key components in regulation of inflammatory signaling cascade and to initiate a cellular response. 1 –3 Among three distinct subgroups of MAPK pathways in mammalian cells (p38MAPK, extracellular-signal-regulated kinase (ERK), and c-Jun N-terminal kinase (JNK) pathways), the p38MAPK pathway has been identified as an important regulator in coordinated release of cytokines in response to inflammatory signal by immunocompetent cells. 4 –8 Activation of p38 kinase is followed by phosphorylation of downstream MAPK-activated protein kinase 2 (MK2) leading to the production of inflammatory molecules through posttranscriptional regulation and production of pro-inflammatory cytokines, such as tumor necrosis factor (TNF)-α, interleukin (IL)-6, IL-8, and IL-1β. Inhibition of p38 kinase leads to significant reduction of these pro-inflammatory mediators and consequent blockade of the inflammatory process. 3,9,10 Due to its vital role in inflammation, p38MAPK has been one of the most explored small molecule therapeutic targets for treatment of chronic inflammatory diseases including rheumatoid arthritis, inflammatory bowel diseases, psoriasis, asthma, and chronic obstructive pulmonary diseases. 10 –16 However, none of the p38MAPK inhibitors developed so far have reached clinic due to unexpected inefficacy and serious toxicity concerns such as liver toxicity, central nervous system toxicities, skin rash, gastrointestinal tract symptoms, and opportunistic infections. Literature reports have attributed association and activation of pro-inflammatory loops (e.g., activation of JNK and Transforming Growth Factor beta (TGFβ)-activated protein kinase-1, TAK1) as the reason of unexpected inefficacy and hepatic toxicity shown by p38MAPK inhibitors (such as BIRB796 and SB203580). 11,17 –22 In addition, p38 signaling regulates numerous downstream substrates and initiates activation of distal kinases (MK2, MK3, Mitogen And Stress Activated Kinase 1/2 (MSK1/2), and MAPK-interacting Protein Kinase 1/2 (MNK1/2)), transcription factors (Cyclic Adenosine Monophosphate-Response Element Binding-Protein (CREB) and Activating Transcription Factor-2 (ATF-2)). 20,22,23 Thus, p38 kinase is thought to be involved in pro- and anti-inflammatory functions and has a wide-ranging regulatory role; hence its inhibition might disrupt the pro- and anti-inflammatory balance.
MK2 is one of the several downstream kinases that are exclusively regulated by direct p38 phosphorylation in response to stress and inflammatory stimuli and has a direct and pivotal role in regulation of pro-inflammatory signaling induced through p38MAPK. 24 –26 Mice deficient in MK2 have showed significant reduction in the lipopolysaccharide (LPS)-induced biosynthesis of TNF-α, IL-1β, IL-6, and nitric oxide (NO), which suggests critical role of MK2 in the production of pro-inflammatory molecules. 24,25,27 In contrast to p38, MK2 has not been reported to be associated with numerous substrates and has a pro-inflammatory role in p38MAPK pathway (Figure 1).

Schematic representation of p38MAPK pathway in production of pro-inflammatory cytokines.
The objective of the present study was to evaluate a comparative pharmacological profile and to understand the relative potency and molecular regulation of pro-inflammatory biomarkers by comparing the effect of the p38 inhibitor (BIRB796, SB203580, and VX745) with an MK2 inhibitor (PF3644022) in various cell-based in vitro assays. We have also made an effort to evaluate the adverse toxic effect related to p38 inhibitors such as alteration in pJNK levels, production of caspase-3, and elevation in the level of liver enzymes using relevant cell-based in vitro models and compared with PF3644022, the literature-reported MK2 inhibitor.
The compound, BIRB796, is reported to be one of the most potent, selective allosteric inhibitors of p38α isoform and binds with slow association and dissociation rates. BIRB796 is a more potent inhibitor of p38MAPK isoform (α- and β-isoforms) than SB203580. It interacts with p38MAPK in a manner distinct from that exhibited by SB203580 and its binding induces a slow conformational change that locks the protein into an inactive conformation. SB203580 is a potent and selective inhibitor of p38 MAPK and it inhibits catalytic activity of p38 by binding to the adenosine triphosphate (ATP)-binding pocket. In contrast with SB203580, BIRB796 does not inhibit CK1δ, GSK3β, RIP2, or Cyclin G-Associated Kinase (GAK) in vitro. However, unlike SB203580, BIRB796 also inhibits p38γ, p38δ isoforms, and JNK2. 28,29 VX745 is a highly potent p38α inhibitor and selective over closely related MAPKs, including ERK1, MK2, and JNK. PF3644022 represents one of the first potent and selective MK2 inhibitors, with oral efficacy in both acute and chronic models of inflammation. 30 This ATP-competitive compound potently inhibited MK2 enzyme activity in cell-free and cell-based assays. In literature, it has been shown to effectively inhibit LPS-stimulated cytokine production in ex vivo and in vivo systems. It also exhibited good pharmacokinetic properties, demonstrating efficacy in the streptococcal cell wall–induced arthritis model. 30
Materials and methods
All chemicals and reagents used were from Sigma-Aldrich, unless otherwise stated. The p38 kinase inhibitors, BIRB796 (chemical name: N-[3-(1,1-dimethylethyl)-1-(4-methylphenyl)-1H-pyrazol-5-yl]-N′-[4-[2-(4-morpholinyl)ethoxy]-1-naphthalenyl]urea, CAS No: 285983-48-4, catalogue no: 5989), SB203580 (chemical name: 4-[5-(4-fluorophenyl)-2-[4-(methylsulfonyl)phenyl]-1H-imidazol-4-yl]pyridine, CAS No: 152121-47-6, catalogue no: 1202), VX745 (chemical name: 5-(2,6-dichlorophenyl)-2-[2,4-difluorophenyl)thio]-6H-pyrimido[1,6-b]pyridazin-6-one, CAS No: 209410-46-8, catalogue no: 3915), and MK2 kinase inhibitor, PF3644022 (chemical name: (10R)-9,10,11,12-tetrahydro-10-methyl-3-(6-methyl-3-pyridinyl)-8H-[1,4]diazepino[5′,6′:4,5]thieno[3,2-f]quinolin-8-one, CAS No: 1276121-88-0, catalogue no: 4279) were procured from Tocris Bioscience (Bristol, UK). The p38α kinase (catalogue no: 04-152; Carna Biosciences, Inc., Japan) and MK2 kinase (catalogue no: 02-142; Carna Biosciences, Inc.) assays were performed using myelin basic protein (MBP) as substrate in ADP Glo assay (catalogue no. V9101, Promega Corporation, Madison, Wisconsin, USA). NO production was measured using Griess reagent (catalogue no. G-7921; Molecular Probes (Oregon, USA)). The enzyme-linked immunosorbent assay (ELISA) kits used for measurement of cytokines in cell-based assays were from eBiosciences (Affymetrix, Santa Clara, California, USA). Aspartate aminotransferase in HepG2 cells was measured using kit from BioVision (Milpitas, California, USA; catalogue no. K753).
HepG2 cells are commonly used model for assessment of cell viability studies and evaluation of hepatic enzymes due to their ability to secrete liver enzymes 31,32 (e.g. aspartate aminotransferase) on induction with inflammatory stimuli (e.g. LPS); therefore, we have used these cells in our studies. For measurements of pro-inflammatory markers, we have used RAW264.7 cells (macrophage origin), THP1 and U937 (monocytic origin), and human peripheral blood mononuclear cells (hPBMCs; human cells having a mixed population of macrophage, monocytes, and T-cells), because these cells are widely reported in literature as LPS-induced models of cellular inflammation due to their immunological origin. 6,28,30
THP-1 and U937 cells were maintained in suspension culture in RPMI-1640 (Invitrogen, California, USA) supplemented with 10% (v/v) heat-inactivated fetal bovine serum (FBS, Gibco, Massachusetts, USA) at 37°C in a humidified atmosphere of 5% CO2. RAW264.7 and HepG2 cells were cultured in Dulbecco’s modified Eagle medium (Gibco, Massachusetts, USA) supplemented with 10% (v/v) FBS, 100 µg mL−1 penicillin, and 10 µg mL−1 streptomycin (Gibco, Massachusetts, USA) at 37°C in 5% CO2. Suspension of RAW264.7 and HepG2 cells was produced from confluent cultures using trypsin/EDTA solution before cell counting.
Blood was isolated from healthy human volunteers in pre-heparinized tubes after informed written consent according to the protocol approved by Institutional Ethics Committee. Isolation of hPBMCs from blood was performed by density gradient centrifugation on Histopaque-1077 (Sigma St. Louis, Missouri, USA). Briefly, human blood was mixed at a ratio of 1:1 with RPMI-1640 media at room temperature and this mixture was layered over Histopaque-1077 in a ratio of 2:1. The tubes were then centrifuged at 1180 × g for 30 min at 20°C without break; buffy coats were collected, pooled, and resuspended in complete RPMI-1640 medium.
In vitro kinase assay for p38 and MK2 enzyme
p38α and MK2 kinase assay was performed by ADP Glo assay. Briefly, p38α (catalogue no: 04-152; Carna Biosciences, Inc.; final assay concentration: 1.80 nM) or MK2 (catalogue no: 02-142, Carna Biosciences, Inc.; final assay concentration: 1.37 nM) was preincubated for 15 min at 25°C with either a test compound or vehicle (dimethyl sulfoxide (DMSO), 1% final concentration) in the presence of ATP (10 μM). After that, the substrate, 10 μg mL−1 MBP, was added to the reaction mix and further incubated for 60 min at 25°C (50 mM HEPES (4-(2-Hydroxyethyl)-1-Piperazine-Ethanesulfonic Acid), pH 7.4, 20 mM MgCl2, 0.2 mM Na3VO4, 1 mM DTT (Dithiothreitol)). The amount of ATP consumed for phosphorylation of MBP substrate was determined by ADP Glo assay.
Effect of p38 and MK2 kinase inhibitors on the cell viability and pro-inflammatory biomarker expression
Cell viability was determined using colorimetric cell counting kit-8 (CCK-8) containing WST-8 dye (catalogue no. CK04-11; Dojindo Molecular Technologies, Rockville, Maryland, USA). Briefly, adherent HepG2 cells (1 × 104 cells per well) were cultured in a 96-well plate with or without various concentrations of p38 or MK2 inhibitor for 24 h. After 24 h incubation, 10 µL of CCK-8 solution was added to each well of the plate and incubated at 37°C for further 4 h. The color developed by dehydrogenase activity of cells is directly proportional to the number of living cells. After 4 h, the absorbance was measured at 450 nm using an ELISA microplate reader, and the percentage of cell viability was calculated.
The concentration of NO in culture supernatant was measured in the form of nitrite, a major stable product of NO using Griess reagents in a colorimetric assay at 548 nm. Briefly, RAW264.7 macrophage cells (2 × 105 cells per well) were seeded in a 96-well plate and incubated in the presence or absence of various concentrations of inhibitors (0.01, 0.1, 1, and 10 µM) for 1 h. The cells were further stimulated by the addition of LPS (1 µg mL−1) for 24 h. After incubation, culture supernatant was collected, 100 µL of culture supernatant was mixed with equal amount of Griess reagents, and the NO production was measured by monitoring the absorbance at 548 nm in an ELISA microplate reader.
The effects of the p38 or MK2 inhibitors on the levels of LPS-induced release of cytokines were evaluated using human monocytic cell line THP-1. Briefly, THP-1 cells were plated at a density of 1 × 105 cells per well in a 96-well plate. The cells were pretreated with the inhibitors for 30 min followed by stimulation with LPS (1 μg mL−1) and then incubated further at 37°C in a CO2 incubator for 16 h. Cell culture supernatants were harvested and evaluated for the respective cytokines by colorimetric ELISA at 450 nm using kits from eBiosciences.
The hPBMCs isolated from fresh human blood were plated at a density of 2×105 cells per well in a 96-well plate. The effect of the p38 or MK2 inhibitors on the levels of phytohemagglutinin (PHA)-induced release of interferon (IFN)-γ (Th1 cytokine) and IL-17 (Th17 cytokine) was evaluated in these cells. The cells were pretreated with the inhibitor for 30 min followed by stimulation with PHA (10 μg mL−1) and then incubated further at 37°C in a CO2 incubator for 48 h. Cell culture supernatants were evaluated for the release of respective cytokines by colorimetric ELISA at 450 nm using kits from eBiosciences.
Effect of p38 and MK2 kinase inhibitors on the toxicity biomarkers in cell-based studies
For measurement of pJNK levels, human monocytic cell line U937 cells (12 h serum-starved, 2.5 × 105 cells per well in a 6-well plate) were pretreated with inhibitors for 1 h followed by further stimulation with LPS for 30 min. After that, the cell lysates were prepared and analyzed for the levels of pJNK using phospho-JNK 1/2 (Thr183/Tyr185) InstantOne™ ELISA kits from eBiosciences.
To study TNF-α-induced caspase-3 activity, PMA-differentiated U937 cells were pretreated with inhibitors for 30 min followed by induction with TNF-α at 5 ng mL−1 for 4 h. The caspase-3 protease activity in U937 cells was measured using a luminescence-based caspase-3 assay kit (Promega Corporation) according to the manufacturer’s instructions. Briefly, U937 cells were placed in a white-walled 96-well plate in complete RPMI-1640 media, followed by treatment with various concentrations of inhibitors for 30 min. After that, the cells were induced with TNF-α at 5 ng mL−1 and incubated further for 4 h at 37°C in a humidified CO2 incubator. After the incubation period, equal volume of Caspase-Glo reagent (catalogue no. G8092; Promega Corporation) was added. The reaction contents were gently mixed using a plate shaker at 300–500 r min−1 for 30 s and incubated further at room temperature for 1 h. The luminescence of each sample was then read in a plate-reading luminometer.
Literature suggests that in vitro hepatotoxicity can be directly determined by measuring levels of hepatic transaminase release into the culture medium of HepG2 cells. 31,32 Therefore, hepatic aspartate aminotransferase (AST) levels in human hepatocellular cell line and HepG2 culture medium were measured using a commercially available assay kit (BioVision). Briefly, HepG2 cells (2 × 105 cells per well) were plated overnight in a 24-well plate. Next day fresh media was replaced and the cells were incubated in the presence or absence of various concentrations of p38 or MK2 inhibitors (3, 10 and 30 µM) for 1 h and then stimulated with LPS (1 µM) for further 48 h. Control cells received 1% of DMSO instead of inhibitors. After incubation, culture supernatant was used to measure the secreted AST levels using a commercial kit (BioVision) in an ELISA microplate reader at 450 nm according to the supplier instructions.
Statistical analysis
The results were expressed as mean ± standard error of the mean of the three independent experiments. Statistical analyses of the results were made using one-way analysis of variance followed by Bonferroni’s test for multiple comparisons and Student’s t-test for single comparison.
Results
In vitro kinase inhibition assay
The in vitro kinase inhibition potency of p38 inhibitors and MK2 inhibitors was confirmed using MBP as the substrate in ADP Glo assay. The IC50 value of BIRB796, SB203580, and VX745 for inhibition of p38 kinase was found to be 34.3 nM, 53.1 nM, and 24.3 nM, respectively, under the assay conditions used. The literature-reported MK2 inhibitor, PF3644022, yielded a potency of 4.08 nM under similar conditions for inhibition of MK2 kinase (Figure 2).

Dose–response curves of p38 and MK2 inhibitors in kinase inhibition assay. MK2: mitogen-activated protein kinase-activated protein kinase 2.
Both p38 and MK2 kinase inhibitors showed a dose-dependent inhibition of pro-inflammatory biomarkers without affecting cell viability
The p38 and MK2 inhibitors tested in cell viability assay did not show any significant induction of cell death in HepG2 cells up to the highest concentration tested (30 µM; Figure 3).
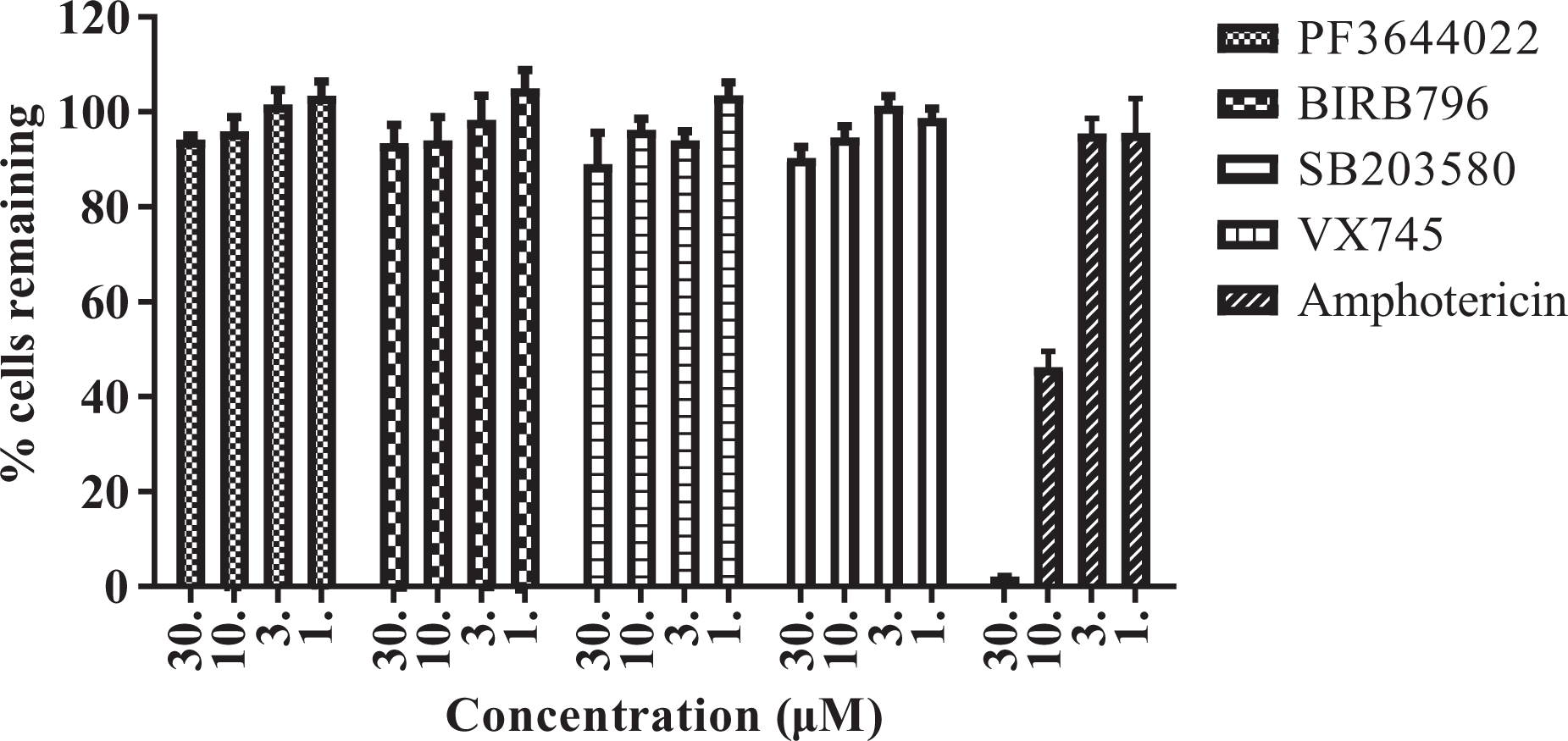
Cell viability assay for p38 and MK2 inhibitors in CCK-8 assay. MK2: mitogen-activated protein kinase-activated protein kinase 2; CCK: cell counting kit.
In the present study, we have observed that treatment of RAW264.7 macrophage cells with LPS (1 μg/ml) exposure produced significant amount of NO in cultured supernatant. Pretreatment of RAW264.7 cells with either p38 inhibitors or MK2 inhibitors (0.01–10 µM) resulted in inhibition of LPS-induced NO release in a dose-dependent manner (Figure 4). To our knowledge, this is the first report indicating that p38 inhibitors and MK2 inhibitors could inhibit LPS-induced NO production in cultured RAW264.7 macrophages.

Inhibition of NO production in LPS-induced RAW cells by p38 and MK2 inhibitors. NO: nitric oxide; LPS: lipopolysaccharides; MK2: mitogen-activated protein kinase-activated protein kinase 2.
We have checked the potency of these inhibitors as a quantitative measure of inhibition of stimulated cytokine release in a relevant cellular context. The human monocytic cell line THP-1 was exposed to LPS in the presence or absence of either p38 inhibitors or MK2 kinase inhibitors.
In line with the literature reports, the p38 inhibitor, BIRB796, was found to be the most potent compound with an IC50 of 5.99 nM, 3.94 nM, and 11.4 nM for inhibition of TNF-α, IL-1β, and IL-8, respectively, in LPS-induced THP-1 cells under these assay conditions. The MK2 inhibitor, PF3644022, showed a potency of 36.2 nM, 20.6 nM, and 119 nM for inhibition of TNF-α, IL-1β, and IL-8, respectively, under similar conditions. The dose-dependent percentage inhibition displayed by the p38 and MK2 inhibitors has been shown in Figure 5, and IC50 values of these inhibitors for these cytokines are reported in Table 1.

Inhibition of (a) TNF-α, (b) IL-1β, and (c) IL-8 production in LPS-induced THP-1 by p38 and MK2 inhibitors. TNF: tumor necrosis factor; IL: interleukin; LPS: lipopolysaccharides; MK2: mitogen-activated protein kinase-activated protein kinase 2.
IC50 values of p38 and MK2 inhibitors in various cell-free and cell-based assays.
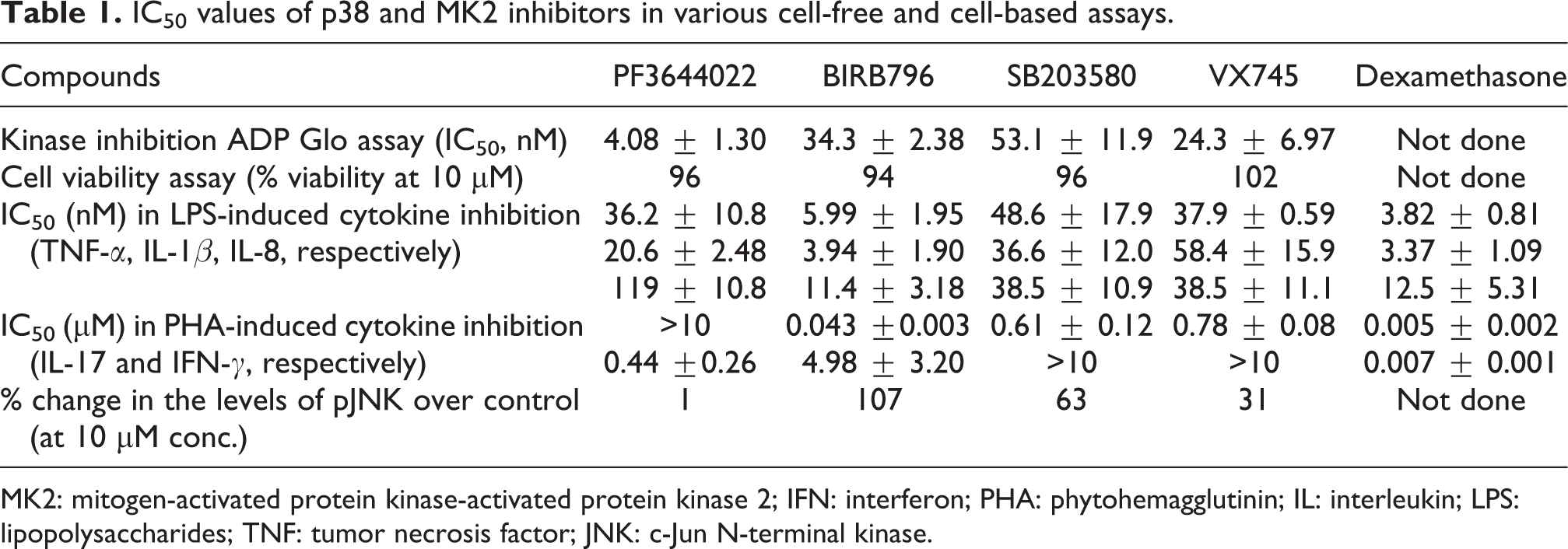
MK2: mitogen-activated protein kinase-activated protein kinase 2; IFN: interferon; PHA: phytohemagglutinin; IL: interleukin; LPS: lipopolysaccharides; TNF: tumor necrosis factor; JNK: c-Jun N-terminal kinase.
Since the role of p38MAPK pathway has not much been studied on T-cells, we aimed to evaluate the inhibitory activity of the p38 and MK2 kinase inhibitors in PHA-induced hPBMCs. For this, we selected an optimal concentration of PHA (10 μg/ml) for 48 h induction in these cells that would allow measurement of the release of IFN-γ and IL-17 cytokines (Figure 6(a) and (b)). The p38 inhibitors (BIRB796, SB239063, and VX745) and MK2 inhibitor (PF3644022) inhibited PHA-induced cytokines release from hPBMCs in a concentration-dependent manner (Figure 7). However, MK2 inhibitor, PF3644022, showed comparatively better potency for inhibition of Th1 cytokine, IFN-γ, whereas it was not markedly inhibited by p38 inhibitors at the concentrations tested (0.001–10 μM). Dexamethasone was used as a positive control in this assay, and it showed an IC50 ranging between 5 nM and 10 nM in these assays (Figure 7 and Table 1). This is the first study to report that MK2 inhibitor can inhibit release of IFN-γ (Th1 cytokine) more potently than p38 inhibitors in PHA-induced T-cell cytokines regulation in human cells.

Time and dose-dependent effect of PHA on the release of (a) IL-17 and (b) IFN-γ cytokines in hPBMCs by ELISA at 450 nm. PHA: phytohemagglutinin; IL: interleukin; IFN: interferon; hPBMC: human peripheral blood mononuclear cell; ELISA: enzyme-linked immunosorbent assay.
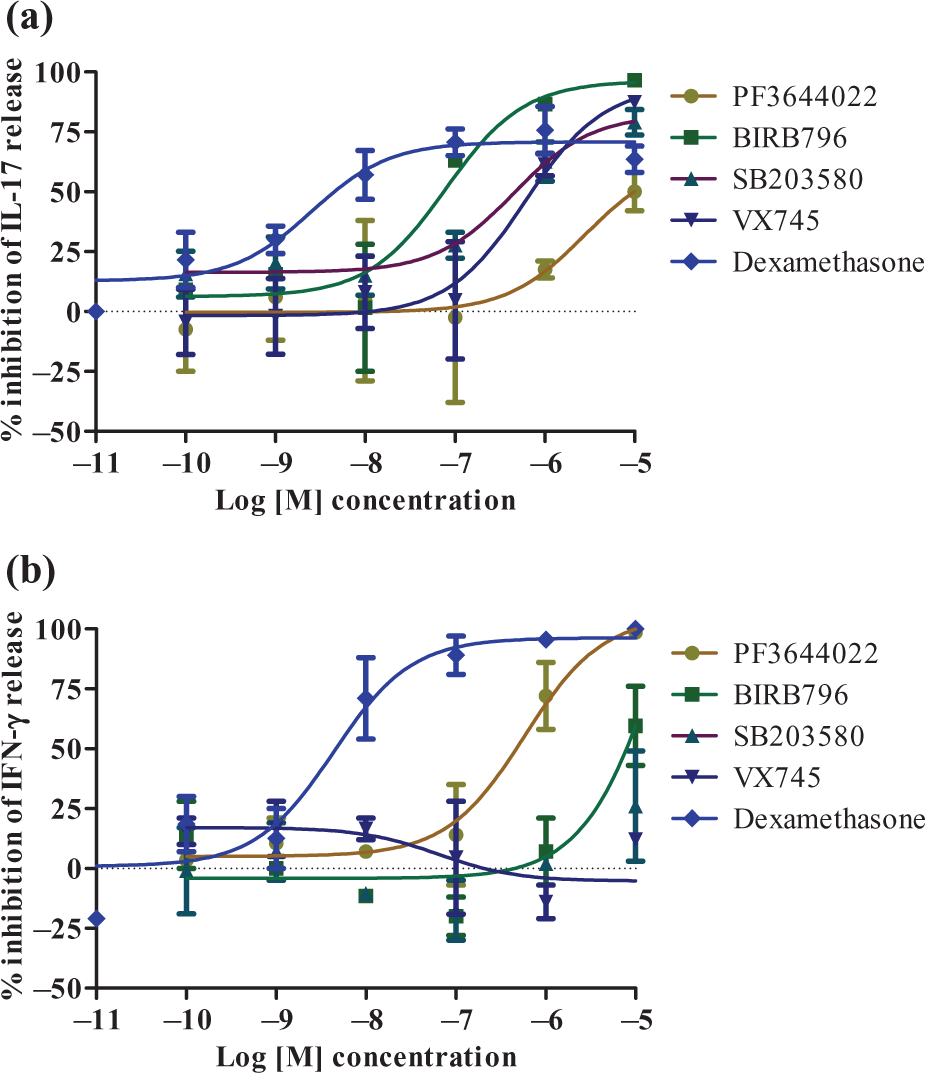
Dose-dependent inhibition of (a) IL-17 and (b) IFN-γ in PHA-induced hPBMCs by p38 and MK2 inhibitors. IL: interleukin; IFN: interferon; PHA: phytohemagglutinin; hPBMC: human peripheral blood mononuclear cell; MK2: mitogen-activated protein kinase-activated protein kinase 2.
Activation of toxicity biomarkers was shown by the p38 inhibitors but not by MK2 inhibitor
Effect of p38 inhibitors (BIRB796, SB203580, and VX745) and MK2 inhibitor (PF3644022) was evaluated on JNK activation in U937 cells induced with LPS. LPS challenge resulted in increased phosphorylation of JNK protein as compared to control cells. All the p38 inhibitors tested at 10 µM concentration showed significantly increased phosphorylation of JNK protein over LPS-treated cells. However, preincubation with MK2 inhibitor, PF3644022, did not show such effects up to 10 μM under similar conditions (Figure 8).

Alteration in LPS-induced pJNK protein in U937 cells by p38 and MK2 inhibitors (**p < 0.01 and ***p < 0.001 versus LPS-positive control). LPS: lipopolysaccharides; JNK: c-Jun N-terminal kinase; MK2: mitogen-activated protein kinase-activated protein kinase 2.
We evaluated the effect of p38 inhibitors and MK2 inhibitors on the LPS-induced release of liver enzymes such as AST in HepG2 cells as a measure of in vitro hepatotoxicity. Incubation of hepatic cells (HepG2) with LPS (1 µg/ml) increased the levels of AST in the HepG2 cell culture supernatant for 48 h, as compared to control cells. The p38 inhibitors tested (BIRB796, SB203580, and VX745) showed a marked increase in the levels of AST as compared to LPS-treated cells. However, MK2 inhibitor, PF3644022, did not show any elevation in the levels of AST enzyme in HepG2 cells as compared to LPS alone (Figure 9).
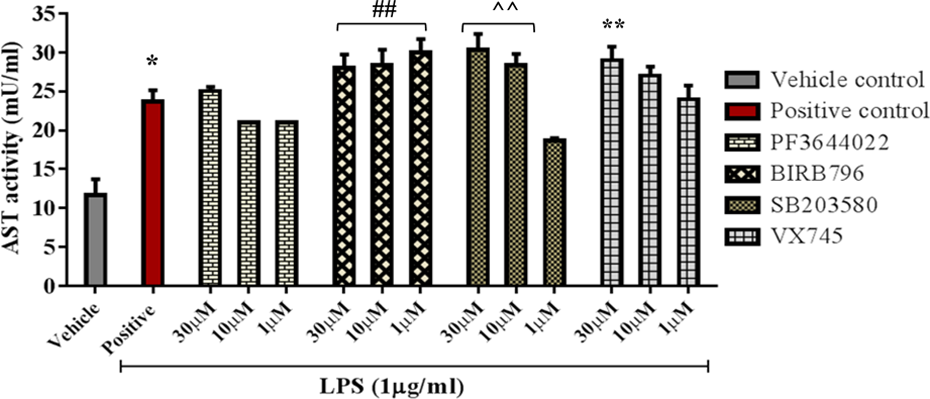
Change in hepatic AST enzyme induced by p38 and MK2 inhibitors in HepG2 cells (*p < 0.05 versus vehicle control; ##, ⁁⁁, **p < 0.01 versus LPS positive control). MK2: mitogen-activated protein kinase-activated protein kinase 2; LPS: lipopolysaccharides.
The effect of PF3644022 and BIRB796 was also evaluated on TNF-α-induced caspase-3 activation in PMA-differentiated U937 cells. TNF-α (5 ng mL−1) caused a significant increase in the caspase-3 activity in U937 cells at 4 h, and the p38 inhibitor, BIRB796, caused dose-dependent overactivation of TNF-α-induced caspase-3 activity, which can be linked to increased pJNK activity shown above. However, PF3644022 (MK2 inhibitor) has not shown any significant activation of caspase-3 over TNF-α alone (Figure 10).

Effect of p38 and MK2 inhibitors on TNF-α-induced caspase-3 activity in U937 cells (**p < 0.01 and ***p < 0.001 versus TNF-α-induced positive control). MK2: mitogen-activated protein kinase-activated protein kinase 2; TNF: tumor necrosis factor.
Discussion
LPS stimulation has been shown to mediate oxidative stress and to release pro-inflammatory cytokines through p38MAPK pathway activation in monocytes and macrophages; 30,33,34 however, the regulation of PHA-induced IFN-γ (Th1) and IL-17 (Th17) cytokines release by p38MAPK pathway is not much clear. Therefore, in this study, we investigated the regulation of PHA-induced IFN-γ and IL-17 cytokine release in human cells, in addition to the other pro-inflammatory cytokines (TNF-α, IL-1β, and IL-8) by p38 and MK2 kinase inhibitors.
The results of our studies clearly demonstrated a dose-dependent inhibition of LPS-induced oxidative stress biomarkers (NO release), release of pro-inflammatory cytokines (TNF-α, IL-1β, and IL-8), and T-cell-mediated release of cytokines (IL-17 and IFN-γ) by the commercially available structurally diverse p38MAPK inhibitors (BIRB796, SB203580, and VX745) and MK2 inhibitor (PF3644022) tested in a cell-based system. The p38MAPK inhibitor, BIRB796, was found to be the most potent inhibitor in this assay, which is in line with the previously published literature data. 25,30,35,36 In addition, the p38 inhibitors inhibited PHA-induced release of IL-17 more potently, whereas MK2 inhibitor did not show IL-17 inhibition. In contrast, MK2 inhibitor, PF3644022, inhibited the release of Th1 cytokine, IFN-γ, more potently than p38 inhibitor. Although both p38 and MK2 inhibitors inhibited LPS-induced release of pro-inflammatory cytokines with similar potency, this is the first report documenting the distinct potent activity of MK2 kinase inhibitor in PHA-induced release of IFN-γ as compared to p38 inhibitors tested. Aberrant IFN-γ expression is associated with a number of inflammatory and autoimmune conditions. Therefore, MK2 inhibitor can have a distinctive action as compared to p38 inhibitors under these pathological conditions.
Furthermore, we also evaluated the adverse toxic effect related to p38 inhibitors such as alteration in pJNK levels, production of caspase-3, and elevation in the level of liver enzymes using cell-based in vitro studies and compared with PF3644022, the literature-reported MK2 inhibitor. We found that inhibition of p38 kinase significantly activates and induces the level of pJNK activity in LPS-stimulated human cells, which is in agreement with previous studies, 18,20,22 whereas PF3644022 did not show any such activation in the pJNK protein level in U937 cells. Activation of pJNK levels has been linked to increased caspase-3 activation, which could contribute to the liver toxicity. Therefore, we aimed to study caspase-3 activation by these inhibitors in human cells. We concluded from our study that BIRB796 significantly and dose-dependently induces caspase-3 activation, which could be mediated through induced levels of pJNK. However, PF3644022 did not induce caspase-3 activation. The activation of pJNK levels by p38 inhibitors observed can be explained on the basis of a negative feedback control of p38α on TAK1. TAK1 is reported to be an activator of p38 and JNK and is negatively regulated by p38. Hence, inhibition of p38 kinase by BIRB796 may lead to increased phosphorylation and activity of JNK protein through activated TAK1. This increased pJNK activity may contribute to liver toxicity and tumor growth and other side effects shown by p38 inhibitors. 17 In contrast to p38α, MK2 kinase does not participate in the feedback signaling loop to TAK1 and this might be the reason that, PF3644022, the MK2 inhibitor did not show any significant induction of either pJNK or caspase-3.
LPS has serious effects on several organs, including the liver. It can also lead to endotoxic shock and death. LPS-induced hepatotoxicity is characterized by excessive oxidative stress and elevation of hepatocellular enzymes (such as Aspartate Aminotransferase (AST) and Alanine Aminotransferase (ALT)), leading to liver injury. Furthermore, human hepatocellular HepG2 cells present a valuable in vitro model and have been reported to be used in many toxicity studies to screen for hepatotoxic compounds. Interestingly, one of the major reported side effects associated with p38 inhibitor is the elevation in the levels of hepatic enzymes and hepatotoxicity; 18,36,37 therefore, we made an effort to evaluate such effects in vitro using human hepatocellular HepG2 cell line as an in vitro cellular model of hepatic injury to check the levels of AST enzyme. The p38 kinase inhibitors tested in LPS-induced HepG2 cells clearly showed an elevation in the levels hepatic AST enzyme as compared to LPS alone, whereas PF3644022 (MK2 inhibitor) did not display such adverse effects.
These observations suggest that although targeting downstream MK2 kinase in p38MAPK pathway can lead to similar or better anti-inflammatory effects as compared to p38 kinase inhibition, but importantly it can avoid the adverse toxic effects shown by targeting the upstream kinase, p38. Thus, inhibition of MK2 kinase can be a preferred strategy over direct p38 kinase inhibition in p38MAPK pathway.
It has now been clear from the literature reports that inhibition of p38 kinase in p38MAPK pathway has been associated with various limitations such as transient efficacy and toxicity concerns. 24 Due to such limitations, the downstream targets to p38 kinase, such as MK2, has drawn attention of researchers and has recently become more interesting for anti-inflammatory therapy. This is because, being downstream, it may spare the substrates/feedback loops associated with p38 and only inhibits the pro-inflammatory arm of the p38MAPK pathway. In contrast to p38α, MK2 does not participate in the feedback signaling loop to TAK1. Furthermore, the MK2 knockout animals are viable and fertile and do not display any significant abnormality, as shown by p38 knockout animals. 17,18,21
Conclusion
Taken together, these data indicate that p38 MAPK is involved in the production not only of pro-inflammatory cytokines but also has normal housekeeping functions through the various substrates associated with it. Targeting downstream kinase, MK2 for inflammatory therapy is attractive because therapeutic modulation of MK2 can tackle a key control point in the regulation of inflammation. In addition, there is mounting evidence that MK2-mediated pathways are activated only under inflammatory conditions. Our study indicates that inhibition of MK2 may act in a selective manner in inflammatory conditions. The current literature about MK2 inhibitors and knockout studies favor MK2 kinase inhibition as a potential and safe alternative therapy for various inflammatory diseases. In summary, we propose that targeting MK2 kinase as an anti-inflammatory approach would be a better therapeutic strategy over direct p38 kinase inhibition to avoid the toxic effects associated with it and warrants further investigation.
Footnotes
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.