
Abstract
The rat has been the preferred rodent toxicology species since before regulatory requirements have been in place, and there exists in the pharmaceutical industry and the regulatory agencies a significant amount of historical data for the rat. The resulting experience base with the rat makes the possibility of replacing it with the mouse for regulated toxicology studies untenable for all but the most extreme circumstances. However, toxicologists are very familiar with the mouse as a model for chronic carcinogenicity studies, and there exist multiple preclinical mouse models of disease. The authors evaluated the use of the mouse for early in vivo toxicology signal generation and prioritization of small molecule lead compounds prior to nomination of a development candidate. In five-day oral gavage studies with three test agents in the mouse, the authors were able to identify the same dose-limiting toxicities as those identified in the rat, including examples of compound-mediated hemolysis as well as microscopic lesions in the alimentary canal, kidney, and pancreas. Performing early signal generation studies in the mouse allows for earlier assessment of the safety liabilities of small molecules, requires significantly less compound, and allows evaluation of more compounds earlier in the project’s life cycle.
Introduction
Toxicity continues to be a primary source of small molecule pharmaceutical candidate attrition at all stages of drug development (Kola and Landis 2004). Pharmaceutical and biotechnology companies have increasingly begun to incorporate preclinical safety assessment earlier into the discovery process, both in the form of new technology (Ulrich and Friend 2002) as well as the earlier application of more standard testing paradigms (Kramer 2008). Although significant attention has been given to human-specific and idiosyncratic toxicity, a significant proportion of toxicity-related attrition occurs preclinically, and toxicity continues to be a primary contributor to attrition. Thus, the earlier identification of preclinical toxicities that are generally predictive of human toxicity (Olson et al. 2000; Greaves, Williams, and Eve 2004) could save time, money, and effort spent on compounds with little chance of becoming marketed pharmaceuticals. The costs associated with discovery and preclinical evaluation of a single drug candidate have been estimated to be greater than $25 million. It has also been suggested that an improvement in predicting failure prior to the initiation of clinical trials could save hundreds of millions in development costs per drug (Boston Consulting Group 2003; Woodcock 2004). Approaches to identify preclinical safety liabilities earlier in the process could lead to the design and selection of lead candidates with an increased probability of becoming marketed drugs. Such an approach, including earlier application of single- and repeat-dose in vivo rodent toxicology studies, would have a profound impact on overall attrition, cost, and time to first in human clinical trials.
Regulatory requirements for nonclinical toxicology assessment of small molecule drug candidates prior to first in human clinical studies include in vitro and in vivo studies for mutagenicity, safety pharmacology, and general toxicology (ICH 1997). Mammalian toxicology studies are required to be performed in both a rodent and a nonrodent species and must be of at least equal duration to that intended for use in human trials. The rat has historically been the rodent species of choice for the majority of nonclinical safety assessment studies. There is significant knowledge of spontaneous lesions, many years of accumulated data on baseline clinical chemistry and hematology ranges, and a prodigious database of compounds that have been assayed in single- and repeat-dose rat toxicology studies. The volume of background data on the rat makes the use of the mouse for GLP toxicology studies unattractive for all but the most compelling cases. However, there are a number of reasons the mouse might be considered as a suitable model for the very earliest in vivo toxicology signal generation studies. One obvious advantage of the mouse over the rat is the significantly lower compound requirement to conduct in vivo toxicology testing. This is of particular value early in drug discovery, when a project team may have multiple lead candidates and compound supply is at a premium. Additionally, scaling up syntheses of multiple leads to provide the 10-20 g or more to support repeat-dose rat toxicology studies would be a significant distraction from ongoing efforts to understand structure-activity relationships and identify lead candidates with optimal efficacy and Absorption, Distribution, Metabolism, and Elimination (ADME) properties. Early in vivo toxicology signal generation allows for the identification and characterization of dose-limiting toxicity (Fielden and Kolaja 2008) and an early estimation of a preclinical safety margin (Kramer, Sagartz, and Morris 2007). We have evaluated the mouse as an early in vivo toxicology signal generation model for identification and characterization of dose-limiting toxicity and selection amongst discovery lead candidates. Both our own experience and an evaluation of the literature demonstrate that the mouse is a suitable model for early preclinical toxicology studies.
Materials and Methods
General Animal Handling and In Vivo Methodologies
All procedures were conducted according to applicable standard operating procedures and with the approval of the Institutional Animal Care and Usage Committee. Male Sprague-Dawley rats were obtained from Charles River Laboratories and were placed in quarantine for 3 weeks following receipt until serology results were negative (Charles River, Wilmington, MA). Male B6(Cg)-Tyr<c-2j> (formerly C57BL6J-Tyrc-2J/J) mice were obtained from the Lexicon Pharmaceuticals, Inc. breeding facility. All animals were acclimated to study rooms (maintained at 71-72°F and a 12 hr light/dark cycle) and routine handling for at least 3 days prior to the initiating in vivo studies. Animals were randomized by body weight prior to being assigned to dosage groups at the outset of each study. Except as noted, animals were weighed daily as an indication of response to the test agents and for the purposes of calculating daily dosages. Additionally, animals were observed regularly throughout the day on days in which they were dosed, and these clinical observations were recorded. Exposure was assessed using saphenous or jugular vein bleeds in unanaesthetized animals, except for terminal exsanguination, which was performed using cardiac puncture immediately following CO2 asphyxiation. To avoid compromising hematology parameters, toxicokinetic (TK) time points were generally kept to a minimum. In all cases, vehicle control animals were bled at the same time points as compound-treated animals, except for the rat study with LP-499399 (see below), which included separate TK animals.
Clinical and Histopathology
Tissues collected for microscopic evaluation were fixed in formalin, embedded in paraffin, and cut in and stained with hematoxylin and eosin using standard methodologies. Lesions were scored for severity on a scale of 1 to 5 as minimal, mild, moderate, marked, or severe. Whole blood from each animal was collected in gel/clot activator tubes and used to prepare serum for clinical chemistry assessed on a COBAS integra 400. An additional aliquot of whole blood from each animal was collected in K2-EDTA tubes for complete blood counts using a Cell-Dyn 3500R, except for reticulocytes, which were assessed using a Becton Dickinson FACSCalibur.
Toxicokinetics
For exposure assessment, toxicokinetic samples were collected from the retro orbital sinus or saphenous vein. Approximately 25 μL of blood was added to K2-EDTA tubes, which were spun following each time point. Ten microliters of the resulting plasma was transferred to fresh polypropylene tubes, and exposure was assessed by mass spectrometry. PK parameters were calculated using WinNonLin.
Single-Dose Toxicokinetic Studies
To assess the toxicokinetics of pCPA in rodents, single-dose oral and intraperitoneal injection toxicokinetic studies were performed in mice and rats. Groups of 3 male C57/B16 mice were administered para-chloro phenylalanine (pCPA, Sigma-Aldrich, St. Louis, MO) formulated in 0.25% methylcellulose/0.1% Tween 80 at 10 mL/kg body weight via a single intraperitoneal injection of 30, 100, or 300 mg/kg; or via oral gavage at 30, 100, 300, 1,000, or 2,000 mg/kg. Blood was collected for exposure assessment from the saphenous vein at 0.5, 1, 2, 4, 6, and 24 hrs postdose. In a separate study, a single dose of 1 mg/kg pCPA formulated in 0.1% Tween 80 was administered via intravenous injection and blood collected at 5, 15, and 30 minutes and 1, 2, 4, 6, and 24 hrs postdose to determine pharmacokinetic parameters for pCPA in the mouse. To assess the toxicokinetics of pCPA in rats, a single dose of 30 or 300 mg/kg was administered via oral gavage or intraperitoneal injection to groups of 3 male Sprague-Dawley rats, then TK bleeds were collected from the saphenous vein at 0.5, 1, 2, 4, 6, and 24 hrs postdose. The pCPA was administered in 0.25% methylcellulose/0.1% Tween 80 at 10 mL/kg body weight to groups of rats via oral gavage or intraperitoneal injection. In a separate study, a single dose of 1 mg/kg Tween 80 was administered via intravenous injection and blood collected at 5, 15, and 30 minutes and 1, 2, 4, 6, and 24 hrs postdose to determine pharmacokinetic parameters for pCPA in the rat.
Repeated-Dose Toxicology Study Parameters
Compound/study-specific details of individual repeat-dose toxicology studies are provided in Table 1 and the following. Methotrexate (MTX; Sigma-Aldrich, St. Louis, MO) was formulated in phosphate buffered saline and administered daily via oral gavage to groups of 5 male C57 black 6 albino mice at 5 mL/kg body weight and 10, 30, and 100 mg/kg body weight for 5 consecutive days. On the final study day at approximately Tmax (1 hr postdose), animals were euthanized by CO2 followed by exsanguination. Methotrexate was formulated and administered daily to groups of 5 male Sprague Dawley rats at 10 mL/kg body weight via oral gavage at 30 mg/kg body weight for 5 consecutive days. On the final study day, animals were euthanized ~1 hr after the final dose by CO2 followed by exsanguination.
In vivo study design.
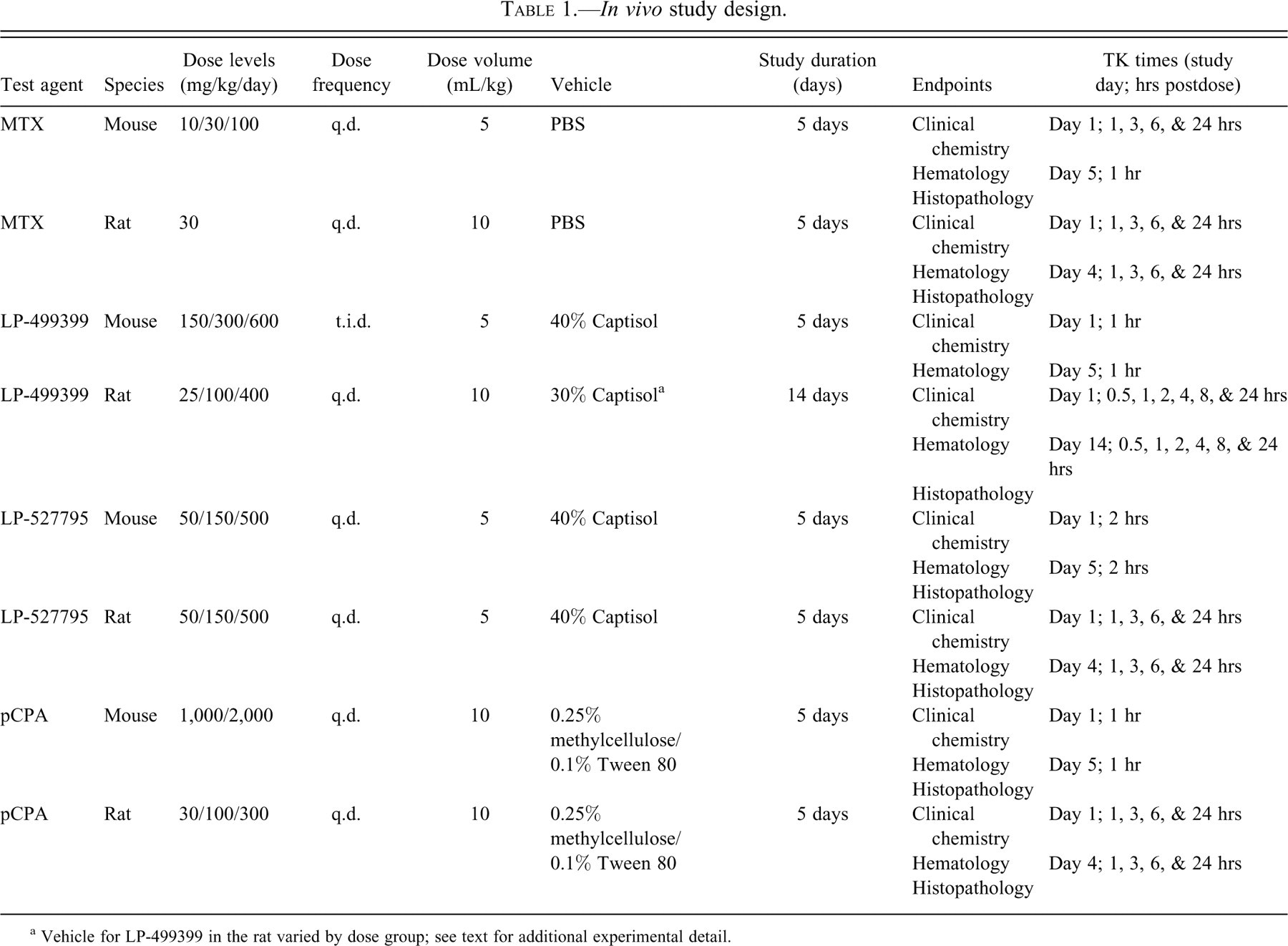
a Vehicle for LP-499399 in the rat varied by dose group; see text for additional experimental detail.
Lexicon oncology compounds LP-499399 and LP-527795 (Figure 1 ) were formulated in 40% captisol and dosed at 5 mL/kg by oral gavage in male C57 black 6 albino mice. LP-499399 was administered t.i.d. to ensure adequate exposure for 13 total doses over 5 days at 50, 100, and 200 mg/kg/dose (150, 300, and 600 mg/kg/day). Plasma exposure was assessed at 1 hr after the first and final doses. LP-527795 was administered once daily with doses of 50, 150, or 500 mg/kg body weight for a period of 5 days. Exposure was assessed at approximately Tmax (2 hrs) following the first and last dose. For both agents, surviving animals were euthanized at Tmax after the final dose by CO2 followed by exsanguination. Groups of 10 male Sprague Dawley rats were dosed orally with LP-499399 once per day for 14 consecutive days at 25, 100, or 400 mg/kg. Compound was formulated in captisol, where the final concentration of captisol in the formulation was 30% for vehicle and 400 mg/kg animals, 1.875% for 25 mg/kg animals, and 7.5% for 100 mg/kg animals. Rats were weighed at least once during the predose phase, on the first day of dosing, and weekly thereafter. On the final study day, animals were euthanized ~24 hrs after the final dose by CO2 followed by exsanguination. Exposure was assessed in separate TK animals, which were bled at 0.5, 1, 2, 4, 8, and 24 hrs following the first and final doses. Groups of 4 male Sprague Dawley rats were dosed orally with LP-527795 formulated in 40% captisol once per day for 5 consecutive days at 50, 150, or 500 mg/kg and 5 mL/kg. Exposure was assessed at 1, 3, 6, and 24 hrs following the first and last dose. Surviving animals were euthanized 1 hr after the final dose by CO2 followed by exsanguination.
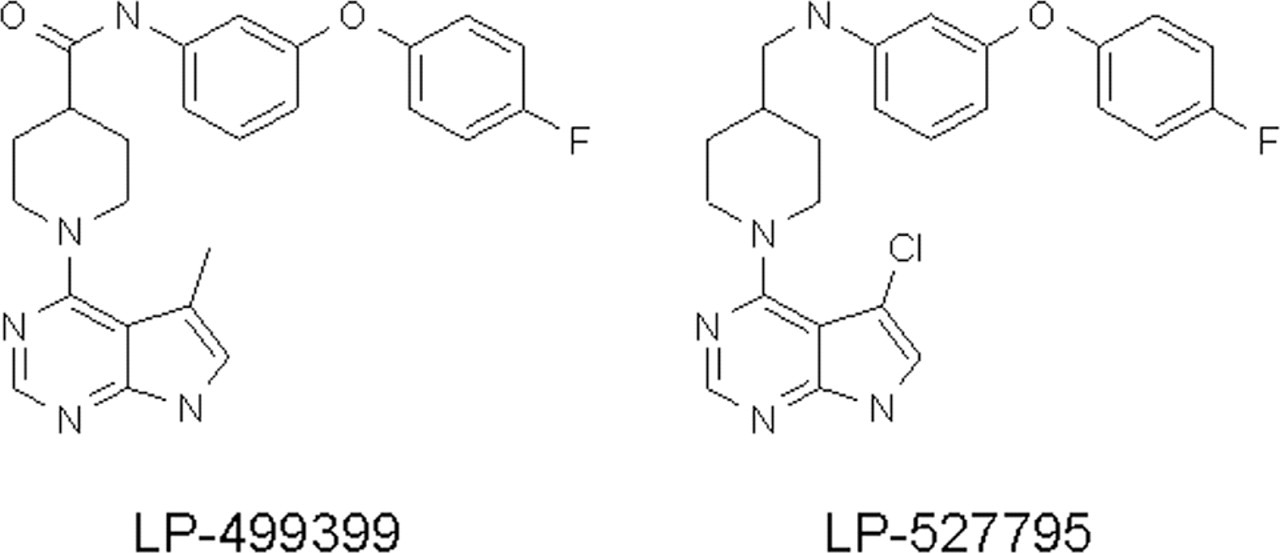
LP-499399 and LP-527795 structures. The compound LP-499399 was being advanced at Lexicon Pharmaceuticals for oncology indications. LP-499399 caused hemolytic anemia in mice and rats, and the margins were insufficient to continue development of the compound. A short-term repeat-dose mouse in vivo study was incorporated into the project team’s testing funnel, and this assay identified LP-527795 as a suitable backup compound that did not cause hemolytic anemia.
The safety and tolerability of repeated dosing of pCPA was assessed in mice using doses of 1,000 or 2,000 mg/kg dosed via oral gavage at 10 mL/kg in 0.25% methylcellulose/0.1% Tween 80. A limited set of tissues (heart, kidney, liver, thymus, spleen, brain, pancreas, testis, urinary bladder, and the entire GI tract) were collected at necropsy for microscopic analysis. Rats were administered doses of 30, 100, or 300 mg/kg once daily for 5 consecutive days via oral gavage at 10 mL/kg in 0.25% methylcellulose/0.1% Tween 80.
Statistical Analysis
All clinical pathology data (body and organ weights, clinical chemistry, hematology, urine chemistry) were evaluated for statistical significance using one-way analysis of variance (ANOVA). If the ANOVA demonstrated statistical significance (p < 0.05), a post hoc Dunnett’s t-test was used for comparisons of each treatment group versus the appropriate control group.
Literature Review
Approximately 2,200 citations were generated using a PubMed search with the key words “rat,” “mouse,” “in vivo,” and “toxicity.” These were reviewed, and those that provided informative and detailed data for compound-mediated toxicity in the mouse and rat are summarized in tabular form. Additionally, selected individual searches for separate mouse and rat reports on several well-characterized toxicants were also reviewed.
Results
Methotrexate in the Mouse and Rat
In the mouse MTX study clinical observations were limited to reduced grooming on later study days in the 30 and 100 mg/kg groups. Though not dose proportional, exposure (as assessed by AUC0-24hr) increased with dose, whereas the Cmax was not significantly different between the three doses (data not shown). Spleen weights were significantly reduced at all 3 dose levels (Table 2 ). The clinical chemistry data revealed modest changes in several parameters, including calcium, creatinine, and potassium (decreased at 100 mg/kg); and chloride (elevated at 30 and 100 mg/kg). Hematology (Table 3 ) revealed that red blood cell counts, hematocrit, and hemoglobin were lower at all doses. Histopathology demonstrated moderate to marked extramedullary hematopoiesis (EMH) in the spleen of control group animals, whereas there was a reduction in EMH in the spleen of MTX-treated animals, and a moderate decrease in hematopoietic cells was observed in the bone marrow (Figure 2 ). Cytologic alterations of crypt cells, villous atrophy, and supperative inflammation in the mucosa were observed in the small intestine of compound-treated mice. The findings were most severe (mild to moderate) in the duodenum and jejunum. There were numerous cytological changes in crypt epithelial cells, including apoptosis and degeneration of individual cells, and loss of granules and other features of differentiation. Mild depletion of thymic cortical lymphocytes was found in the high-dose treatment group only.
Clinical chemistry, body and spleen weight in mouse and rat in response to daily dosing with MTX.
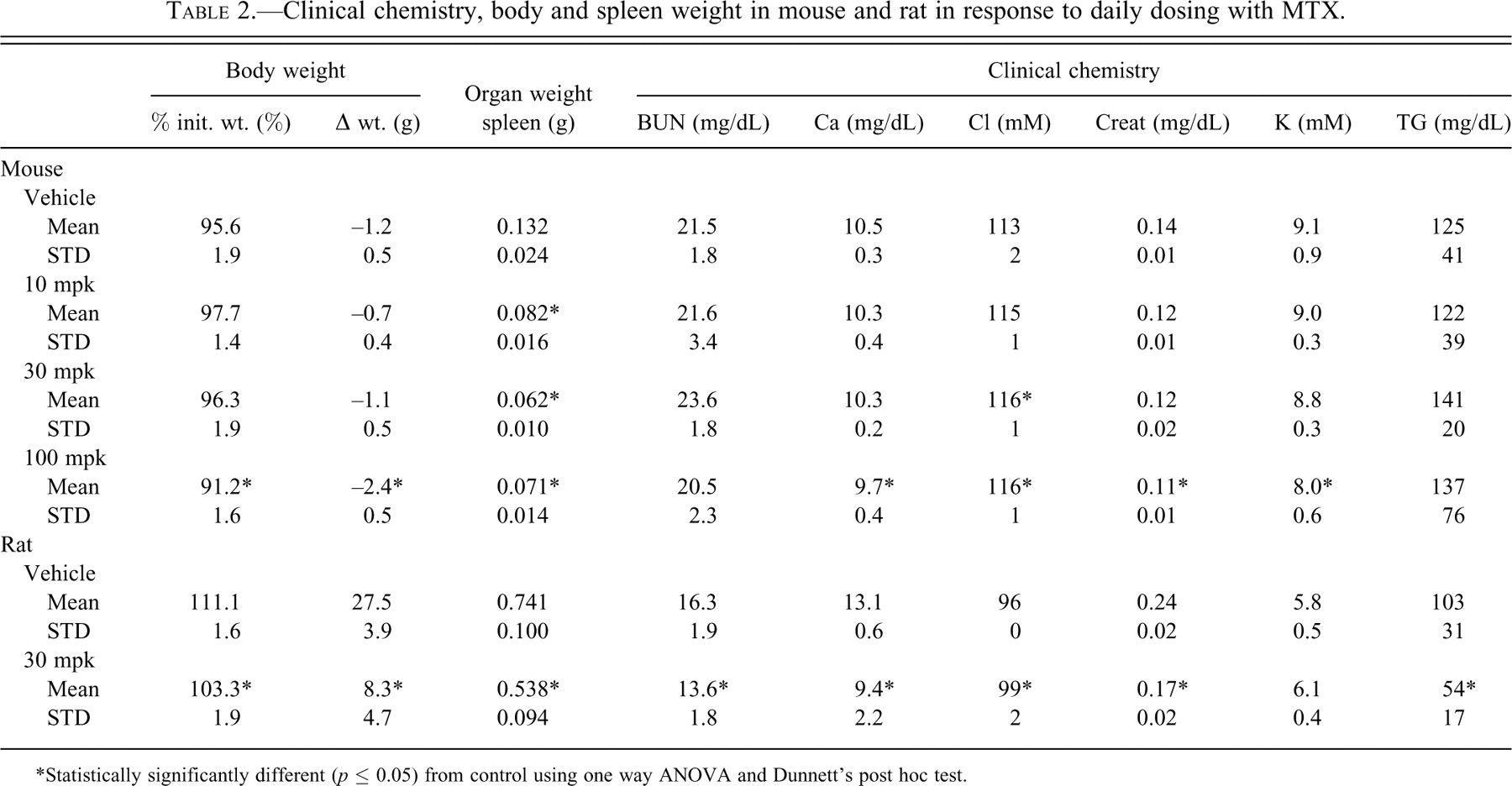
*Statistically significantly different (p ≤ 0.05) from control using one way ANOVA and Dunnett's post hoc test.
Hematology in mouse and rat in response to daily dosing with MTX.
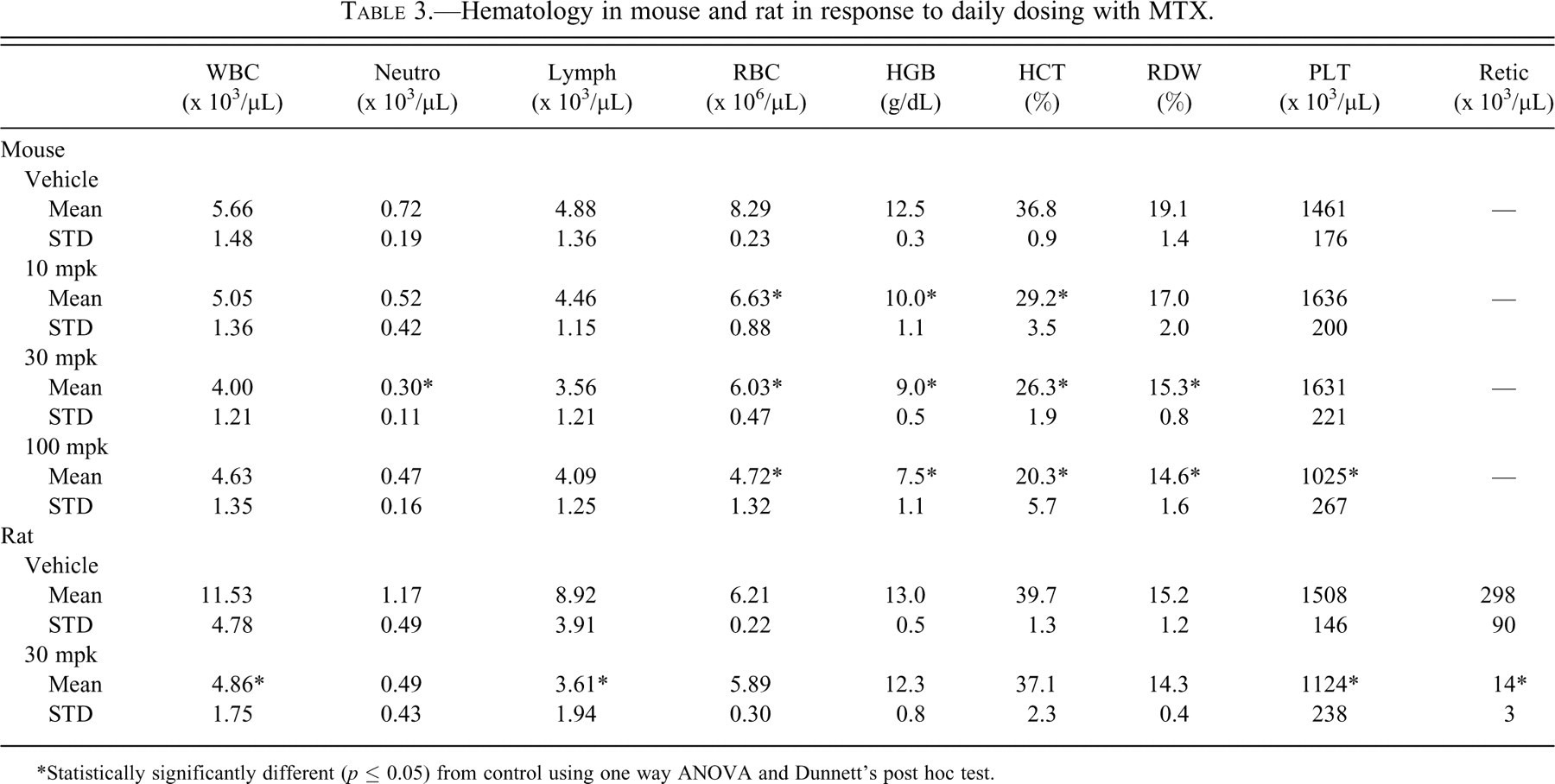
*Statistically significantly different (p ≤ 0.05) from control using one way ANOVA and Dunnett’s post hoc test.
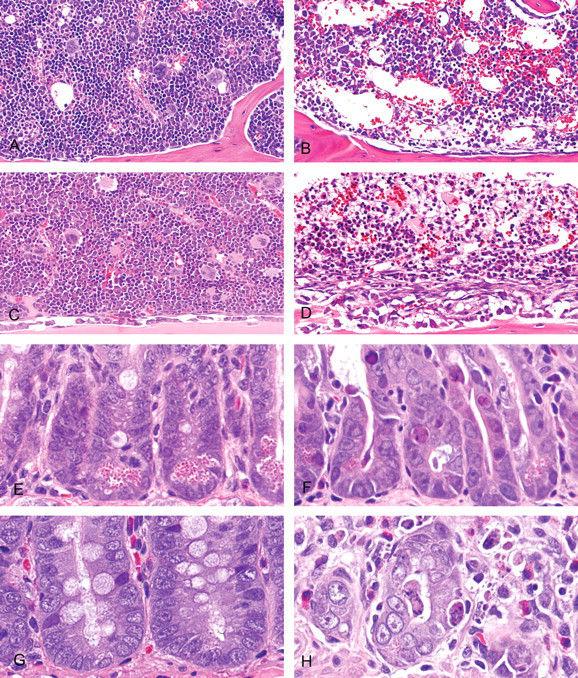
Dose-limiting bone marrow and small intestine lesions observed in the mouse and rat with methotrexate. Mice and rats were administered vehicle or MTX at 10, 30, or 100 mg/kg body weight and 30 mg/kg body weight, respectively, via oral gavage, once per day, for 5 days. (A) Mouse, bone marrow; 0 mg/kg methotrexate. Inconspicuous blood vessels in marrow are surrounded by tightly packed hematopoietic cells. (B) Mouse, bone marrow; 100 mg/kg methotrexate. Dilated blood vessels are expanded to fill the marrow space left vacant by markedly reduced numbers of hematopoietic cells. (C) Rat, bone marrow; 0 mg/kg methotrexate. Essentially identical appearance to that seen in the normal mouse, the marrow space is filled with tightly packed hematopoietic cells. (D) Rat, bone marrow; 30 mg/kg methotrexate. Drug toxicity is similarly manifested by dilation of blood vessels and markedly reduced numbers of hematopoietic cells. (E) Mouse, duodenum; 0 mg/kg methotrexate. Normal intestinal crypt epithelium. (F) Mouse, duodenum; 100 mg/kg methotrexate. Scattered apoptotic and necrotic epithelial cells are present in crypts lined by irregular epithelium showing nuclear changes and cytoplasmic basophilia. (G) Rat, duodenum; 0 mg/kg methotrexate. Essentially identical appearance to that seen in the normal mouse. (H) Rat, duodenum; 30 mg/kg methotrexate. Intestinal crypt epithelial lesions are essentially identical to those present in similarly treated mice. H&E stain.
In rats treated with 30 mg/kg methotrexate, the main clinical observation was rough coat and significantly reduced weight gain (Table 2). Exposure and mean residence time at 30 mg/kg in the rat was quite similar to that seen in the mouse at the same dose (Figure 3 ). Necropsy observations included a statistically significant decrease in spleen weight. Clinical chemistry data revealed significant decreases in calcium, creatinine, and triglycerides. Hematology data demonstrated significant reductions in total white blood cells, lymphocytes, and eosinophils, along with reductions in platelets and reticulocytes (Table 3). The primary microscopic lesions associated with administration of methotrexate in rats were marked bone marrow hypocellularity (decrease in hematopoietic cells) and mild to moderate mucosal necrosis and villous atrophy in the duodenum and jejunum (Figure 2).
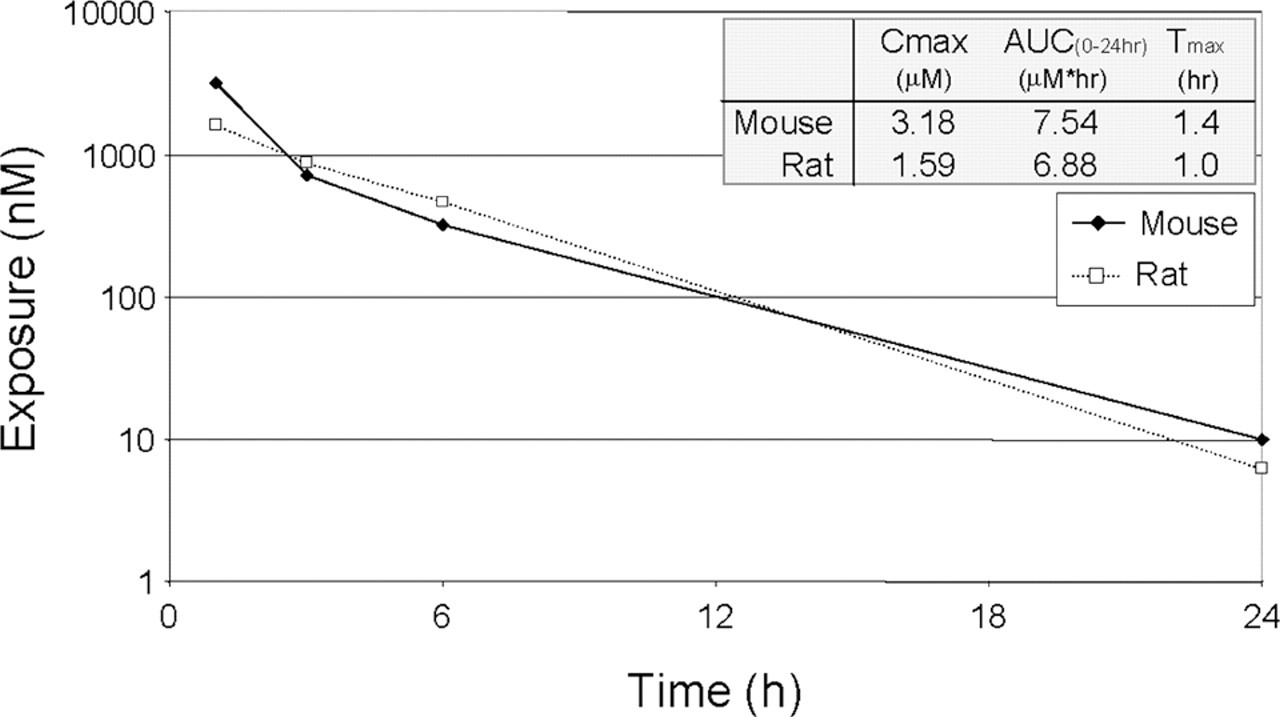
Plasma exposure of methotrexate in the mouse and rat. Exposure of methotrexate following a single oral dose of 30 mg/kg was similar in the mouse and rat. Although clearance is often greater with most compounds in the mouse than in the rat, adequate exposure for the identification of dose-limiting toxicity can usually be achieved in the mouse using significantly less compound than would be required in the rat.
Lexicon Pharmaceuticals Oncology Compounds in the Mouse and Rat
LP-499399 was dosed by oral gavage in the mouse, t.i.d. to ensure adequate exposure, for 13 total doses over 5 days at 50, 100, and 200 mg/kg/dose (150, 300, and 600 mg/kg/day). The most prominent clinical signs observed at higher doses (300 and 600 mg/kg/day) were hunched posture, lethargy, labored breathing, and loose stool. Exposure increased with dose, but in a nonlinear fashion above 300 mg/kg/day (data not shown). At necropsy all groups including the vehicle-treated mice had frothy liquid in the intestines. The spleen was enlarged in the 150 and 300 mg/kg/day groups, while a decrease in spleen weight was observed at 600 mg/kg/day. Hematology data revealed a decrease in red blood cells, hemoglobin, and hematocrit at 300 and 600 mg/kg/day and a concomitant increase in mean corpuscular volume, mean corpuscular hemoglobin, and mean corpuscular hemoglobin concentration (Table 4 ). Histopathology revealed increased EMH in the spleen at 150 mg/kg/day and a marked decrease in EMH in the 300 and 600 mg/kg/day animals. Additionally, minimal to marked hemosiderosis, histiocytosis, and apoptosis was observed in the spleen of all compound treated groups.
Hematology in mouse and rat in response to daily dosing with experimental oncology compounds.
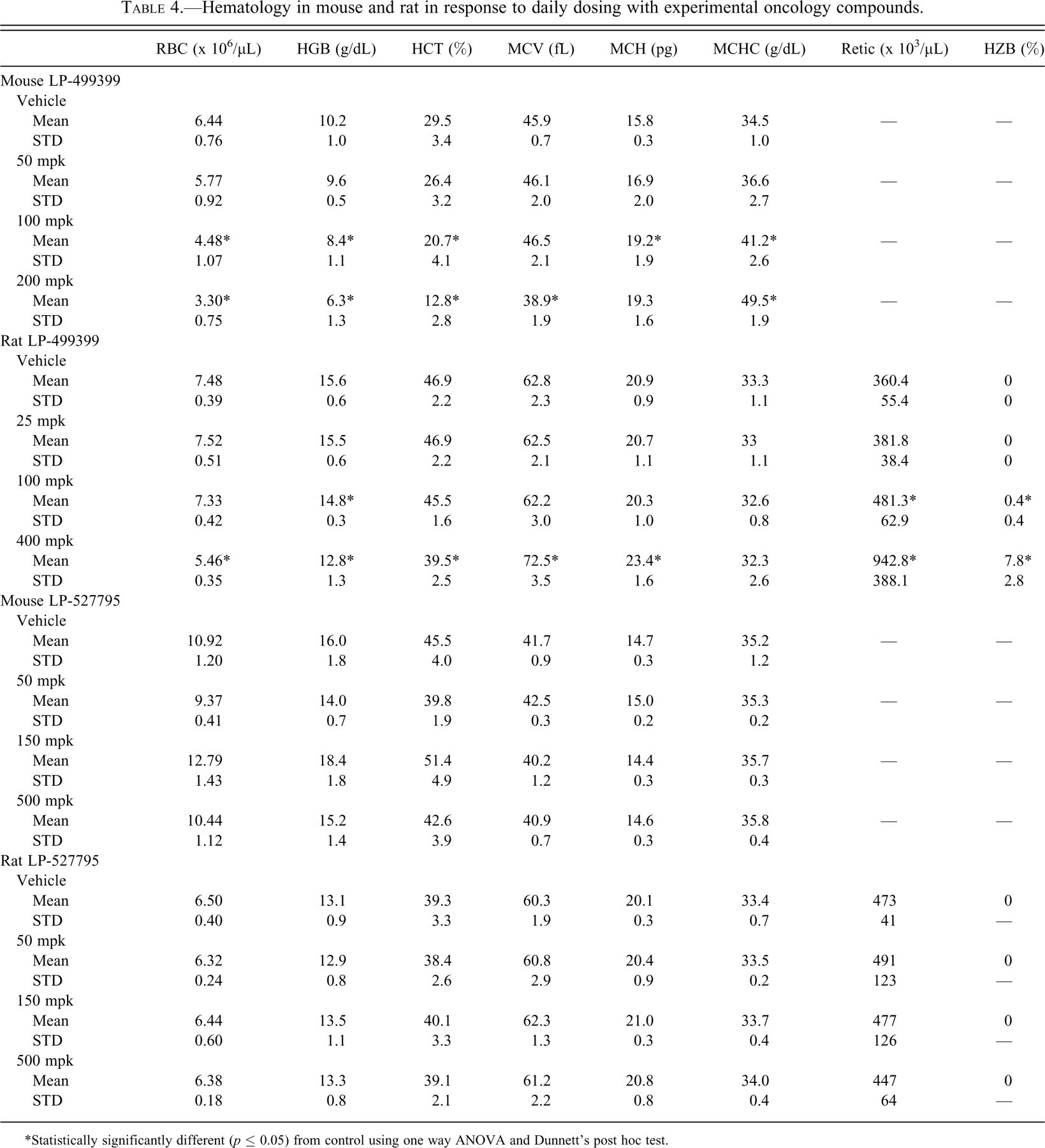
*Statistically significantly different (p ≤ 0.05) from control using one way ANOVA and Dunnett’s post hoc test.
Exposure to LP-499399 in the rat generally increased with the increase in dose level from 25 to 400 mg/kg (data not shown). Decreased body weight gain was noted at 400 mg/kg, and significantly increased liver and spleen weights were observed at 100 and 400 mg/kg. The most prominent test-article-related effects were consistent with Heinz body hemolysis (Table 4). Red cell mass (i.e., red blood cell count, hemoglobin, and hematocrit) was mildly to moderately reduced for animals given 100 or 400 mg/kg, and reticulocytes were elevated. The increased liver and spleen weights corresponded microscopically with increased extramedullary hematopoiesis and hemosiderin deposits in both organs. Additional findings included minimal to moderate degeneration of renal tubular cells and increased tubular cell pigment deposits.
In a preliminary mouse dose-range-finding study with LP-527795, male C57B16 mice were treated via oral gavage at 50, 150, or 500 mg/kg for 5 days. The 500 mg/kg group animals were sacrificed moribund after the 3rd dose, and the 150 mg/kg group were sacrificed moribund following the 4th dose. Greater than 10% body weight loss was observed in all mid- and high-dose group animals by the time of sacrifice, and animals displayed labored breathing, rough coats, and reduced activity. Upon necropsy, the stomachs in the mid- and high-dose groups were dilated, and the intestines were filled with frothy, yellow fluid. Spleen and thymus weights were dose-dependently and statistically significantly reduced in all groups. Total white blood cells and lymphocytes were reduced (data not shown); whereas red blood cell count, hemoglobin, and hematocrit were not affected at any dose of LP-527795 (Table 4). Microscopic evaluation revealed the absence of histopathologic lesions in the liver, but mild renal tubule degeneration was observed at 500 mg/kg, and dose-dependent mild to marked lymphoid depletion with mitotic arrest was noted at all dose levels in the thymic cortex and the red pulp of the spleen (data not shown).
The compound LP-527795 was relatively well tolerated in the rat up to 500 mg/kg, although the high-dose group gained significantly less weight than the control. Plasma exposures similar to those observed in the mouse at the same dose levels (data not shown). Clinical chemistry data revealed dose-dependent elevations in serum transaminases and alkaline phosphatase. Hematology data revealed changes in white blood cell populations (data not shown); but no red cell parameters were significantly affected (Table 4), and Heinz bodies were not observed. Histopathology revealed minimal to mild lymphoid depletion with mitotic arrest at all dose levels in the thymus and spleen. Histopathology was not assessed in the liver or kidney, as the program was de-prioritized for strategic reasons.
Para-Chloro Phenylalanine in the Mouse and Rat
pCPA was dosed via intravenous injection, intraperitoneal injection, or oral gavage in single dose mouse and rat toxicokinetic studies (Figure 4 ). Although total exposure was higher in the rat than the mouse using both i.p. and p.o. dosing routes, the difference between i.p. and p.o. dosing within each species was minimal (Table 5 ). Repeat-dose studies were therefore performed via oral gavage, although higher dose levels were used for the mouse. In the mouse, red blood cells, hemoglobin, hematocrit, and numerous clinical chemistry parameters were statistically significantly elevated (Tables 6 and 7 ). Microscopic analysis of both mouse and rat showed prominent lesions of exocrine pancreas acinar cells. Two of 4 mice dosed at 2,000 mg/kg demonstrated moderate apoptosis of pancreatic acinar cells (Figure 5 ), and the remaining animals showed mild to moderate depletion of exocrine granules in the pancreas. One of 5 mice dosed at 1,000 mg/kg was scored as minimal for pancreatic acinar cell apoptosis, while the other animals showed minimal to mild degranulation. Apoptosis of pancreatic acinar cells was associated with rounding and shrinkage of cells, nuclear pyknosis, and karyorrhexis/karyolysis.
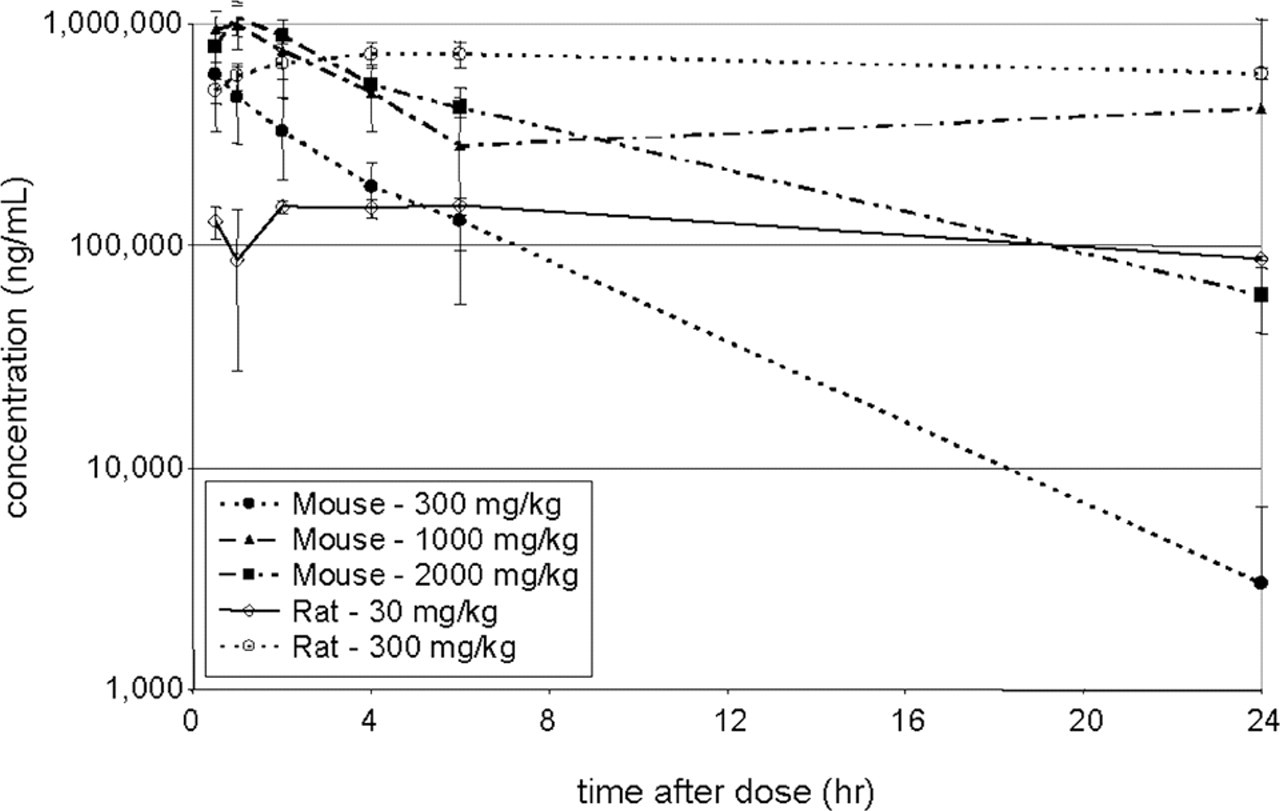
Plasma exposure in mouse and rat following oral doses of pCPA. Plasma concentration was determined in the mouse and rat following single oral doses of pCPA. Observed and calculated pharmacokinetic parameters are given in Table 7. pCPA was administered via oral gavage to mice at 300 (filled circles, dotted line), 1,000 (triangles, dashed line), and 2,000 (filled squares, dashed line) mg/kg; and to rats at 30 (open diamonds, solid line) and 300 (open circles, dotted line) mg/kg.
Toxicokinetic parameters of pCPA in the mouse and rat.
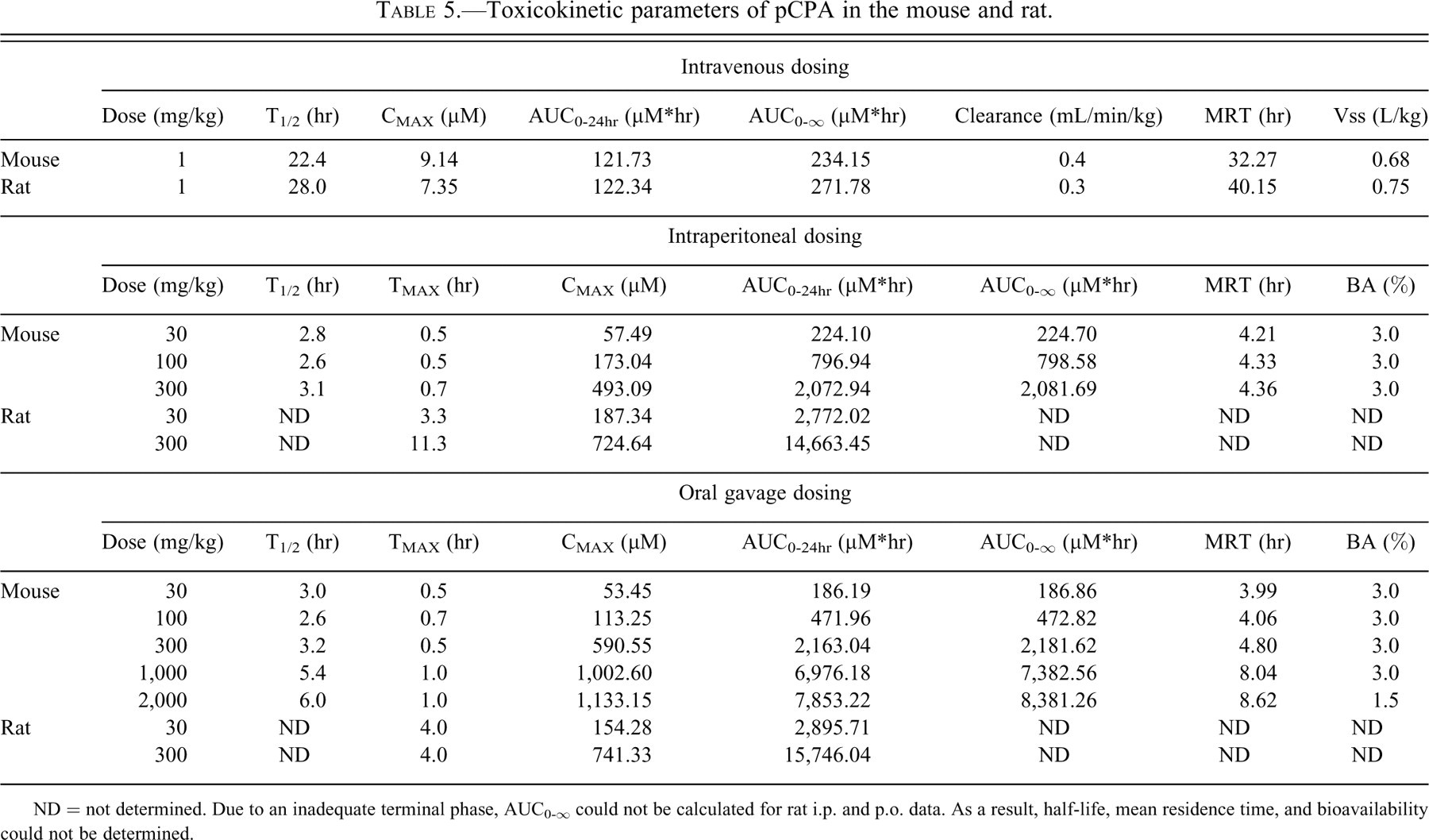
ND = not determined. Due to an inadequate terminal phase, AUC0-∞ could not be calculated for rat i.p. and p.o. data. As a result, half-life, mean residence time, and bioavailability could not be determined.
Clinical chemistry in mouse and rat in response to daily dosing with pCPA.
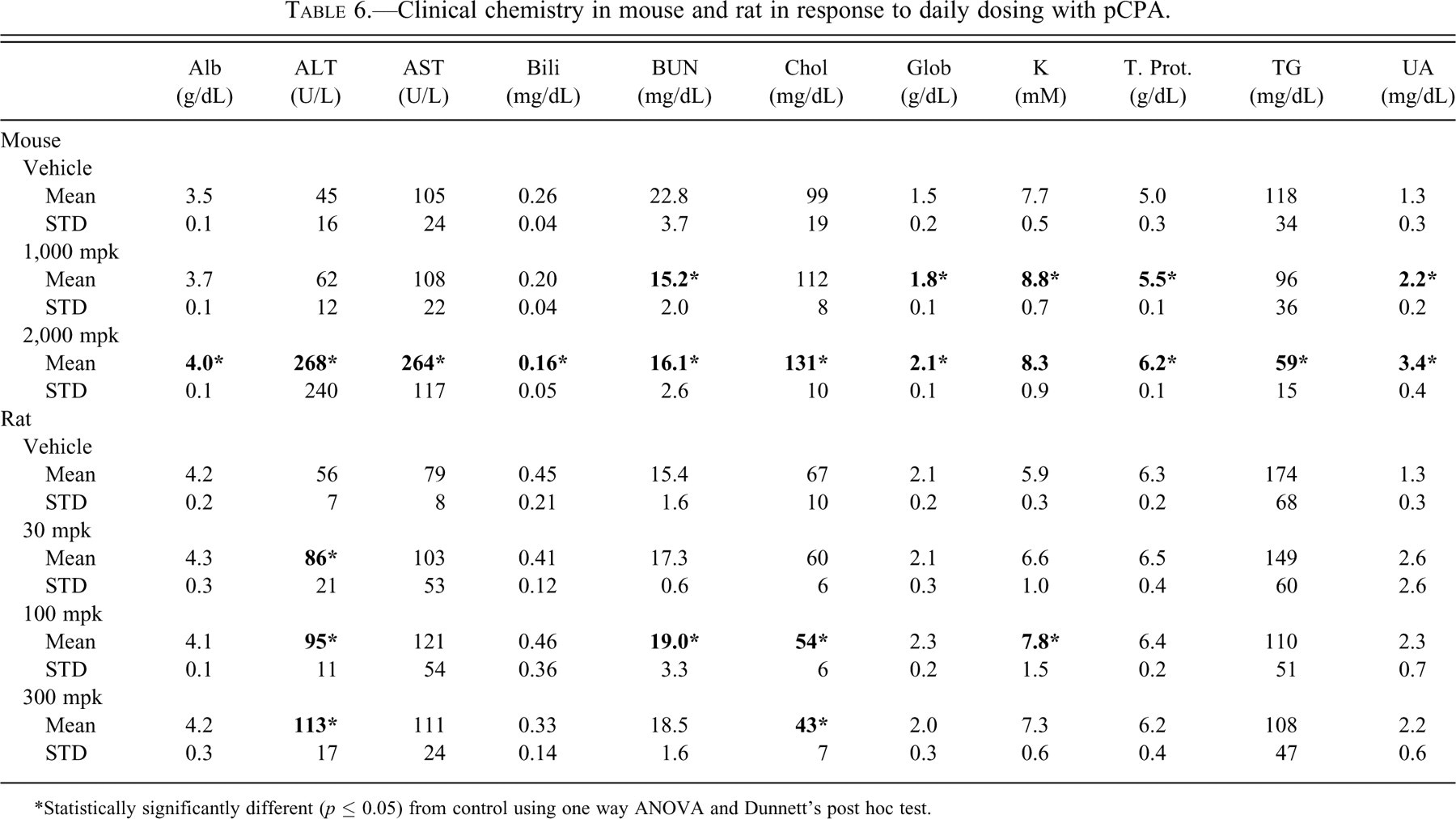
*Statistically significantly different (p ≤ 0.05) from control using one way ANOVA and Dunnett’s post hoc test.
Hematology in mouse and rat in response to daily dosing with pCPA.
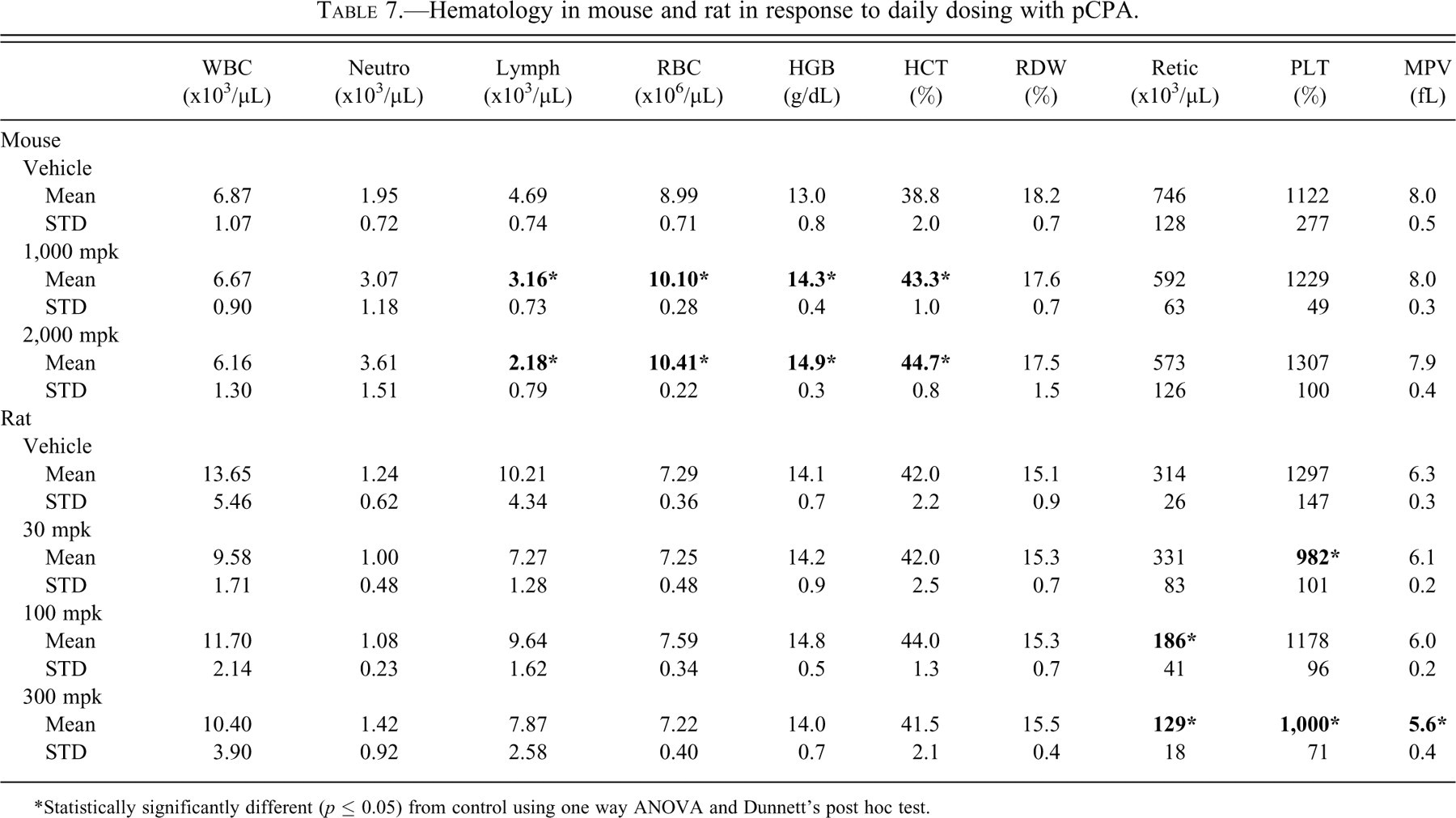
*Statistically significantly different (p ≤ 0.05) from control using one way ANOVA and Dunnett’s post hoc test.
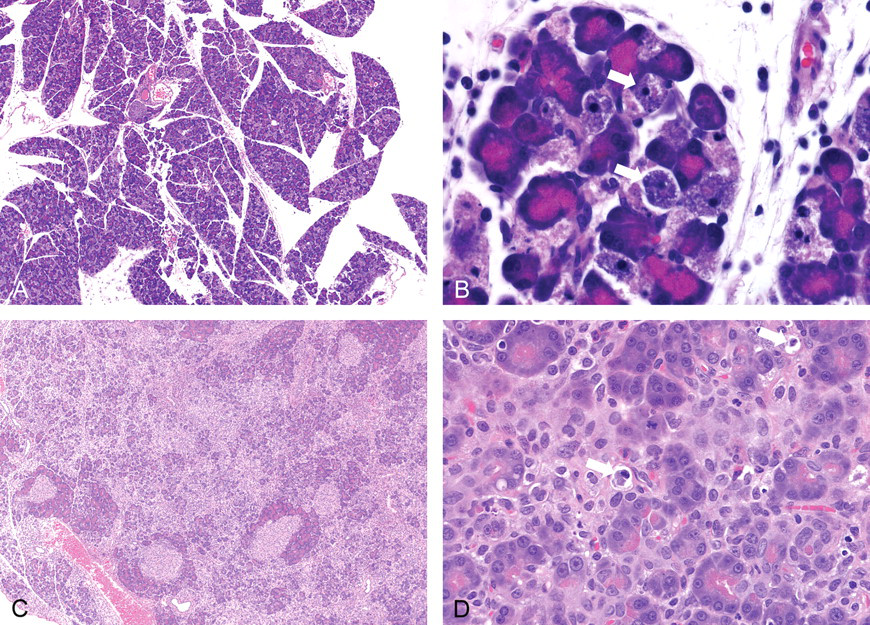
Pancreas toxicity of pCPA in the mouse and rat. Microscopic analysis of both mouse and rat showed prominent apoptosis of exocrine pancreas acinar cells. In the mouse, acinar cell apoptosis was observed in 2 of 4 animals dosed at 2,000 mg/kg and 1 of 5 mice dosed at 1,000 mg/kg. Apoptosis of pancreatic acinar cells was associated with rounding and shrinkage of cells, nuclear pyknosis and karyorrhexis/karyolysis (white block arrows). A similar lesion was observed in the rat, involving dose-related apoptosis of pancreatic exocrine cells causing degeneration and expansion of the interstitium with protein, histiocytes and infiltration of polymorphonuclear cells. (A) Mouse pancreas, 2,000 mg/kg, 20X magnification. (B) Mouse pancreas, 2,000 mg/kg, 40X magnification. (C) Rat, 300 mg/kg, 20X magnification. (D) Rat, 300 mg/kg, 40X magnification.
In the rat, treatment with pCPA caused statistically significant increases in ALT at all 3 dose levels assayed, and several other parameters were increased or decreased (Table 6). There were also reductions in reticulocytes, platelets, and mean platelet volume; whereas red cell parameters were unaffected (Table 7). Microscopic analysis revealed a dose-related apoptosis of pancreatic exocrine cells causing degeneration and expansion of the interstitium with protein, histiocytes and infiltration of polymorphonuclear cells. The lowest dose (30 mg/kg) showed rare apoptotic foci, scored as minimal; but at 100 mg/kg and above pancreases showed either early (mild to moderate) lesions of disseminated foci of exocrine apoptosis or later-stage lesions consisting of degeneration and inflammation described above (Figure 5).
Discussion
Although significant advances have been made in the development and application of in vitro toxicity assays, in vivo safety evaluation remains the most useful tool for identifying target organ toxicity (Fielden and Kolaja 2008). The primary goals of early safety evaluation are to characterize dose-limiting toxicity and to get an early estimate of the preclinical safety margin. Because essentially all compounds are toxic at sufficiently high doses, an early determination of a safety margin (i.e., the ratio of the lowest toxic exposure relative to an exposure that provides efficacy in a validated preclinical model of disease) is critical to positioning the identified dose-limiting toxicity. Identifying and characterizing the dose-limiting toxicity early, while the synthetic chemistry effort is active, allows the project team to address and minimize toxicity. Dose-limiting toxicity can be divided into three broad types of effect: adverse or exaggerated pharmacology, secondary pharmacology (or off-target toxicity), and compound- or template-mediated toxicity (Kramer, Sagartz, and Morris 2007), each of which is approached in a different manner. Overviews of the safety-related attrition suggest that a significant proportion of all compounds that failed because of toxicity were because of mechanism-based or “on-target” toxicities (Car 2006). Given its role as a common pharmacology model, it seems clear that the mouse may also be a suitable toxicology model for on-target toxicities. We sought to assess whether the mouse can faithfully predict adverse/exaggerated primary pharmacology as well as compound-mediated toxicity and off-target effects.
Adverse Primary Pharmacology: Methotrexate in the Mouse and Rat
Methotrexate was first approved by the FDA in 1953 and has been a standard of care for numerous oncology and autoimmune indications. It is believed that MTX functions by blocking nucleotide synthesis and the cell cycle, mediated by inhibition of dihydrofolate reductase activity (Genestier et al. 2000). Methotrexate has significant toxicity in humans and preclinical species associated with its primary mode of action. In the present studies, exposure and mean residence time were similar in both species at 30 mg/kg, and the terminal half-life appeared greater in the mouse than in the rat. In both the mouse and the rat, spleen weight and overall body weight were similarly affected at similar doses and exposure levels. Although methotrexate caused reductions in platelets in both species, effects on lymphocytes and on red blood cell parameters were not perfectly correlated between the species. However, although reductions in lymphocyte counts were not observed in mice after 5 days of dosing, lymphoid depletion in the thymic cortex was observed microscopically, suggesting that the effect on lymphocytes would not have been overlooked were a discovery candidate similar to MTX evaluated in the mouse. Similarly, reticulocyte numbers were profoundly lower after 5 days of dosing in the rat, suggesting an antiproliferative effect on the rapidly dividing progenitor cells. Reticulocyte assessments were not performed in the mouse MTX study; however, effects on a number of red cell parameters are consistent with reduction in reticulocytes in the mouse. Clinical chemistry results were generally similar between the two species, with the most obvious effect being modest but statistically significant effects on blood electrolytes. Microscopic lesions observed in both species included bone marrow hypoplasia and lesions in the small intestine. In the mouse, a reduction in extramedullary hematopoiesis was observed in the control group spleen, likely due to a regenerative hematopoietic response due to the fact that 5 TK bleeds were performed over 5 days instead of the usual 2. This effect was markedly reduced in the MTX-treated mice, indicating that MTX inhibited normal proliferation of hematopoietic cells. EMH was generally not observed in control rats, probably because of greater blood volume and smaller volume perturbation caused by TK sampling. Although not perfectly matched between mouse and rat, the results clearly demonstrate that the mouse did faithfully predict the nature of the dose-limiting toxicity of this compound (lesions attributable to decreased cell division in mitotically active tissues), demonstrating that the mouse successfully identified toxicity related to MTX’s primary mode of pharmacologic action.
Off-Target Effects: Lexicon Pharmaceuticals Oncology Compounds in the Mouse and Rat
LP-499399 was being studied at Lexicon for oncology indications before being terminated preclinically for insufficient safety margins. Hematology and pathology data from studies with LP-499399 in mice and rats indicated the presence of hemolytic anemia, as indicated by a dose-dependent decrease in RBC counts, hematocrit, and hemoglobin levels, and enlarged spleens with extramedullary hematopoiesis. Reticulocyte assessments were not performed in the mouse study at the time; however, effects on a number of red cell parameters were consistent with an effect on reticulocytes in the mouse that would parallel the findings in rat (Table 4). These findings were consistent with the destruction of circulating erythrocytes and demonstrated that LP-499399 was not well tolerated at doses necessary for efficacy.
LP-527795 was among a number of compounds identified as candidate backup molecules for LP-499399. Because the mouse had successfully predicted hemolysis with LP-499399, a short-term, repeat-dose in vivo mouse assay was incorporated into the team’s testing funnel to identify backup candidates that either lacked the hemolytic activity or had suitable safety margins for the effect, which was both reversible and monitorable. Several backup candidates were evaluated in the mouse, and LP-527795 was shown not to cause hemolysis (Table 4), a finding that was later confirmed in the rat. Running the backup candidates in mouse rather than rat allowed for more compounds to be evaluated in a shorter time frame, because of reduced compound requirements. In this example, the mouse faithfully predicted off-target adverse effects, while requiring significantly smaller amounts of compound than similar studies in the rat.
Compound-Mediated Toxicity: pCPA in the Mouse and Rat
pCPA has been studied in the clinic for treatment of carcinoid syndrome (Engelman, Lovenberg, and Sjoerdsma 1967) and as an antiemetic in cancer patients (Alfieri and Cubeddu 1995). Among its dose-limiting effects in animal models is an inhibition of pancreatic secretion (Bieger, Wacker, and Forssmann 1971) and an associated pancreatic toxicity (Furuse et al. 1993; Chen et al. 1996). The effect is independent of pCPA’s primary pharmacologic activity but rather appears to be due to inhibition of intracellular transport by “nonnatural” aromatic amino acids (Bieger and Kern 1975) and/or inappropriate incorporation into proteins and peptides (Forssmann and Bieger 1973).
In the present studies, pCPA was first assessed in single-dose toxicokinetic studies to determine its oral bioavailability in the mouse and rat, as much of the literature has used i.p. dosing in rats. Although AUC0-∞ and therefore oral bioavailability could not be calculated in the rat because of insufficient terminal phase, the TK studies demonstrated that whereas exposure in the mouse was significantly lower than in the rat, i.p. and p.o. administration provided similar exposure (Table 5). Clinical pathology data for the two compounds was not well conserved, in that there were a large number of parameters that were significantly different from control in the mouse that were unaffected in the rat (Tables 6 and 7). Many of these parameters, including red blood cells, hemoglobin, hematocrit, albumin, globulin, total protein, and uric acid, may reflect intravascular depletion in the mouse that was apparently not present in the rat. The pancreas was the primary target organ of pCPA as seen in both species, and even at 1,000 and 2,000 mg/kg the mouse required less compound than the rat at 100 and 300 mg/kg. These results show that the mouse can faithfully predict compound/template-mediated toxicity.
Other Examples of the Comparability between Mouse and Rat
Table 8 provides a summary of adverse findings in the mouse and rat for a number of small molecules. Although many of the compounds included in Table 8 are environmental contaminants and industrial chemicals, the results were still considered relevant to a comparison of toxicologic responses in mice and rats. For the purposes of summarizing the data, attempts were made wherever possible to highlight the toxicity that occurred at lowest doses (i.e., the dose-limiting adverse effects) in Table 8. Studies were categorized based on duration as “acute” (single-dose studies; panel A), “subchronic” (3-day to 4-week studies; panel B), and “chronic” (8- to 14-week studies; panel C). In the case of many of the acute studies, the main endpoint was often an LD50. In those cases where only an LD50 was reported, there was generally good agreement between the two species (Figure 6 ). A comparison of rat and mouse LD50s for 36 of the compounds in Table 8, panel A demonstrated a correlation coefficient of .9378. Although this may be of limited use in identifying dose-limiting toxicity or understanding toxicity mechanisms, acute toxicity studies provide valuable dose-setting information in advance of longer term studies. Additionally, in those examples where histopathology was assessed in the acute studies, there was generally good agreement between mouse and rat with respect to both the dose-limiting adverse finding and the dose/exposure at which the effects were observed.
Toxicologic effects of various compounds in rat and mouse.
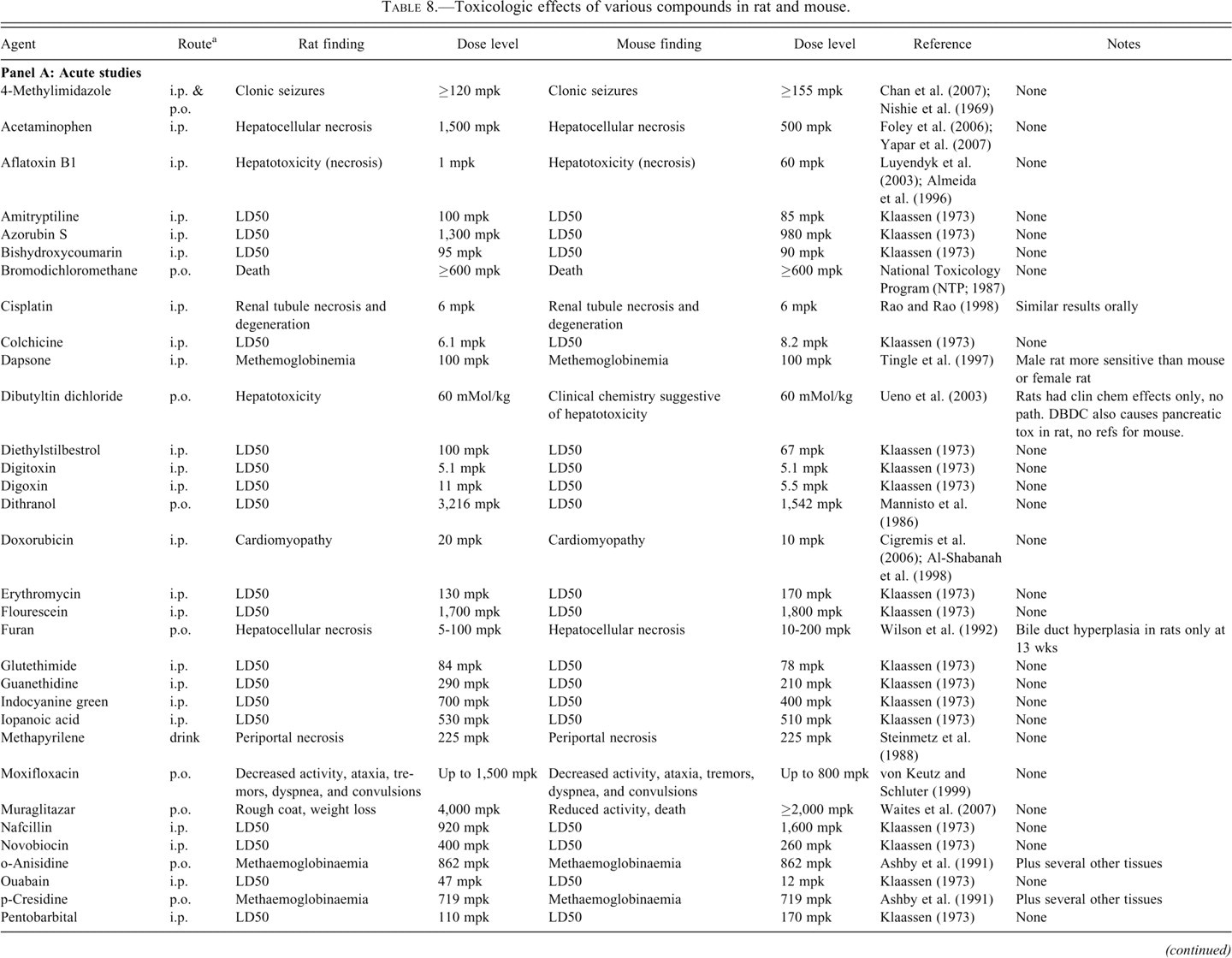
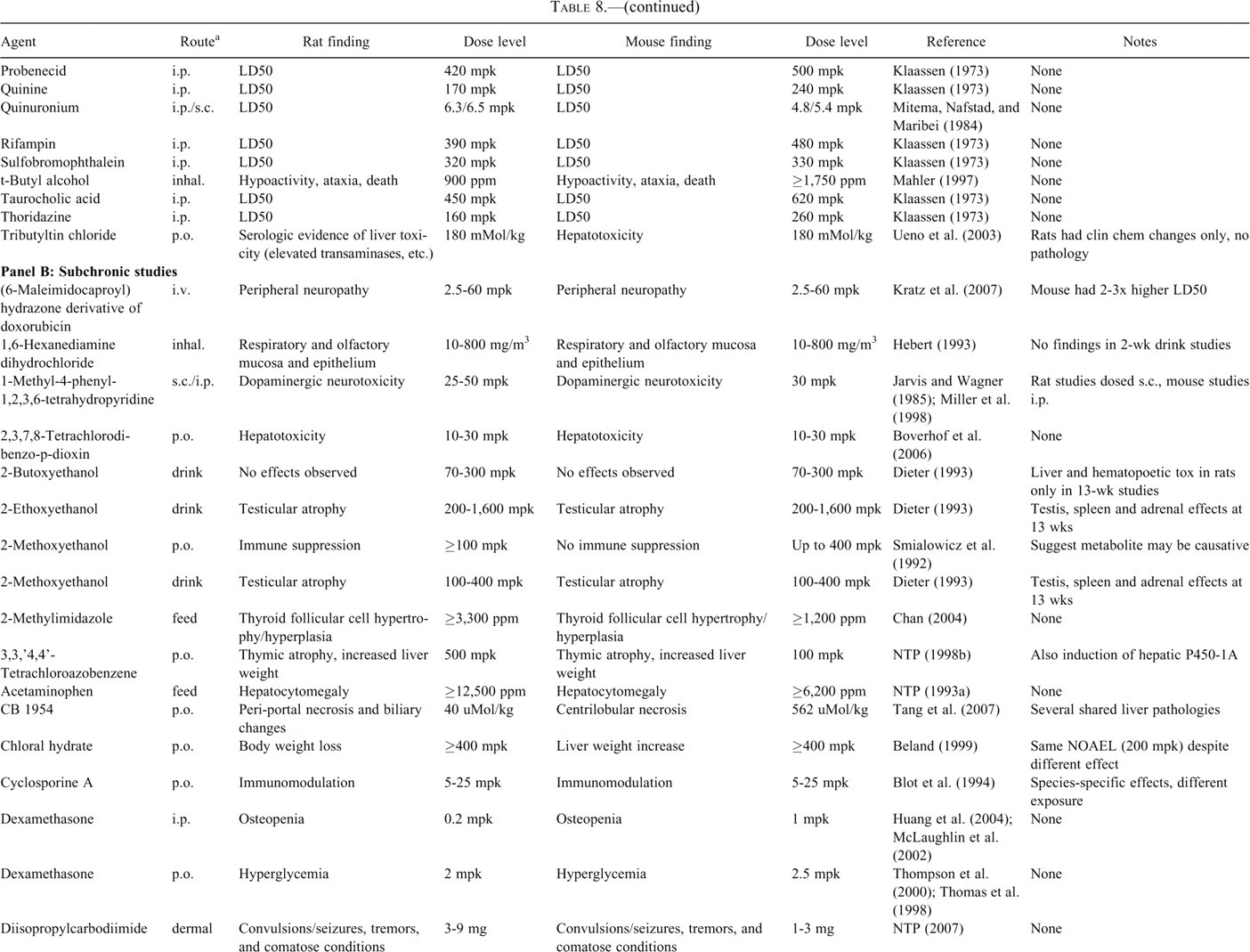
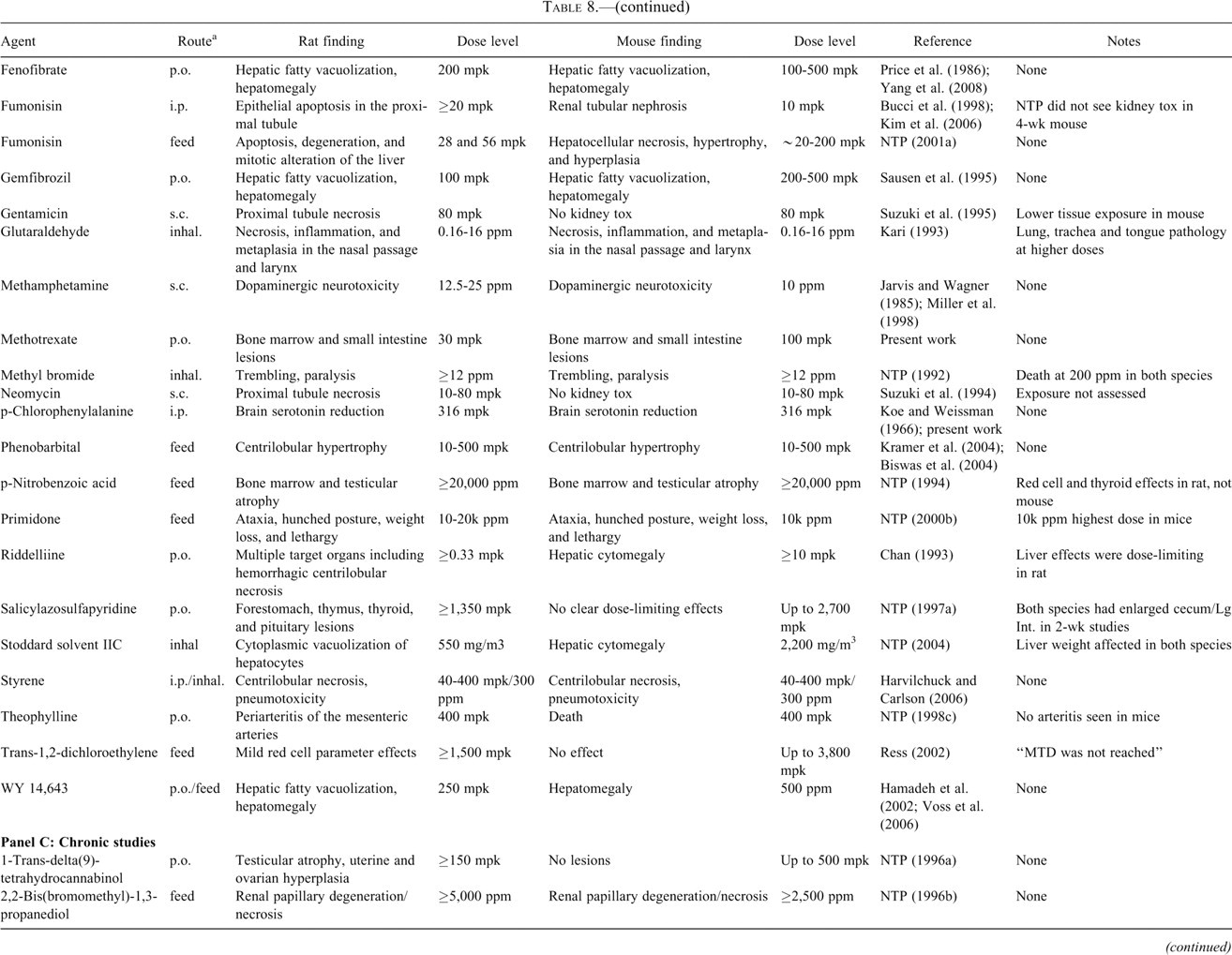
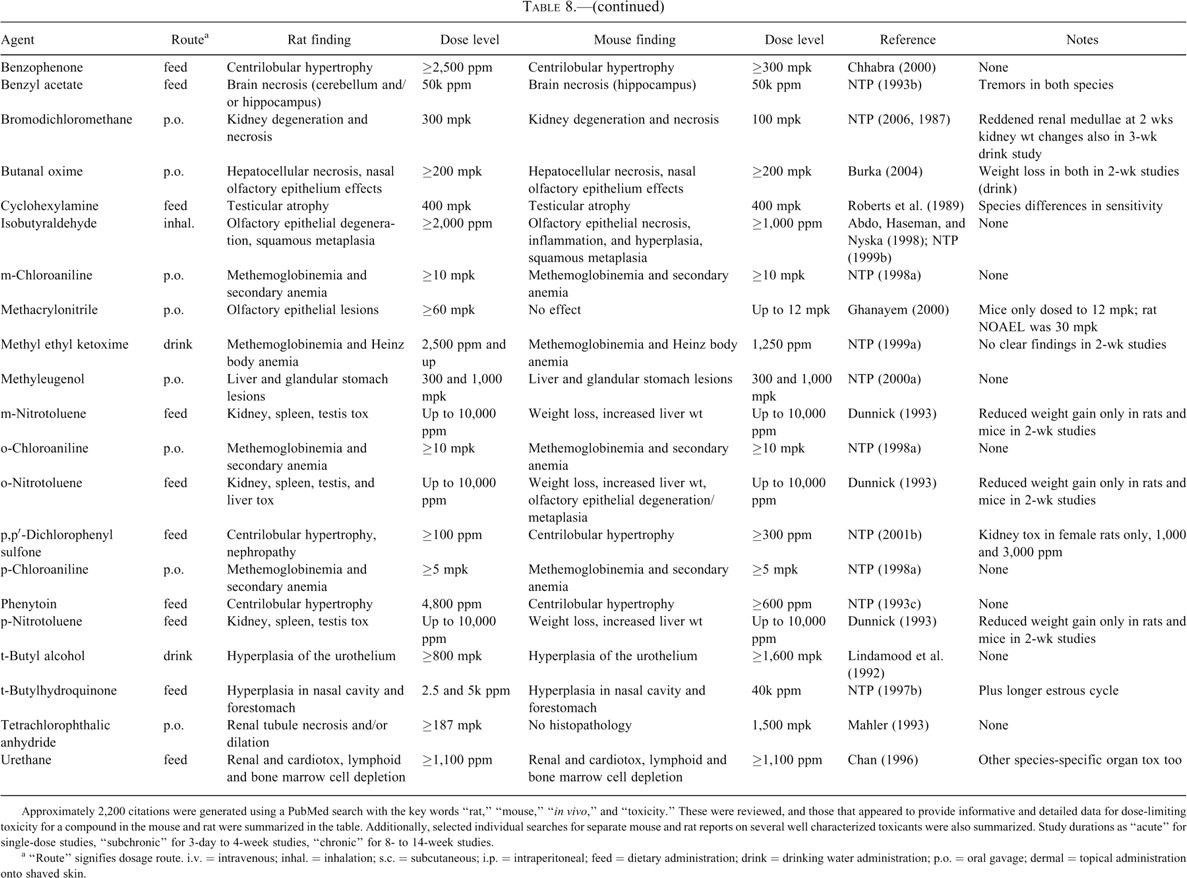
Approximately 2,200 citations were generated using a PubMed search with the key words “rat,” “mouse,” “in vivo,” and “toxicity.” These were reviewed, and those that appeared to provide informative and detailed data for dose-limiting toxicity for a compound in the mouse and rat were summarized in the table. Additionally, selected individual searches for separate mouse and rat reports on several well characterized toxicants were also summarized. Study durations as “acute” for single-dose studies, “subchronic” for 3-day to 4-week studies, “chronic” for 8- to 14-week studies.
a “Route” signifies dosage route. i.v. = intravenous; inhal. = inhalation; s.c. = subcutaneous; i.p. = intraperitoneal; feed = dietary administration; drink = drinking water administration; p.o. = oral gavage; dermal = topical administration onto shaved skin.
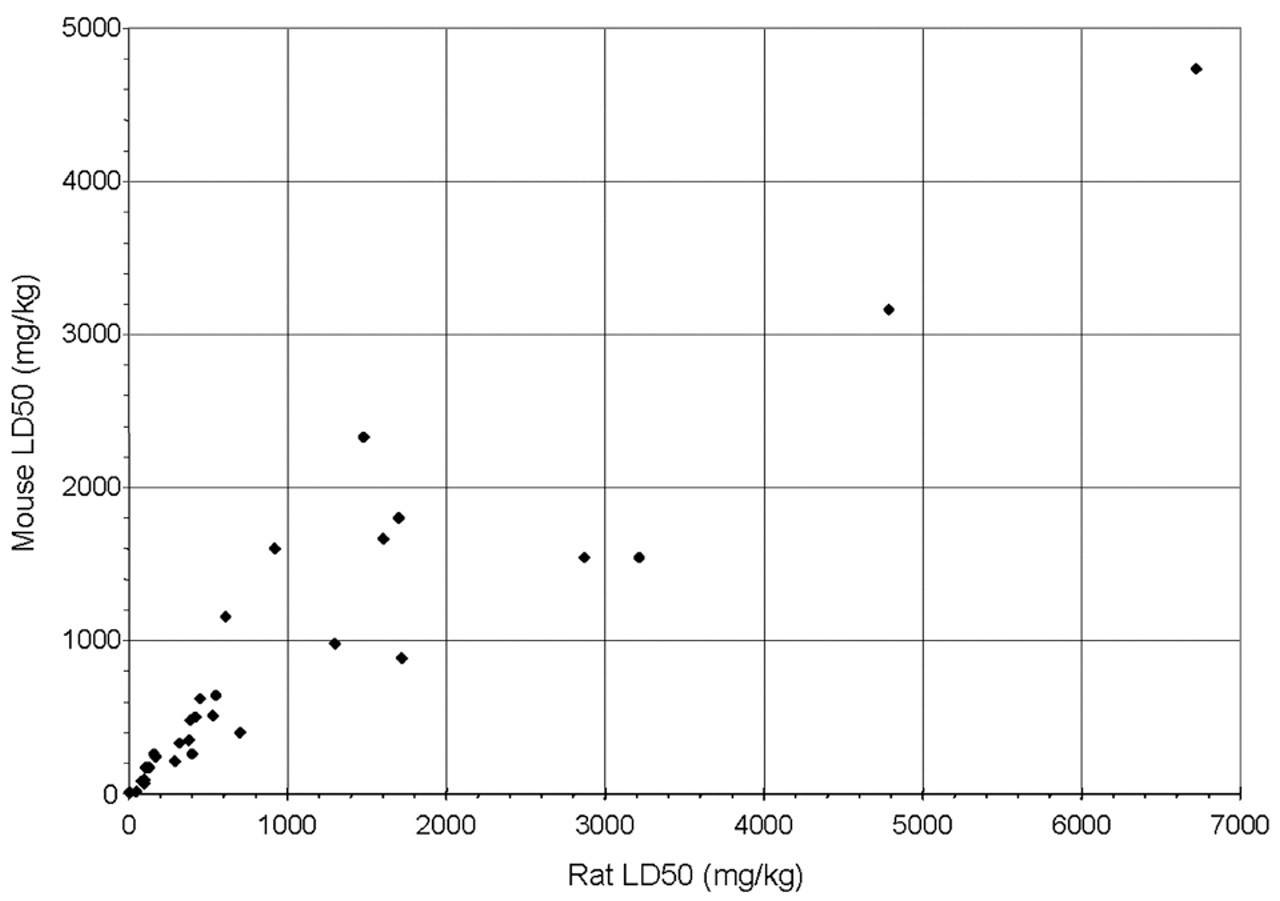
LD50s in the mouse and rat. LD50s in the rat and mouse were gathered for those compounds from Table 8 for which the data were available in both species. The data include p.o., i.p., and s.c. dosing, but comparisons were in all cases limited to like dosing routes. The correlation coefficient for the 36 comparisons was .9378, demonstrating that the mouse is a suitable model for early dose setting in the rodent.
There is also good agreement between the mouse and the rat for many of the compounds assessed in 3-day to 4-week studies. This is exemplified by our own experience with MTX, LP-499399, LP-527795, and pCPA. Table 8, panel C summarizes the results of a number of compounds that were evaluated chronically (8-14 weeks) in both the mouse and the rat. As with the subchronic studies, there was generally good agreement between the species with respect to dose-limiting target organs, although there were notable exceptions (ortho-, meta-, and para-nitrotoluene, tetrachlorophthalic anhydride, and 1-trans-delta(9)-tetrahydrocannabinol) in which mouse was not predictive of rat.
Among those compounds for which there was not concordance between the rat and mouse, the differences can in some cases be explained by discrepancies in exposure, species-specific metabolism, or failure to adequately escalate the dosage. Gentamicin dosed once daily subcutaneously for 7 consecutive days caused proximal tubule necrosis in the rat at 80 mg/kg but caused no such effect at the same dose in mice (Suzuki et al. 1995). Although higher dose studies were not performed, Suzuki et al. (1995) did demonstrate higher tissue exposure of gentamicin in the rat versus the mouse, suggesting that mouse may simply have had lower exposure to the agent. Methacrylonitrile caused olfactory epithelial lesions in the rat, but no adverse effects were reported in the mouse; however, upon closer inspection, the report states that a NOAEL for the rat was 30 mg/kg, whereas the mouse was not dosed above 12 mg/kg (Ghanayem 2000). In studies with salicylazosulfapyridine (National Toxicology Program 1997a) and trans-1,2-dichloroethylene (Ress 2002), no clear dose-limiting effects were observed in the mouse despite dosing up to 2,700 and 3,800 mg/kg, respectively; whereas dose-limiting tissue lesions were identified in the rat at 1,350 and 1,500 mg/kg, respectively. However, it is important to note that even in the rat, these compounds had to be dosed near a limit-dose before toxicity was observed, showing that the mouse did faithfully demonstrate that both compounds were comparatively benign. Although the toxicity of a prospective drug candidate is best interpreted in light of its relation to efficacy (i.e., safety margin), the demonstration of a very large margin early in a program’s chemistry effort would be much more easily achieved in the mouse than in the rat. Finally, in a small number of examples, there was no obvious reason the mouse was not predictive. In the case of 2-methoxyethanol, immune suppression was observed in rats upon treatment by oral gavage following 10 daily doses of 100 mg/kg and above, whereas no such effects were observed in mice up to 400 mg/kg (Smialowicz et al. 1992). Smialowicz et al. (1992) note that whether the differences are because of genuine immunologic differences or pharmacokinetic or metabolic differences remains to be determined.
Conclusions
In the majority of the examples assessed, the mouse faithfully identified the dose-limiting toxicities and target organs identified in the rat in single and short-term repeat-dose studies. Perhaps the greatest weaknesses of the mouse as an early preclinical toxicology model are related to exposure and blood volume. Principles of allometric scaling suggest that weight/surface area normalized clearance is often higher in the mouse than in the rat, resulting in a shorter half-life and lower overall exposure. In many of the examples in Table 8 where the mouse was less sensitive than the rat (i.e., dose-limiting toxicity occurred at higher doses in the mouse than in the rat), lower exposure was likely a primary cause. Indeed this appeared to be the case with pCPA in our hands. However, even if compounds must be administered in higher dosages in the mouse to achieve dose-limiting toxicity, lower body weight/surface area in the mouse typically translates to significant savings in compound requirement at a time when compound availability is highly limiting. Blood volume was also a limiting but not insurmountable factor in the use of the mouse for early safety assessment. Given the small blood volume of a mouse, repeated TK sampling has an impact on clinical pathology parameters and may not be commensurate with institutional animal care and usage blood volume guidelines. Although this has caused us to limit TK sampling in repeat-dose studies, we have found that a single-dose tolerability study with rich TK sampling serves as a useful dose-range-finding study in advance of the repeat-dose study and allows for collection of all of the usual pharmacokinetic parameters. Additionally, we have limited coagulation endpoints to rat studies and on occasion (e.g., instances of compound-mediated intravascular depletion) have had insufficient blood volume for the full set of the clinical pathology endpoints. In such cases, we have sacrificed measurement of reticulocytes, as red cell polychromasia and increased mean corpuscular volume can hint at an increase in reticulocytes in the absense of actual data.
It should also be noted that most studies use outbred lines of rats such as Sprague-Dawley, although inbred strains such as F344 are occasionally used. Additionally, whereas many mouse studies use F1 hybrids of inbred strains, the CD-1 outbred mouse line is perhaps most commonly used for safety assessment in the mouse. Genetic differences between inbred strains can cause considerable variation in response to toxicants. The use of multiple strains to avoid genetic bias has been proposed, although the practicality of testing in multiple strains has obvious limitations. In the present studies we chose to use the inbred C57 black 6 mouse line for the simple reason that all of the preliminary pharmacology data were generated in this mouse line. A primary consideration when characterizing the safety liabilities of small molecules is to characterize the dose-limiting toxicity and assess the potential for the compound to advance into regulatory studies and on into the clinic. A suitable therapeutic window or preclinical safety margin is a key to assessing the significance of the identified toxicity. Although there are undoubtedly differences between mouse lines with respect to exposure and sensitivity, we had no reason to believe that any particular strain of mice was more or less faithful in predicting toxicity observed in other species. We have found that by taking systemic exposure into account, the inbred mouse line used in this study was quite predictive of toxicities observed in the rat. The fact that an outbred mouse line was predictive suggests that an inbred line such as CD-1 may perform even better.
Although the rat has been the species of choice for the vast majority of preclinical toxicology studies performed in the evaluation of pharmaceutical candidates, the mouse can be a suitable model for very early safety assessment. In the past 4 years at Lexicon Pharmaceuticals, 8 compounds have been advanced into development, and many more molecules have been evaluated in both mouse and rat. These studies have demonstrated a high level of agreement between the mouse and the rat for both toxicology and pharmacology endpoints. An added advantage of performing early safety assessment in the mouse is the wide availability of mouse genetic models. The ability to assess the toxicity of a compound in a mouse model in which the compound’s primary pharmacology target has been knocked out provides an outstanding way to distinguish primary adverse pharmacology from secondary pharmacology and chemically mediated toxicity. Thus, in spite of blood volume limitations and generally higher clearance, we have found the mouse to be exceptionally useful both for lead prioritization and for dose-range finding in advance of rat studies. A testing paradigm that uses the mouse for early toxicity signal generation allows for the identification of toxicity liabilities earlier in a project’s life cycle. The earlier identification of safety issues facilitates the prioritization of discovery lead compounds and allows additional time in which to perform mechanistic toxicology studies in order to understand, manage, and possibly address issues while active structure-activity relationships are being developed.
Footnotes
Acknowledgments
The authors wish to thank Charles Cruz, Jianghong Jiang, Aixia Sun, and Liam Moran for bioanalytical support; and David Rawlins and Jane Almstead for synthetic chemistry support. We also thank Billy Hampton, Michelle Holmes, Ryan Vance, and June Wingert for necropsy support; and Kristen Loh, Brian Spanhel, Melissa Vetter, and Mary Wendt for clinical pathology support.