
Abstract
BMS-645737, an inhibitor of vascular endothelial growth factor (VEGF) receptor-2 and fibroblast growth factor (FGF) receptor-1, has anti-angiogenic activity and was evaluated in nonclinical studies as a treatment for cancer. This article characterizes the BMS-645737-induced clinical, gross, and histologic lesions of incisor teeth in Sprague-Dawley (SD) rats. Rats received 0 800 mg/kg BMS-645737 in a single-dose study or consecutive daily doses of 0 20 mg/kg/day in a 1-month study. The reversibility of these effects was assessed in the 1-month study. White discoloration and fracture of incisors were observed clinically and grossly in the 1-month study. In both studies, dose-dependent histopathologic lesions of incisors were degeneration and/or necrosis of odontoblasts and ameloblasts; decreased mineralization of dentin; inflammation and necrosis of the dental pulp; and edema, congestion, and hemorrhage in the pulp and periodontal tissue adjacent to the enamel organ. Partial recovery was observed at lower doses after a two-week dose-free period in the one-month study. Drug-induced incisor lesions were considered to be related to the pharmacologic inhibitory effects on VEGF and FGF signaling, that is, inhibition of growth and maintenance of small-diameter vessels that support the formation of dentin and enamel in growing teeth and/or to perturbances of function of odontoblasts and ameloblasts or their precursors.
Keywords
Introduction
Angiogenesis, the process of new blood vessel formation from preexisting ones, occurs in various physiologic and pathologic processes including embryogenesis, growth and development, pregnancy, endochondral bone formation, wound healing, inflammation, and neoplasia (Carmeliet 2003; Dai and Rabie 2007; Detmar et al. 1995; Frank et al. 1995; Presta et al. 2005; Reynolds, Grazul-Bilska, and Redmer 2002; Roy, Bhardwaj, and Yla-Herttuala 2006). Angiogenic factors, such as vascular endothelial growth factors (VEGFs) and fibroblast growth factors (FGFs), along with others, interact to promote angiogenesis by stimulating endothelial cells to grow, migrate, develop into vessels, and remodel. VEGFs and FGFs elicit their pro-angiogenic effects by binding to high-affinity cell surface tyrosine kinase receptors that stimulate signal transduction pathways leading to endothelial cell proliferation, migration, and survival (Ho and Kuo 2007; Roy, Bhardwaj, and Yla-Herttuala 2006; Zachary and Gliki 2001). In the adult, angiogenesis is not common during health, as formed blood vessels are stable and quiescent in most tissues (Shojaei and Ferrara 2008; Shweiki et al. 1993). Exceptions include the formation of new blood vessels during cyclic ovarian follicular and luteal development, cyclic endometrial changes and pregnancy, wound healing, and inflammation (Byrne, Bouchier-Hayes, and Harmey 2005; Demir, Seval, and Huppertz 2007; Frantz et al. 2005; Fraser 2006; Gerber et al. 1999; Girling and Rogers 2005; Reynolds, Grazul-Bilska, and Redmer 2002; Roy, Bhardwaj, and Yla-Herttuala 2006; Torry et al. 2007).
VEGF-A, a major mediator of angiogenesis and one of a group of heparin-binding growth factors, was initially described as a vascular permeability factor produced by carcinoma cells and subsequently as an endothelial cell mitogen (Connolly et al. 1989; Senger et al. 1983). In addition to VEGF-A, the VEGF family includes VEGF-B, -C, -D, -E, -F, and placental growth factor (Dvorak 2005; Ferrara, Gerber, and LeCouter 2003; Roy, Bhardwaj, and Yla-Herttuala 2006). VEGF-A, which binds to at least three receptor tyrosine kinases, VEGFR-1 (Flt-1), VEGFR-2 (Flk-1/KDR), and VEGFR-3 (Flt-4), is considered to be the most important angiogenic growth factor of this family and is commonly referred to as simply VEGF (Dvorak 2005; Homsi and Daud 2007; Otrock, Makarem, and Shamseddine 2007; Shojaei and Ferrara 2008). Although VEGFR-1, -2, and -3 are all expressed on endothelial cells, the angiogenic activity of VEGF is mediated primarily through VEGFR-2, whereas VEGFR-3 plays an important role in lymphangiogenesis (Ho and Kuo 2007; Roy, Bhardwaj, and Yla-Herttuala 2006; Zachary and Gliki 2001). VEGFR-1 may function as a negative regulator of VEGFR-2 and is associated with the VEGF-mediated chemotaxis of monocytes/macrophages (Clauss et al. 1990; Shen et al. 1993). Additionally, in the presence of both VEGF and platelet-derived growth factor (PDGF), VEGFR-2 inhibits pro-angiogenic events through suppression of pericyte/vascular smooth muscle function and vessel maturation (Greenberg et al. 2008).
During embryonic and postnatal development, VEGF plays a critical role in angiogenesis, vasculogenesis, and lymphangiogenesis and is expressed in most tissues (Ferrara 1999; Ho and Kuo 2007; Matsumota and Claesson-Welsh 2001). The importance of VEGF in the developing fetus is illustrated by the observations that a mutation in the VEGFR-1 or VEGFR-2 gene or deletion of a single VEGF allele in mice results in embryonic lethality (Carmeliet et al. 1996; Ferrara et al. 1996; G.-H. Fong et al. 1995; Shalaby et al. 1995).
Although VEGF is necessary for tissue development, growth, and repair, it is generally regarded that established vasculature in adults is less dependent on VEGF for maintenance (Shojaie and Ferrara 2008). However, other data suggest that the developed vasculature of some tissues, including pancreatic islets, pituitary gland, and small intestinal villi, is dependent on VEGF for maintenance, without which the capillary network undergoes regression (Ferrara and Davis-Smyth 1997; Kamba et al. 2006; Monacci, Merrill, and Oldfield 1993). Additionally, VEGF inhibition has been associated with the development of emphysema in rats and a reduction in the number of tracheal capillaries in mice (Baffert et al. 2004; Kasahara et al. 2000).
VEGF has been implicated in the pathogenesis of several diseases including iris or intraocular neovascularization, psoriasis, endometriosis, chronic obstructive pulmonary disease, osteoarthritis, rheumatoid arthritis, and neoplasia (Adamis et al. 1996; Aiello et al. 1995; Carvalho, Blank, and Shoenfeld 2007; Gille et al. 2001; Kanazawa 2007; McLaren et al. 1996; Murata, Yudoh, and Masuko 2008; Ng, Krilleke, and Shima 2006).
The FGF family in mice and humans consists of at least twenty-two distinct molecules and additional isoforms that have a conserved 120 amino acid core domain (Itoh 2007; Itoh and Ornitz 2004; Ornitz and Itoh 2001; Presta et al. 2005). Like the VEGF family, the FGF family is a group of heparin-binding growth factors. The prototypical FGFs, acidic FGF (FGF-1) and basic FGF (FGF-2), were originally identified as mitogens for cultured fibroblasts (Gospodarowicz 1974; Itoh 2007). FGFs mediate cellular responses by binding to four receptor tyrosine kinases, FGFR-1, -2, -3, and -4 (Eswarakumar, Lax, and Schlessinger 2005). The functions of FGF/FGFR ligand/receptor pairs are less well characterized than those of VEGF/VEGFR pairs (Auguste, Javerzat, and Bikfalvi 2003).
The FGFs are broad-spectrum mitogens, induce cell migration and differentiation, and have a role in a number of physiologic processes including embryogenesis and development, angiogenesis, neuronal development and maintenance, wound healing, and hematopoiesis (Grose and Dickson 2005; Miller et al. 2000). The diverse and extensive functions of the FGF family reflect the observation that FGFRs are expressed on essentially all cell types (Cao, Cao, and Hedlund 2008). In vitro and in vivo, FGFs stimulate endothelial cell proliferation, migration, and differentiation (Auguste, Javerzat, and Bikfalvi 2003; Itoh 2007). The role of FGFs in embryogenesis and early development has been demonstrated by targeted disruption of individual FGF genes in mice. However, the phenotypes of these mice range from embryonic lethality to subtle changes in adults, suggesting redundancy of the FGF family or expression of individual FGFs in specific tissues (Auguste, Javerzat, and Bikfalvi 2003; Itoh and Ornitz 2004; Miller et al. 2000). Disruption of the FGF-1 or FGF-2 gene (single or double knockout) results in viable offspring without a defective vascular phenotype, whereas disruption of the FGFR-1 or FGFR-2 gene results in death around the time of implantation, prior to development of the vascular system (Arman et al. 1998; Deng et al. 1994; Dono et al. 1998; Miller et al. 2000).
Genetic alterations of FGF or FGFR genes have been associated with skeletal disorders, neoplasia, and other conditions (Eswarakumar, Lax, and Schlessinger 2005; Itoh 2007). Mutations in FGFR-1, -2, and -3 have been identified in craniosynostosis, and mutations in FGFR-3 have been identified as causes of dwarfism (Webster and Donoghue 1997; Wilkie 1997). Mutations in FGF-10, -14-, and -23 are causes of lacrimal and salivary gland aplasia, hereditary spinocerebellar ataxias, and autosomal dominant hypophosphatemic rickets, respectively (ADHR Consortium 2000; Entersarian et al. 2005; van Swieten et al. 2003). Increased expression of FGFs has been identified in several types of cancer, including prostate, bladder, kidney, testicular, and lung cancers (Cronauer et al. 2003; Dorkin et al. 1999; Marek et al. 2009). Ectopic expression or mutations of FGFRs have been associated with endometrial cancer and multiple myeloma (Byron et al. 2008; Paterson et al. 2004).
VEGF and FGF are important in tumor growth and metastasis and are overexpressed or aberrantly expressed in many types of neoplasms relative to non-neoplastic tissues (Boocock et al. 1995; Brahimi-Horn and Pouyssegur 2006; Cappellen et al. 1999; Chesi et al. 1997; Dorkin et al. 1999; Ferrara and Davis-Smyth 1997; Homsi and Daud 2007; Marsh et al. 1999; Valve et al. 2000; van Rhijn et al. 2001). While VEGF contributes to tumor growth primarily as an inducer of angiogenesis, FGFs may contribute to tumor growth as angiogenic agents, mitogens for neoplastic cells, or inhibitors of apoptosis (Cronauer et al. 2003). The increased expression of VEGF and/or FGF in neoplastic tissues along with the increased expression of VEGFR in tumor endothelial cells compared to non-neoplastic endothelial cells may help explain the apparent specificity of VEGF/FGF antagonists for neoplastic tissue (L. Brown et al. 1993; Ho and Kuo 2007; Plate et al. 1992, 1994).
Pharmacologic inhibitors of angiogenesis have been shown to provide therapeutic benefit in the treatment of diseases in which angiogenesis plays a major role. This is evidenced by the nonclinical efficacy of inhibitors of receptor tyrosine kinase signaling (FGF, PDGF, and/or VEGF) in suppressing tumor xenograft growth in mice; the clinical use of the tyrosine kinase inhibitors sunitinib, sorafenib, SU6668, and SU5416 in advanced malignancies; and the clinical activity of the anti-VEGF monoclonal antibodies, bevacizumab and ranibizumab, in the treatment of metastatic cancers and neovascular age-related macular degeneration, respectively (T. Fong et al. 1999; Hurwitz et al. 2004; Kieran et al. 2009; Roodhart et al. 2008; Rosenfeld et al. 2006; Sachdev and Jahanzeb 2008; Yang et al. 2003). Therefore, inhibitors of VEGF or FGF signaling continue to be investigated as means to control solid tumor growth and other angiogenesis-mediated diseases (Arora and Scholar 2005; Bergers et al. 2003; Chen and Forough 2006; Kiselyov, Balakin, and Tkachenko 2007; Manetti and Botta 2003; Rusnati and Presta 2007; Zakrzewska, Marcinkowska, and Wiedlocha 2008). A number of strategies to block VEGF/FGF signaling pathways have been explored, including ribonucleic acid aptamers that bind extracellular VEGF (Gragoudas et al. 2004), anti-VEGF-derived antibody binding fragments (Fab) that bind extracellular VEGF (Rosenfeld et al. 2006), monoclonal antibodies against VEGF (Ferrara et al. 2004; Ryan et al. 1999), monoclonal antibodies that bind VEGFR (Brekken et al. 2000), small-molecule inhibitors of VEGFR or FGFR tyrosine kinase activity (Arora and Scholar 2005; Manetti and Botta 2003; Rusnati and Presta 2007; Wedge et al. 2005, 2000), and small molecules that bind extracellular FGF (Manetti, Corelli, and Botta 2000).
The microscopic changes occurring in rats, dogs, and nonhuman primates administered repeat doses of inhibitors of VEGF, FGF, or multiple receptor tyrosine kinases have been reported. In four- and thirteen-week studies in cynomolgus macaques, recombinant humanized anti-VEGF (rhuMAbVEGF), a monoclonal antibody against VEGF, caused physeal dysplasia of the femur and humerus that was characterized by thickening of the growth plate cartilage, retention of hypertrophied chondrocytes, inhibition of vascular invasion into the zone of hypertrophied chondrocytes, and formation of a subchondral bony plate (Ryan et al. 1999). Additional changes in female macaques were decreased ovarian weights, due to a decrease in the number of corpora lutea, and decreased uterine weights in the absence of microscopic correlates. In a two-week repeat-dose study in rats, ZD4190, an inhibitor of VEGF receptor tyrosine kinase activity, induced dose-related increases in the thickness of the epiphyseal zone of hypertrophy in the femur and tibia (Wedge et al. 2000). Similarly, in a 4-week repeat dose study, ADZ2171, an inhibitor of VEGF, PDGFβ, and c-KIT receptor tyrosine kinase activity, caused dose-dependent increases in the hypertrophic chondrocyte zone of the tibial and femoral growth plates in rats and a marked reduction in the number of corpora lutea in the ovary (Wedge et al. 2005). In thirteen-week (rats and cynomolgus monkeys) and six- (rat) and nine-month (cynomolgus monkeys) studies, sunitinib, an inhibitor of multiple receptor tyrosine kinases, caused epiphyseal growth plate thickening, ovarian follicular atresia and decreased number of corpora lutea, and endometrial atrophy (Patyna et al. 2008). Dental caries of rodent incisors have also been observed in rats administered sunitinib, and dental dysplasia was reported in rats administered an unspecified VEGF inhibitor (Hall, Westwood, and Wadsworth 2006; Patyna et al. 2008).
The spectrum of toxic lesions associated with inhibition of FGF signaling overlaps those caused by inhibition of VEGF signaling. PD176067, a small-molecule inhibitor of FGF receptor tyrosine kinase and, at higher concentrations, VEGF receptor tyrosine kinase, induced dose-related physeal dysplasia (increased thickness of the zones of chondrocyte proliferation and hypertrophy) of the femur, tibia, and sternum along with hyperphosphatemia and soft tissue mineralization in seven-week-old and eleven-month-old female rats treated for fourteen days and juvenile dogs treated for up to two weeks (A. Brown et al. 2005; Courtney et al. 2003; Datta, Graziano, and Courtney 2003).
The epiphyseal growth plate and ovarian changes that occur as a result of targeted inhibition of receptor tyrosine kinases or their ligands are recognized as pharmacologic effects of a class of drugs that have anti-angiogenic activity (Hall, Westwood, and Wadsworth 2006; Patyna et al. 2008).
BMS-645737, a small molecule that selectively and competitively inhibits both VEGFR-2 and FGFR-1 tyrosine kinases, has anti-angiogenic activity and was evaluated as a possible treatment of cancer. The gross and microscopic descriptions of degenerative changes in the incisors of rats described in this report were observed during the conduct of a single dose and a one-month repeat-dose toxicity study with BMS-645737 in rats.
Materials and Methods
Test Compound
BMS-645737 was synthesized by the Bristol-Myers Squibb Company Research and Development facility in Candiac, Quebec, Canada. In the single-dose study, the control article and test article carrier (vehicle) were polyethylene glycol (PEG) 400. For the one-month study, the control vehicle was 50% PEG 400 in water and the test article carrier was 50% PEG 400 and 0.01 M methanesulfonic acid (MSA) in water.
Animals
For both the single-dose and the one-month studies with BMS-645737, male and female SD rats were obtained from Charles River Laboratories in Raleigh, North Carolina. On day 1 of the single-dose study, animals were eight weeks old and weighed approximately 240 to 265 grams (males) or 170 to 200 grams (females). SD rats in the one-month study were approximately nine weeks old and weighed approximately 270 to 345 grams (males) or 185 to 240 grams (females) on day 1 of the study.
Animals were randomly assigned to dose groups for each study using a computer-generated random list to achieve similar group mean body weights. Rats were housed individually in stainless steel wire-bottom cages. Rats were provided pelleted food (Harlan Teklad Diet #2018C: Certified 18% Protein Rodent Diet; Harlan Teklad, Madison, WI) daily except during fasting periods that occurred for scheduled necropsy. Purified tap water was provided ad libitum via an automatic watering system. Humidity and temperature were maintained within acceptable ranges (approximately 30–70% and 18–26°C, respectively) for the duration of the study. In addition, study rooms were maintained on a twelve-hour light-dark cycle. The studies were conducted in an Association for the Accreditation and Assessment of Laboratory Animal Care (AAALAC)–accredited facility and in accordance with the National Research Council’s Guide for the Care and Use of Laboratory Animals (Institute of Laboratory Animal Resources, 1996) and facility standard operating procedures (SOPs). Animal care and study procedures were approved by the facility’s Institutional Animal Care and Use Committee.
Experimental Design
In the single-dose study, single doses of 0 (vehicle control), 200, 400, or 800 mg/kg of BMS-645737 were administered by oral gavage to rats (five rats/sex/group) at a dose volume of 20 mL/kg. Scheduled necropsies were conducted on day 15.
In the 1-month study, single daily doses of 0 (vehicle control), 2, 6, or 20 mg/kg of BMS-645737 were administered by oral gavage to fifteen rats/sex/group at a dose volume of 10 mL/kg. The dose for the high-dose group was lowered to 15 mg/kg/day on day 18 (females) or 19 (males) due to the deteriorating condition of the animals. The high dose is hereafter referred to as 20/15 mg/kg/day. End-of-dose necropsies on early-terminated high-dose rats were conducted on days 22 (females) or 23 (males) and on day 30 for the other groups. Following a two-week, dose-free observation period, necropsies were conducted on days 36 (females) or 37 (males) for the high-dose group and on days 43 (females) or 44 (males) for the other groups.
Experimental Procedures
Animals were euthanatized at the designated times; complete necropsies were performed; and representative samples of a complete panel of tissues, including mandibular incisors and molars, were collected at necropsy, fixed in 10% neutral buffered formalin, and processed for light microscopy. After a minimum of forty-eight hours fixation, the mandible and attached teeth were placed in a commercially prepared decalcification solution containing formalin and formic acid (Cal-rite, Richard Allen Scientific, Kalamazoo, MI, USA). Following initial decalcification, a single longitudinal section of the mandible with incisor and three transverse sections of incisor or mandible with molar and incisor were collected (Figure 1 ). Following final decalcification the tissues were embedded in paraffin, sectioned at 4 μm, and stained with hematoxylin and eosin. Data for sections of nonerupted incisors (supported by mandibular bone) and sections of erupted incisors (distal to gingival margin) were tabulated and interpreted collectively. Only the dental clinical observations, mortality data, and gross and microscopic lesions of teeth and select other lesions that have been described for inhibitors of VEFG/FGF signaling pathways are discussed in this communication.
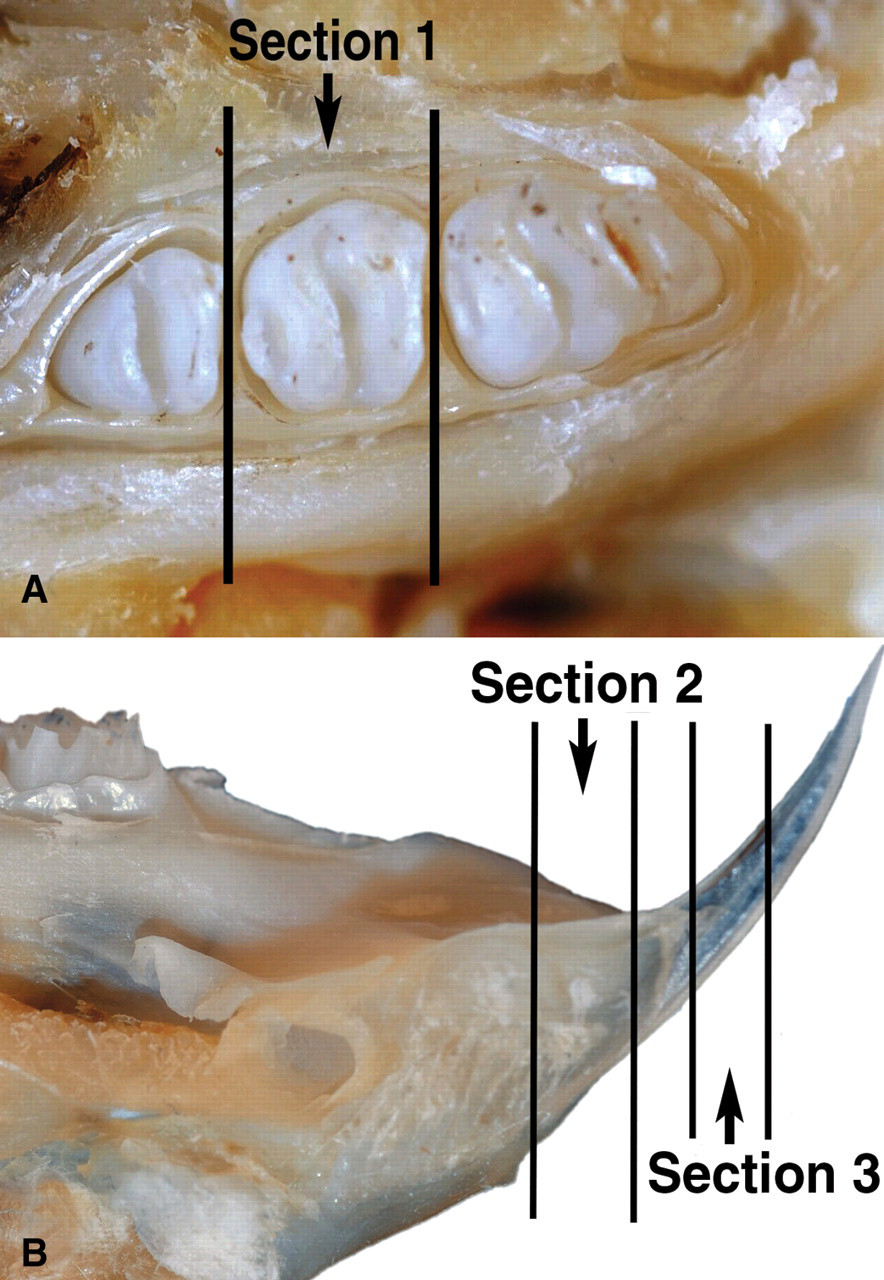
Photograph of rat mandible demonstrating sectioning levels for molar and incisor. (A) Transverse section at level of second molar (section 1). (B) Transverse sections at junction of incisor and gingiva (section 2) and erupted incisor (section 3).
Results
Mortality and Dental Clinical Observations
In the single-dose study with BMS-645737, drug-related mortality occurred on days 3 to 5 in one of five males given 400 mg/kg and 3 of 5 males given 800 mg/kg. Incisor abnormalities were not observed clinically in any animals.
In the one-month study with BMS-645737, drug-related mortality occurred on days 15 to 22 in six of fifteen male and seven of fifteen female rats given 20/15 mg/kg/day. Incisor abnormalities were not observed clinically during the dosing period. However, on days 42 and 43, which were during the two-week dose-free observation period, broken incisors were observed in three of five male rats given 6 mg/kg/day. As noted previously, animals given 20/15 mg/kg/day did not survive beyond day 37.
Gross Pathology
In the single-dose study with BMS-645737, no drug-related macroscopic dental lesions were observed. In the one-month study with BMS-645737, drug-related, macroscopic dental lesions were observed only at necropsies conducted following the two-week postdose observation period. Broken or missing incisors and/or white discoloration of incisors were observed in rats given BMS-645737 at 6 or 20/15 mg/kg/day (Table 1 ).
Incidence of gross incisor lesions in rats administered BMS-645737 for up to one month followed by a two-week dose-free period.
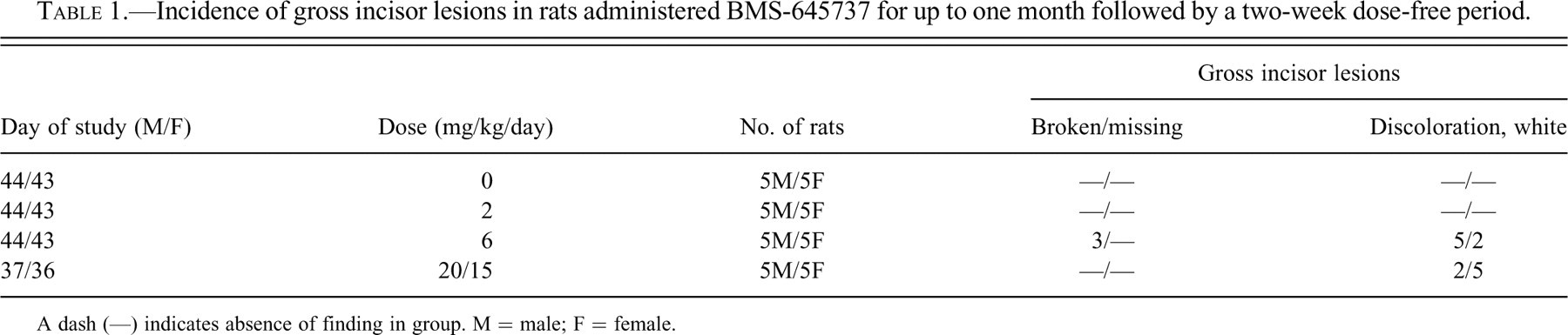
A dash (—) indicates absence of finding in group. M = male; F = female.
Histopathology
Single-Dose Study
Drug-related microscopic changes in the incisors were observed in all dose groups at day 15 following administration of a single dose of BMS-645737. Changes were not observed in controls (Figure 2a) or in rats that died or were euthanatized on days 3 to 5. Dental lesions were observed in 100% of treated females and in four of five, four of five, and two of five males given BMS-645737 at 200, 400, or 800 mg/kg, respectively. Lesions were more prominent in nonerupted than erupted incisors and were not observed in the molars. Lesions occurred primarily in the dentin, along with changes in the odontoblasts and enamel organ. There was thinning or irregular thickness of the dentin layer with disruption of the tubular architecture (Figures 2b-e). The disrupted dentin had a “marbleized” appearance, characterized by amorphous, irregular foci of nonstaining, pale eosinophilic, or amphophilic dentin. Occasionally the abnormal dentin contained inclusions of cellular debris, which were most likely necrotic odontoblasts shed into the newly formed dentin. In transverse sections of incisors, the outer layer of dentin (oldest dentin, formed prior to dosing) appeared normal, whereas the inner layer (dentin formed after dosing) was thin or irregular in thickness. The layer of odontoblasts was thin and populated by fewer and less elongate cells than normal, and cells in the papillary epithelium of the enamel organ were vacuolated.
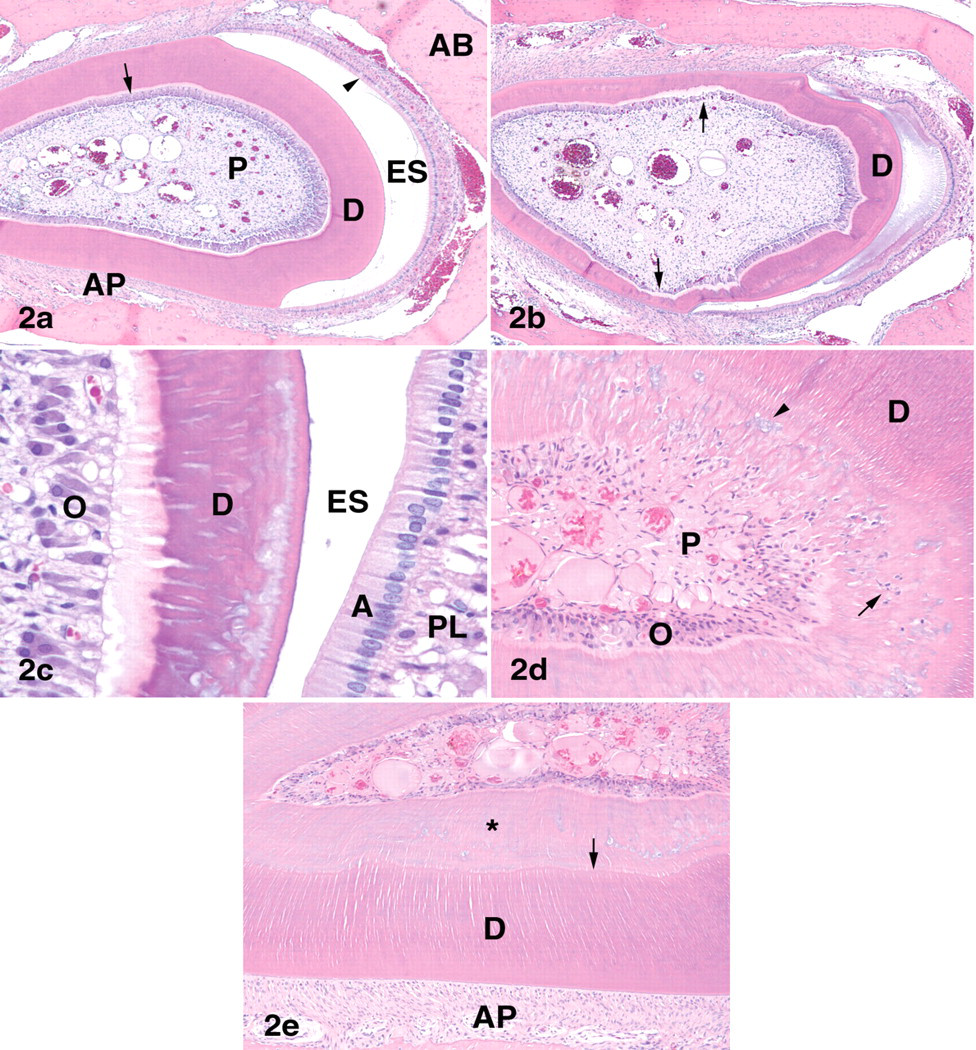
(A) Transverse section of nonerupted mandibular incisor at the level of the second molar, vehicle-treated female rat. Arrow = odontoblasts; arrowhead = ameloblasts. Hematoxylin and eosin (HE) × 50. (B-E) Incisors of rats administered a single dose of BMS-645737 at 800 mg/kg, postdose day 15. (B) Nonerupted incisor. The dentin layer is variable in width and irregularly mineralized. Note thinning of odontoblast layer (arrows). HE × 50. (C) Nonerupted incisor. Note irregular foci of poorly mineralized, amphophilic dentin, vacuolation and decreased size and number of odontoblasts, and vacuolation of papillary layer of enamel organ. Ameloblasts appear normal. HE × 400. (D) Erupted incisor. Dentin contains foci of irregular mineralization (arrowhead) and inclusions of cellular debris (arrow). Note degeneration of odontoblasts. HE × 200. (E) Nonerupted incisor near gingival margin. The outer layer of dentin is normal and distinct from the inner layer (*), which exhibits irregular mineralization. Note distinct line of demarcation between normal and abnormal dentin (arrow). HE × 200. D = dentin; O = odontoblasts; P = pulp; A = ameloblasts; ES = enamel space; PL = papillary layer of enamel organ; AP = alveolar periosteum.
A microscopic finding attributed to the pharmacologic effects of BMS-645737 was increased thickness of the distal femoral physis with chondrocyte hypertrophy. Mineralization of multiple soft tissues, including cardiac muscle, coronary arteries, aorta, kidney, stomach, colon, lung, ovary, and arteries of the lung, mesentery, pancreas, and tongue, was also observed.
One-Month Study
Microscopic lesions were observed in incisors in all dose groups following one month of daily dosing with BMS-645737 but were not observed in control animals. Dental lesions occurred in all rats given BMS-645737 at 6 or 20/15 mg/kg/day and were present in the odontoblasts, dentin, ameloblasts, enamel, and/or dental pulp. There was degeneration, single-cell necrosis, diffuse necrosis, or loss of odontoblasts (Figures 3a-b). Recently formed dentin was markedly thinned; poorly or irregularly mineralized with disruption of the tubular structure; and sharply demarcated from the normal, more mature dentin. There was relative expansion of the pulp to accommodate the increased pulp cavity diameter as a result of thinning of the dentin. At the labial margin of the incisor, in addition to thinning and disruption of dentin and loss of odontoblasts, there were often partial to complete loss of ameloblasts with collapse of the enamel space, or structural deformities of the formed enamel that was present in some sections. Hemorrhage, congestion, and/or edema of the pulp and periodontal ligament and necrosis, neutrophil infiltration, and thrombosis of the pulp were observed in some rats (Figures 3c-e). Neutrophil infiltration of the pulp was most common in the erupted part of the incisor and was considered secondary to incisor fracture. In two male and one female rats given BMS-645737 at 2 mg/kg/day, thinning of the dentin was observed in the absence of other degenerative or necrotic changes.
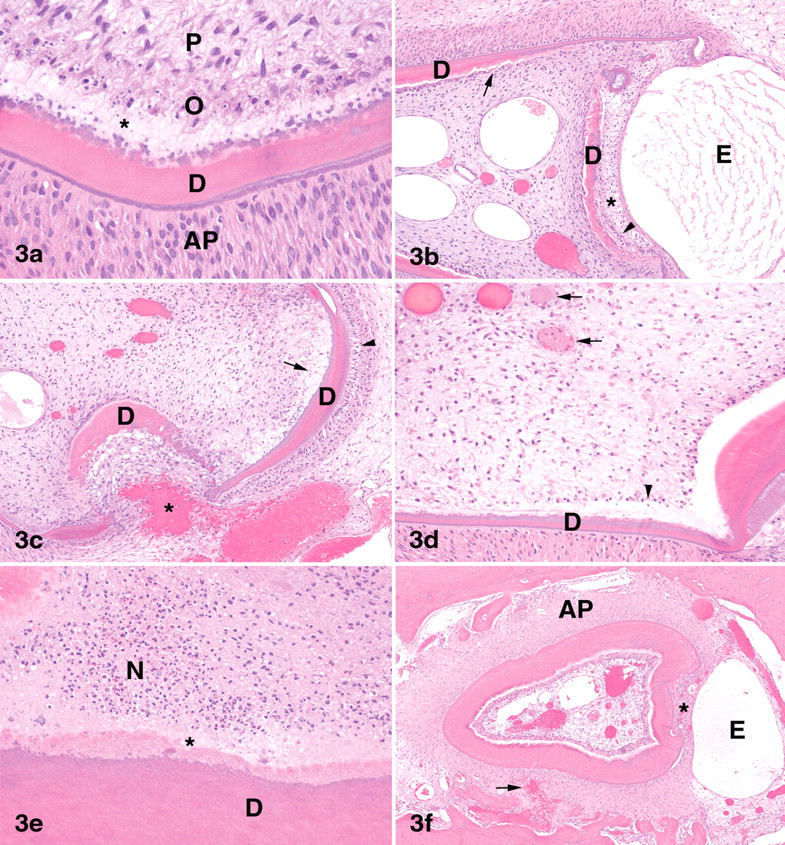
Incisors of rats administered BMS-645737 at 20/15 mg/kg/day for seventeen to thirty days. (A) Nonerupted incisor. Odontoblasts are necrotic. The inner layer of dentin (*) is markedly thinned; poorly mineralized; and sharply demarcated from the normal, previously formed dentin. HE × 400. (B) Nonerupted incisor. Dentin is markedly thin, disrupted, and discontinuous. In some areas there is complete loss of odontoblasts (arrow). Note thin remnant of enamel organ (arrowhead) and collapsed enamel space (*). The alveolar periosteum is edematous. HE × 100. (C) Nonerupted incisor. Periosteal hemorrhage (*) extends through the disrupted dentin and into the pulp cavity. Note necrosis and loss of odontoblasts (arrow) and vacuolation of ameloblasts (arrowhead). The enamel space is collapsed. HE × 100. (D) Nonerupted incisor. Note thrombi in pulp (arrows); necrosis and loss of odontoblasts (arrowhead); and thin, poorly mineralized dentin. HE × 200. (E) Erupted incisor. Large areas of the pulp are necrotic. Note zone of poorly mineralized dentin (*) and normal dentin. HE × 200. (F) Nonerupted incisor of rat administered BMS-645737 at 20/15 mg for 30 days followed by a two-week dose-free period. Odontoblasts have regenerated, and the dentin is thicker and more mineralized than at the end of the dosing period. Note overall decreased size of incisor; expansion of alveolar periosteum, edema, hemorrhage (arrow); and collapse of enamel space and loss of enamel organ (*). HE × 50. D = dentin; O = odontoblasts; P = pulp; AP = alveolar periosteum; E = edema; N = necrosis.
Following a two-week, dose-free observation period, dental lesions were observed in all rats given BMS-645737 at 6 or 20/15 mg/kg/day, but not in rats given 2 mg/kg/day or in controls. Odontoblasts in rats given 20/15 mg/kg/day were increased in number and height, compared to odontoblasts examined two weeks earlier at the end of the dosing period (Figure 3f). The dentin layer was also thicker with increased mineralization following the two-week dose-free period and was nearly equivalent in thickness to control dentin. Although odontoblasts and the dentin layer exhibited partial recovery, the overall diameter of the incisor was greatly diminished at this time point. Ameloblasts had degenerated further during the two-week dose-free period or were absent. Remnants of the papillary epithelium of the enamel organ were occasionally present, but usually absent. The enamel space was collapsed and no longer apparent. The overall shape of the tooth in cross-section was triangular or resembled a blunted arrowhead. Along with the presence of new alveolar bone, the periodontal ligament was thickened, as if filling in the alveolar socket to accommodate for the decreased sized of the incisor. In rats given BMS-645737 at 6 mg/kg/day, degenerative changes in the enamel organ, odontoblasts, and/or dentin at the end of the two-week dose-free period were similar to, but less severe than, those observed in rats given 20/15 mg/kg/day. Compared with changes noted at the end of the one-month dosing period, these changes in the enamel organ, odontoblasts, and/or dentin at 6 mg/kg/day were of similar incidence but of significantly lower severity, indicating partial recovery of these changes at this dose. In rats given 2 mg/kg/day, thinning of the dentin layer, as seen at the end of the one-month dosing period, was not noted at the end of the two-week dose-free period. Based on these findings, degenerative incisors lesions were considered to be generally reversible in rats given BMS-645737 at 2 mg/kg/day, partially reversible at 6 mg/kg/day, but not reversible at a dose of 20/15 mg/kg/day within a two-week dose-free period.
A microscopic finding attributed to the pharmacologic effects of BMS-645737 on inhibition of VEGF and/or FGF signaling was increased thickness of the distal femoral physis with chondrocyte hypertrophy. Mineralization of the stomach and ovary and minimal hyperphosphatemia, changes that have been reported with other inhibitors of FGF signal transduction, were also present (A. Brown et al. 2005; Courtney et al. 2003; Datta, Graziano, and Courtney 2003).
Discussion
Administration of a VEGFR/FGFR inhibitor to rats caused dose-related incisor toxicity, characterized by degeneration or necrosis of odontoblasts and a subsequent decrease in dentin production and degeneration of the enamel organ, resulting in thinner, discolored, and more fragile incisors. These changes were responsible for the broken or missing incisors and white discoloration of incisors that were observed clinically.
Rat incisors erupt during the second week of life, are white, and develop a yellow-brown color that increases in intensity with age (Schour and Massler 1949). The color of mature incisors is attributed to deposition of iron-containing pigments by the ameloblastic epithelium (H. Brown and Hardisty 1990; Kallenbach 1970). The white discoloration of incisors in rats administered a VEGFR/FGFR inhibitor in the present studies was attributed to drug-induced ameloblastic degeneration.
The dentin of rodent incisors is covered by enamel on the labial surface and cementum on the lingual surface of both the erupted and nonerupted portions (Kuijpers, van de Kooij, and Slootweg 1996). Rodent incisors have no roots, grow continuously, and completely renew themselves every forty to fifty days (Schour and Massler 1949). Therefore, gross incisor lesions may not be detected during toxicity studies less than forty days in duration but may become evident during a recovery period. Since incisors are not routinely examined microscopically in toxicity studies, tooth lesions that do not manifest grossly during the study may go undetected. The continuously growing rodent incisors require stem cells capable of differentiating into odontoblasts and ameloblasts (Tummers and Thesleff 2009). In contrast to incisors, rodent molars, which have both a crown and roots, grow little once root development is complete. Although odontoblasts remain active in molars throughout the life of the rodent, once root formation is complete, dentin production continues at a slower rate and that produced is referred to as secondary dentin (Long and Leininger 1999). Enamel production in the first molar is completed at about 10 days of age, which is prior to tooth eruption. The enamel organ subsequently degenerates, and the enamel of molars can not be replaced once damaged (Schour and Massler 1949). Based on these differences between rodent incisors and molars, incisors are likely to be more sensitive than molars to the effects of chemicals and drugs that are toxic to stem cells, odontoblasts, and/or ameloblasts.
Teeth, like other epithelial appendages such as hair and glands, develop through a sequence of reciprocal interactions between epithelial and mesenchymal tissues (Thesleff and Sharpe 1997). The four major families of signaling molecules that regulate tooth development are FGFs, bone morphogenetic proteins (BMPs), sonic hedgehog, and Wnt (Miletich and Sharpe 2003; Thesleff and Aberg 1999; Thesleff and Sharpe 1997). FGF is involved not only in the regulation of tooth development but also in the formation of craniofacial bone, palate, salivary gland, and craniofacial and tongue muscles (Nie, Luukko, and Kettunen 2006). In mice, putative stem cells in the apical end of the unerupted incisor proliferate and differentiate into mesenchyme-derived odontoblasts and epithelial-derived ameloblasts (Harada et al. 1999). Expression of several FGFs and FGFR-1, -2, -3, and -4 have been localized to the epithelial and/or mesenchymal portions of the developing tooth (Cam et al. 1992; Kettunen, Karavanova, and Thesleff 1998; Kettunen and Thesleff 1998; Wilkinson, Bhatt, and McMahon 1989). FGF is expressed by odontoblasts and cementoblasts (Madan and Kramer 2005); and FGF, VEGF, PDGF, and epidermal growth factor (EGF) are found in the dentin matrix (Roberts-Clark and Smith 2000).
FGF-10 is a survival factor for stem cells in developing incisor germs of mice (Harada et al. 2002), and epithelial stem cell proliferation in the cervical loops of rodent incisors is regulated in part by FGF (X. Wang et al. 2007). In FGF-3–/–FGF-10+/– mutant mice, labial cervical loops, where epithelial stem cells reside, are smaller than wild type. The enamel layer is present, although defective, in FGF-3–/– mice, whereas the enamel layer is either thin or absent in FGF-3–/–FGF-10+/– mutant mice. Grossly in both FGF-3–/– and FGF-3–/–FGF-10+/– mutant mice, incisors are white rather than yellow-brown and additionally, in FGF-3–/–FGF-10+/– mutant mice, incisors are thin and frequently broken (X. Wang et al. 2007). These findings in mutant mice are similar to the gross lesions observed in rats administered BMS-645737 in the present studies and support a role of FGF signal transduction inhibition in the pathogenesis of these lesions. The expression of FGFR-1 in both odontoblasts and ameloblasts and of FGFR-2 splice variant IIIb in ameloblasts suggests that FGFs and their receptors have roles in odontoblast and ameloblast differentiation and deposition of their respective secretory products, predentin and enamel matrix (Kettunen, Karavanova, and Thesleff 1998; Russo, Maharajan, and Maharajan 1998; Thesleff, Vaahtokare, and Partanen 1995; Tsuboi et al. 2003). Epithelial tissue-specific inactivation of FGFR-1 in mice had no effect on ameloblast differentiation but resulted in disorganization of formed ameloblasts and compromised ameloblast function, evidenced by defective enamel structure (Takamori et al. 2008). In FGFR-2(IIIb)–/– mice, teeth did not develop beyond the bud stage (De Moerlooze et al. 2000).
In the developing tooth, VEGF and VEGFR-2 are expressed in odontoblasts and the inner enamel epithelium and may regulate odontoblast development and the differentiation of inner enamel epithelium to ameloblasts (Aida et al. 2005; Miwa et al. 2008). VEGF expression in pulp fibroblasts and odontoblasts of human teeth is higher in immature than mature permanent teeth, suggesting a role of VEGF in tooth maturation (Q. Wang et al. 2007). VEGFR-2 is expressed in endothelial cells of the tooth pulp of both primary and permanent human teeth and may function to respond to morphogenetic and survival signals mediated by VEGF (Grando Mattuella et al. 2007). As stated previously, VEGF, as well as FGF, PDGF, and EGF, are found in dentin matrix (Roberts-Clark and Smith 2000).
VEGF is expressed in the dental follicle of rats and stimulates osteoclastogenesis that promotes proper bone resorption and subsequent tooth eruption (Wise 2009; Wise and Yao 2003). Osteopetrotic (op/op) mice, deficient in colony stimulating factor-1 (CSF-1), have defects in macrophage and osteoclast differentiation, leading to the osteopetrotic phenotype, which includes small incisors or failure of incisor eruption (McLean and Olsen 2001; Yoshida et al. 1990). Niida et al. (1999) have shown than VEGF can substitute for CSF-1 in mature op/op mice and stimulate development of osteoclasts. The lesions observed in the present studies cannot be explained by failure of osteoclastogenesis and tooth eruption secondary to VEGFR inhibition, since the incisors continued to erupt, as evidenced by in-life observations of incisor color changes and continued growth. Furthermore, the lesions in the present study were indicative of abnormal tooth formation, rather than defective eruption.
Dental dysplasia in SD rats has been reported as a background finding at an incidence up to 14.5% (Losco 1995). Contributing factors were considered to be the feeding of a powdered diet, leading to overgrowth and malocclusion of incisors, and the repeated clipping of these overgrown incisors. Lesions of dental dysplasia included loss or displacement of odontoblasts and ameloblasts, occlusion of the pulp cavity with dysplastic dentin, and pulpitis/periodontitis. Dental lesions considered background findings were not observed in the present studies.
Numerous chemicals and drugs have been shown to induce lesions in rodent incisors, specifically in odontoblasts and/or ameloblasts and their respective secretory products, predentin and enamel matrix (Table 2 ).
Rodent incisor lesions induced by chemicals and drugs.

Dental lesions in this study were considered to be due to pharmacologic interference with growth and repair of the vasculature supplying the incisors, inhibition of ameloblasts and/or odontoblast differentiation/function, or both, thereby preventing normal growth and remodeling of dental tissues. Since incisors of rats grow continuously, an uninterrupted and sufficient blood supply as well as continued ameloblast and odontoblast function are critical to support their growth and remodeling throughout life. Molars did not exhibit degenerative changes, presumably because rodent molars do not grow continuously and were fully formed at the time of dosing.
In addition to incisor changes, administration of BMS-645737 to rats resulted in pharmacologic inhibition of neovascularization into the epiphyseal growth plate, resulting in a thickening of the zone of chondrocyte hypertrophy. Additionally, soft tissue mineralization and hyperphosphatemia, which have been described with inhibition of FGF signaling, were observed (A. Brown et al. 2005; Courtney et al. 2003; Datta, Graziano, and Courtney 2003).
In tissues of healthy mice, VEGF signaling inhibition resulted in capillary regression in the pancreatic islets, thyroid gland, adrenal cortex, pituitary, choroid plexus, villi of the small intestine, and epididymal adipose tissue (Kamba et al. 2006). Also in healthy mice, VEGF signaling inhibition resulted in a reduction in the number of tracheal mucosal capillaries (Baffert et al. 2004). These findings suggest that, in addition to a role in angiogenesis, growth factors such as VEGF and/or FGF may be necessary for the maintenance of incisor capillary integrity or capillary survival.
Whether dental dysplasia of rat incisors in the present studies was the result of the pharmacologic inhibition of angiogenesis or interference with capillary maintenance, an effect of VEGF/FGF inhibition on ameloblast and/or odontoblast differentiation/function, or both is not certain. There is evidence that both angiogenesis and vasculogenesis are important in the formation of capillaries of the rat enamel organ (Manzke et al. 2005) and, as previously outlined, that VEGF and FGF signaling have roles in tooth development and growth.
Footnotes
Acknowledgments
The authors would like to thank Deborah Litz, Dickran Garbooshian, and the histology and necropsy staffs of Bristol-Myers Squibb for their assistance with necropsies and slide preparation.