
Abstract
Epiphyseal growth plate dysplasia (chondrodysplasia) might be considered as the pathognomonic feature of antiangiogenic treatment in preclinical species as it is reliably and dose-responsively induced in rodents and monkeys with vascular endothelial growth factor receptor (VEGFR) inhibitors, fibroblast growth factor (FGF) receptor inhibitors, matrix metalloproteinase inhibitors, and vascular targeting agents. Here we report epiphyseal growth plate dysplasia in juvenile rabbits treated with an oral spleen tyrosine kinase inhibitor induced by off-target antiangiogenic inhibition of VEGF and FGF family kinase receptors. Epiphyseal growth plate dysplasia resulted in weakening and fracturing of the femoral head physis in 6 of 10 male and 1 of 10 female animals as well as microfracturing and dysplasia of the distal femoral articular cartilage in 1 male animal. Fracture lines ran through the zone of hypertrophic cartilage (as well as adjacent zones), were orientated parallel to the physeal plane, and often involved displacement of the femoral head. We would suggest that the high prevalence of growth plate fracture in the rabbit may represent a potential additional adverse risk to those already established for children treated with antiangiogenic therapy.
Keywords
Introduction
Vascular endothelial growth factor (VEGF; also known as vascular permeabilizing factor; Senger et al. 1983) was first discovered in 1989 (Leung et al. 1989) and since then has become established as one of the pivotal molecules driving angiogenesis through its effects on endothelial proliferation, survival, migration, differentiation (Shibuya and Claesson-Welsh 2006), and permeability (Nagy et al. 2008). As such, VEGF and its cognate VEGF receptors (VEGFRs), Flt-1, kinase insert domain, and Flt-4, are required for survival of the embryo during gestation (reviewed in Hanahan 1997), growth (Gerber and Ferrara 2000), and development of the organism (Gerber, Hillan et al. 1999) as well as ovarian and uterine cycling in the adult (Ferrara et al. 1998).
Partnering developmental angiogenesis is pathological angiogenesis. In this context, angiogenesis acts as a driver for inflammation, wound healing (Howdieshell et al. 2001), and is essential for the development of cancer (Hanahan and Weinberg 2000). In the latter disease, angiogenic hot spots are directly correlated with poor prognosis (Jannink, van Diest, and Baak 1995; Belien, van Diest, and Baak 1999; Ahn et al. 2001; Mineo et al. 2004). It is not surprising therefore that angiogenesis has become an attractive target for drug therapy, resulting in the approval of several small (sunitinib [Sutent]; Pfizer and sorafenib [Nexavar]; Bayer/Onyx Pharmaceuticals) and large (bevacizumab [Avastin]; Genetech/Roche) molecule inhibitors for the treatment of cancer in man and animals. During the safety assessment testing of these and other VEGFR inhibitor drugs, a number of angiogenic-dependent toxicities have emerged that are of preclinical and clinical significance. These toxicities are characterized by vascular insufficiency in target organs such as in the growing bone and rodent incisor, which require continuous angiogenesis to support growth. In other organs, such as the adrenal gland and choroid plexus, VEGF signaling is required for the maintenance of vascular endothelium (Kamba et al. 2006; Maharaj et al. 2008).
In the rodent, therefore, treatment with VEGFR inhibitors results in a constellation of toxicity characterized by incisor tooth dental dysplasia (Patyna et al. 2008), ovarian atrophy (Wedge et al. 2005), dose-responsive epiphyseal growth plate dysplasia (Wedge et al. 2005), adrenal degeneration (Patyna et al. 2008), and choroid plexus inflammation (Patyna et al. 2008) as well as other less well-characterized changes.
In the bone, VEGF couples chondrogenesis with osteogenesis. VEGF inhibition results in the thickening of the epiphyseal growth plate due to retention of hypertrophic chondrocytes in the terminal zone of hypertrophic cartilage of the physis at the chondro-osseous border. The molecular mechanism underlying this is through delayed vascular invasion which results from VEGF inhibition—a process necessary to signal chondrocyte apoptosis prior to conversion to bone (Gerber and Ferrara 2000). Identical changes have been noted in monkeys and rats treated with bevacizumab (a recombinant humanized monoclonal antibody that inhibits VEGF A; Ryan et al. 1999) and fibroblast growth factor receptor (FGFR) inhibitors, respectively (Brown et al. 2005), as well as matrix metalloproteinase (MMP)-9 inhibitors (Vu et al. 1998). FGF is a potent angiogenic molecule modulating endothelial growth, migration, and survival (Cross and Claesson-Welsh 2001), whereas MMP-9 is a key regulator of growth plate angiogenesis and hypertrophic chondrocyte apoptosis. Epiphyseal growth plate dysplasia may therefore be regarded as a target-related toxicity secondary to antiangiogenesis in growing animals.
Here we report epiphyseal growth plate dysplasia and fracture in juvenile rabbits dosed with a spleen tyrosine kinase (SYK) inhibitor, fostamatinib disodium. This effect is mediated by the active metabolite, R406, which inhibits VEGF and FGF family kinase receptors with a similar potency to the intended therapeutic target, SYK (IC50 4–60 nM; Braselmann et al. 2006; Rolf et al. 2015). The dysplastic changes noted in the rabbit growth plate are morphologically identical to those noted in other species treated with VEGFR and FGFR inhibitors and are consistent with the expected mode of action of R406 acting through vascular inhibition. To the authors’ knowledge, this is the first report of epiphyseal growth plate dysplasia in the rabbit and the first case of physeal fracture in a preclinical species resulting from dysplasia, illustrating the potential adverse effects of this change in larger species.
Materials and Methods
Test Material
Fostamatinib was synthesized by AstraZeneca Research and Development, Alderley Park, and administered by oral gavage made up of water containing 0.1% carboxymethylcellulose sodium, 0.1% methylparaben sodium, and 0.02% propylparaben sodium.
Animals and Treatment
All studies were performed in accordance with the standards of animal care and ethics described in “Guidance on the Operations of the Animals (Scientific Procedures) Act 1986” issued by the UK Home Offices and conducted so that any clinical expression of toxicity remained within a moderate severity limit as described by the guidelines agreed with the UK Home Office Inspector.
New Zealand white rabbits substrain Hsdlf:NZW were singly housed in polypropylene cages with aspen chew blocks, cardboard tubes, and soft nesting material, with ad libitum food (Teklad 2930 rabbit diet) and water. Animals were maintained in a 12-hr light/dark cycle at a target temperature and relative humidity of 16°C–20°C and 40–70%, respectively. Animals were acclimatized for at least 7 days before the first dose and were aged 9 weeks at the start of dosing.
Study design
Nine-week-old animals comprising 41 male and 39 female (group size n = 10 except for the controls, where n = 11 for male and n = 9 for female animals due to a sexing error) were administered either vehicle or fostamatinib twice daily by oral gavage at 0, 5, 15, or 30 mg/kg for 28 days. All animals were observed for clinical signs at least once daily, and their body weight was measured twice daily and compared to predose values. The blood samples of 0.3 ml were taken for toxicokinetic sampling on the first and last day of the study to measure the active metabolite, R406. Approximately 1.5 ml of blood was sampled from the marginal ear vein for clinical pathology evaluation on day 25 (males) or day 27 (females).
Clinical pathology
Blood samples were analyzed for red and white blood cell indices as well as plasma chemistry (albumin, alanine aminotransferase [ALT], alkaline phosphatase [ALP], aspartate aminotransferase, bilirubin, calcium, cholesterol, creatinine, globulin, glucose, glutamate dehyrdrogenase, phosphate, potassium, sodium, total protein, triglycerides, and urea). Changes in measured indices were analyzed for statistical significance using pairwise Shirley’s test.
Necropsy and Histology
All animals were killed by an overdose of pentobarbital sodium administered intravenously for scheduled necropsy on the day after the final dose. Animals were necropsied according to standard operating procedures, where any macroscopic abnormalities were recorded. A limited tissue set was sampled which included angiogenic-dependent tissues (femoral head, femorotibial joint, sternum, adrenal glands, lower jaw bone for incisors, and ovaries); hemopoietic tissues (thymus and spleen); as well as the gastrointestinal tract, heart, kidneys, liver, trachea, and lungs. The adrenal glands, liver, ovaries, spleen, and thymus were trimmed and weighed at necropsy. Changes in organ weights were analyzed for statistical significance using PathData statistical package (Dunnett’s test and Dunn’s test). Sampled tissues were immersion fixed in buffered 10% formalin, processed to wax blocks, and then sectioned and stained with hematoxylin and eosin (H&E) for examination by light microscopy according to standard operating principals. Bones were fixed for 2–3 days and then transferred into 10% formic acid for decalcification, until the bones had a slight flex (normally 1–2 weeks). H&E slides were examined and peer reviewed by board-certified pathologists with experience in laboratory animal pathology. All data were Good Laboratory Practice compliant, which was achieved using an electronic data capture system (PathData V6.2d).
Slide Production
The 4 µm-thick formalin-fixed paraffin-embedded tissue sections were cut and mounted on SuperFrost Plus® (Thermo Fisher Scientific, Runcorn, UK) electrostatically charged glass slides. Sections were allowed to dry at 37°C overnight in an incubator and heated in an oven to 60°C for 10 min before being dewaxed and hydrated prior to staining.
Results
Toxicokinetic Evaluation
Toxicokinetic evaluation confirmed the absence of exposure in control animals and approximately dose proportional exposure (Cmax and Area Under the Curve [AUC]) for animals dosed at 15–30 mg/kg twice daily. Exposure for animals dosed at 5 mg/kg twice daily could not be confirmed except for female animals on day 28, since samples were below the lower level of quantification. Free Cmax and free AUC for male and female animals dosed at 30 mg/kg/day was 298 nmol/L and 269 nmol/L and 3,200,000 nM hr/L and 2,590,000 nM hr/L, respectively.
Clinical Signs
One animal was euthanized for welfare reasons on day 28 of the study due to hind leg lameness. Clinical veterinary evaluation noted a click in the right hip joint together with abnormal posture and gait.
All dose levels were well tolerated. Male animals dosed at 30 mg/kg twice daily showed reduced body weight gain (approximately 26% compared to control values between days 25 and 29). This was associated with statistically significant increase in food consumption (p < .05) in male animals between days 18 and 22 dosed at 15 and 30 mg/kg twice daily, but these changes were not significant at the end of the study.
Clinical Pathology
Treatment with fostamatinib resulted in slightly higher red cell distribution widths (6–22%) in male and female animals treated with 15 and 30 mg/kg twice daily and lower white cell counts (24–44%) in male and female animals treated with 30 mg/kg twice daily as well as slightly higher platelet counts (31–43%) in male animals treated with 15 and 30 mg/kg twice daily (summarized in Supplementary Table 1).
Summary Incidence of Pathology in the Femorotibial Joint and Bones of Animals Treated with 5–30 mg/kg Twice Daily.
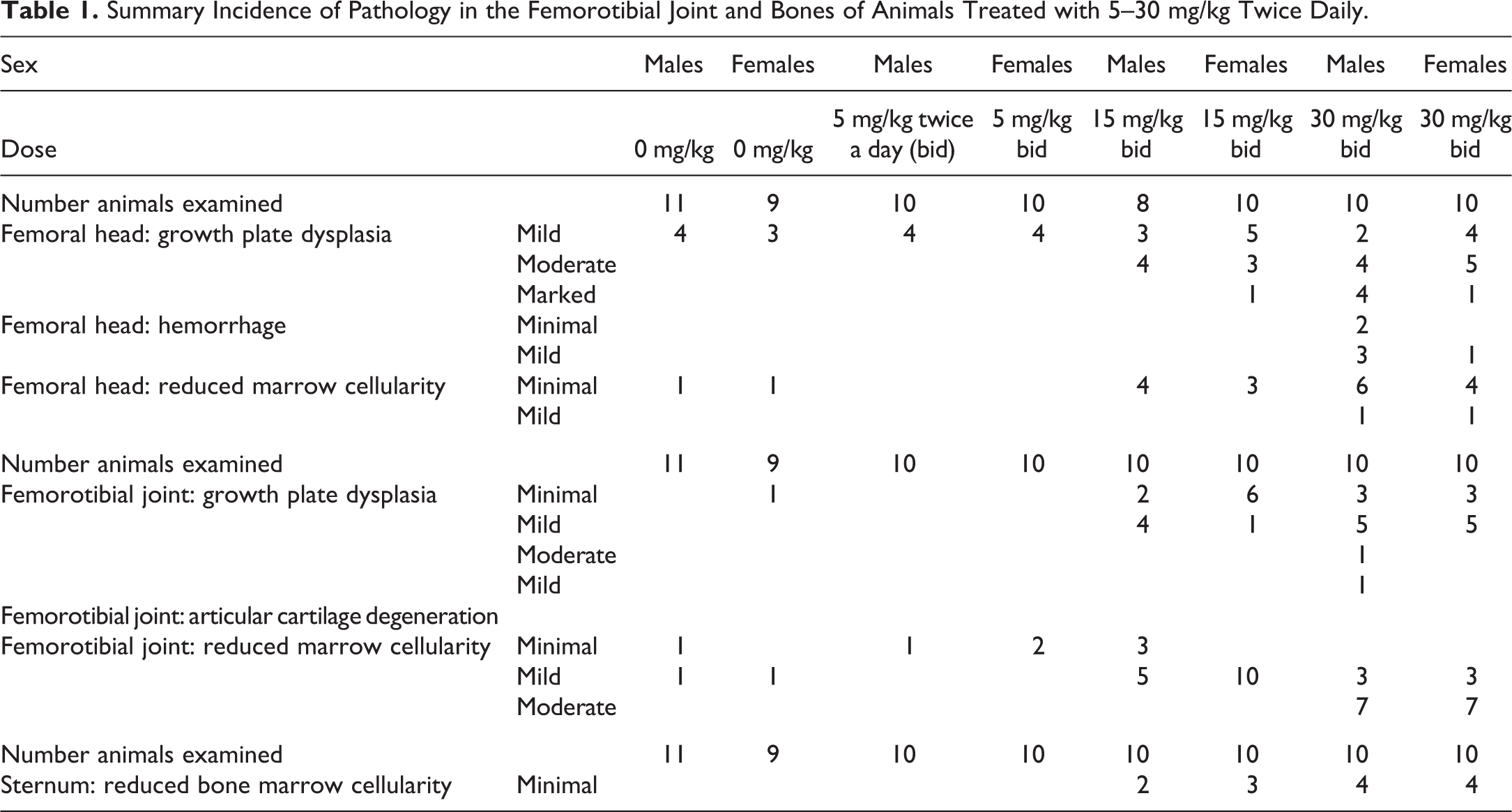
Treatment with fostamatinib also resulted in slightly raised liver indices (ALP 25–49%; total bilirubin 3–5×) for male and female animals dosed with 530 mg/kg twice daily and slightly higher ALT (40–53%) in male and female animals treated with 30 mg/kg twice daily.
Pathology
The only notable macroscopic changes seen at necropsy were bilateral fractures of the left and right femoral heads in 1 male animal treated with 30 mg/kg twice daily and unilateral fracture of the right femoral head in one male animal treated with 30 mg/kg twice daily.
Significant decreases in splenic weight were noted in male animals treated with 15 and 30 mg/kg twice daily and female animals treated with 30 mg/kg twice daily as well as thymic weight in male animals treated with 30 mg/kg/day and female animals treated with 15 and 30 mg/kg twice daily. In addition, significant increases in adrenal gland weight were noted in female animals treated with 5–30 mg/kg twice daily. Changes in thymic, splenic, and adrenal gland organ weight were not associated with any microscopic changes.
At microscopy, a number of dose-responsive and compound-related changes were noted, the most significant of which was epiphyseal growth plate dysplasia and growth plate (physeal) fractures. Growth plate dysplasia and frequency of physeal fracturing were more severe in male animals compared to female animals and more severe in the femoral head compared to the femorotibial joint (Table 1). In the sternum, no changes were noted in the bone or growth plate.
Growth plate dysplasia was characterized by focal to diffuse thickening and expansion of the terminal zone of hypertrophic cartilage due to retention of hypertrophic chondrocytes. In the femoral head, growth plate dysplasia was mild to marked and occurred in male and female animals treated with 15 and 30 mg/kg twice daily. This was accompanied by horizontal microfracturing of the growth plate zone of hypertrophic cartilage in 1 male animal and full thickness fracturing of the growth plate in a further 5 males (one of which was bilateral) and 1 female treated at 30 mg/kg twice daily (Figure 1). Growth plate fractures were variably accompanied by hemorrhage, matrix degeneration (with loss of basophilic staining), chondrocyte necrosis, and displacement of the femoral head due to separation within the zone of hypertrophic cartilage or at the interface between the zone of hypertrophic cartilage and neighboring zones (Figure 2 and Table 2). In 1 male animal treated with 30 mg/kg twice daily, mild degeneration and microfracturing at the interface between the zone of hypertrophic cartilage and mature zone were noted in the distal femoral articular cartilage (Figure 3).
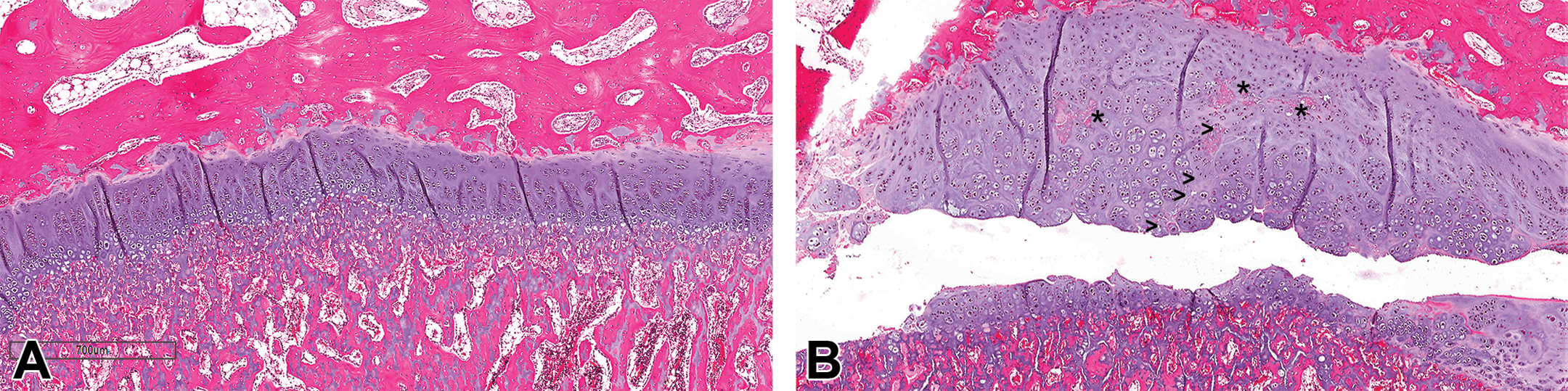
H&E-stained images of femoral head growth plate from (A) control rabbit and (B) rabbit treated with 30 mg/kg twice daily with growth plate dysplasia characterized by the thickening of the growth plate and accumulation of irregular columns of hypertrophic chondrocytes. A prominent fracture line runs through the zone of hypertrophic cartilage and extends into the metaphysis. A second area of matrix and chondrocyte degeneration is visible (*) fragmenting off from the original fracture and extending to the border of the zone of hypertrophic cartilage and mature zone (>).
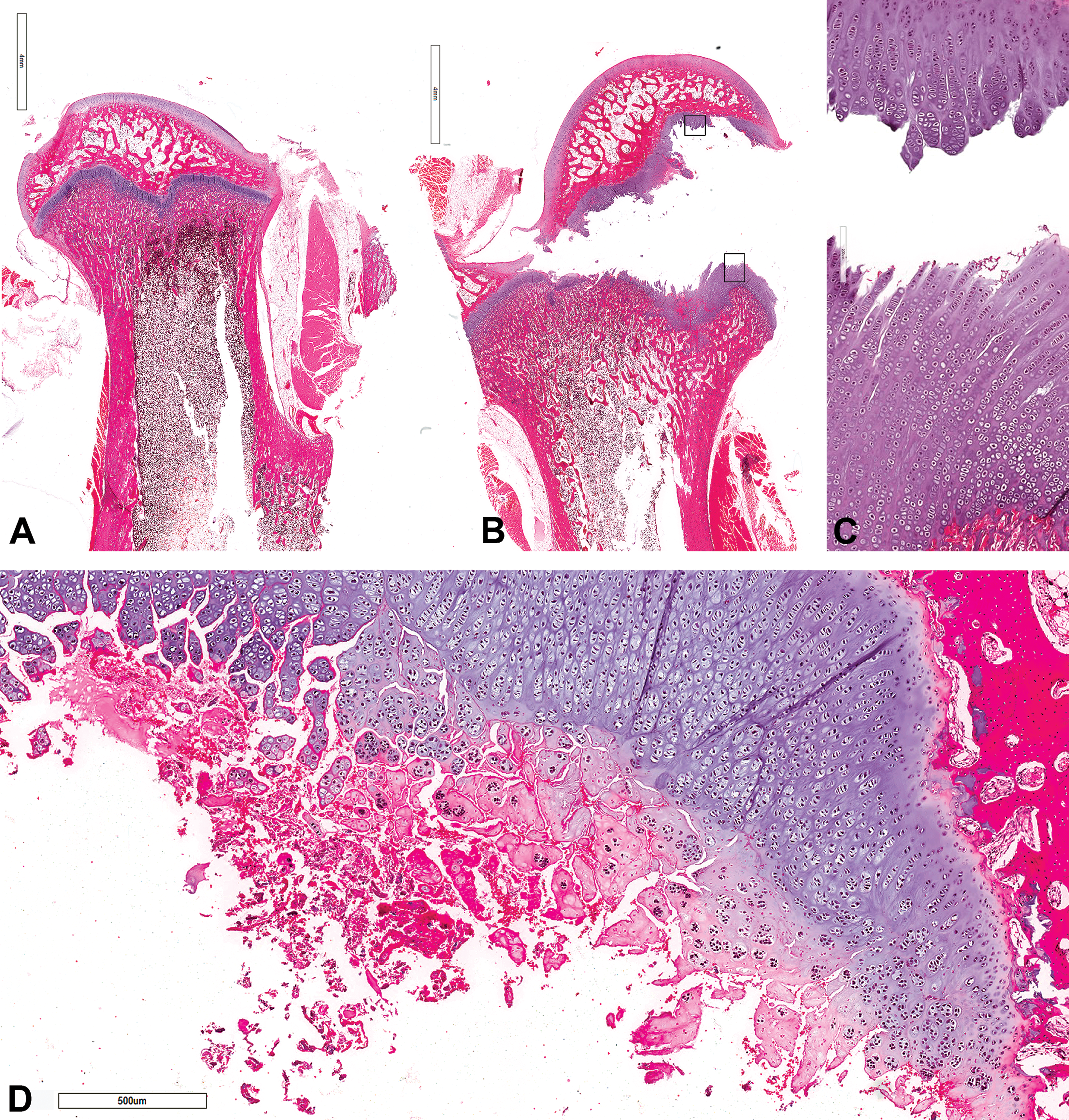
H&E-subgross images of femoral head from (A) control rabbit and (B) rabbit treated with 30 mg/kg twice daily with growth plate dysplasia and displacement of femoral head. (C) High-power view of boxed areas from (B) showing fracture through zone of hypertrophic cartilage and fissuring between columns of chondrons. (D) Image of a rabbit treated with 30 mg/kg twice daily with growth plate dysplasia and fracturing of the growth plate zone of hypertrophic cartilage showing matrix degeneration, chondrocyte necrosis, and hemorrhage.
Incidence and Location of Femoral Head Fractures in Rabbits Treated with 30 mg/kg Twice Daily.
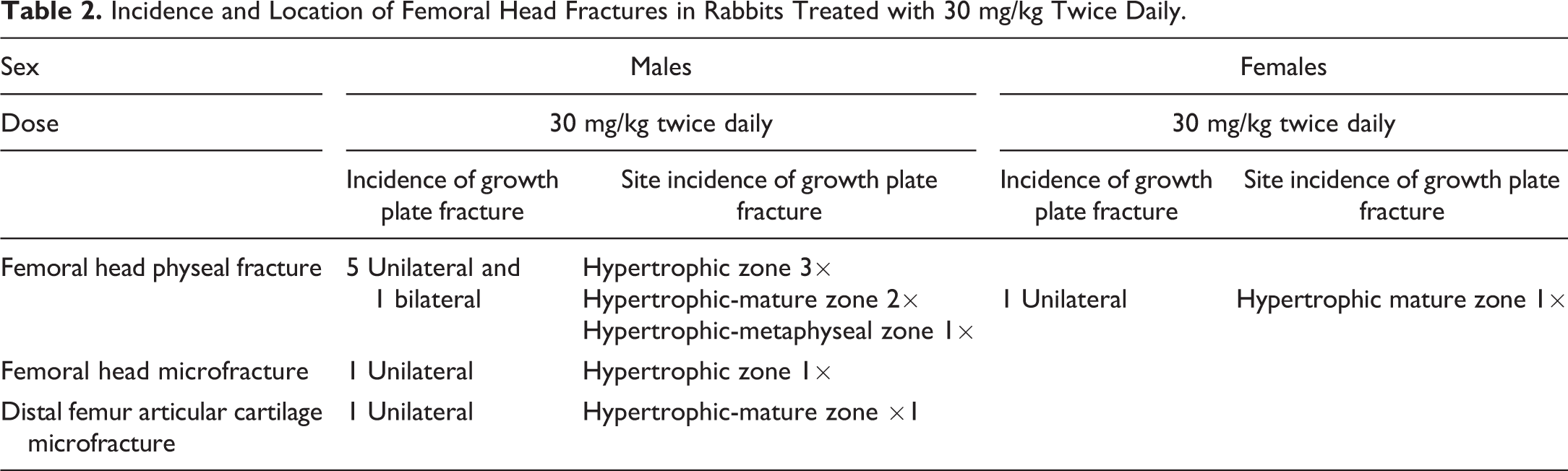
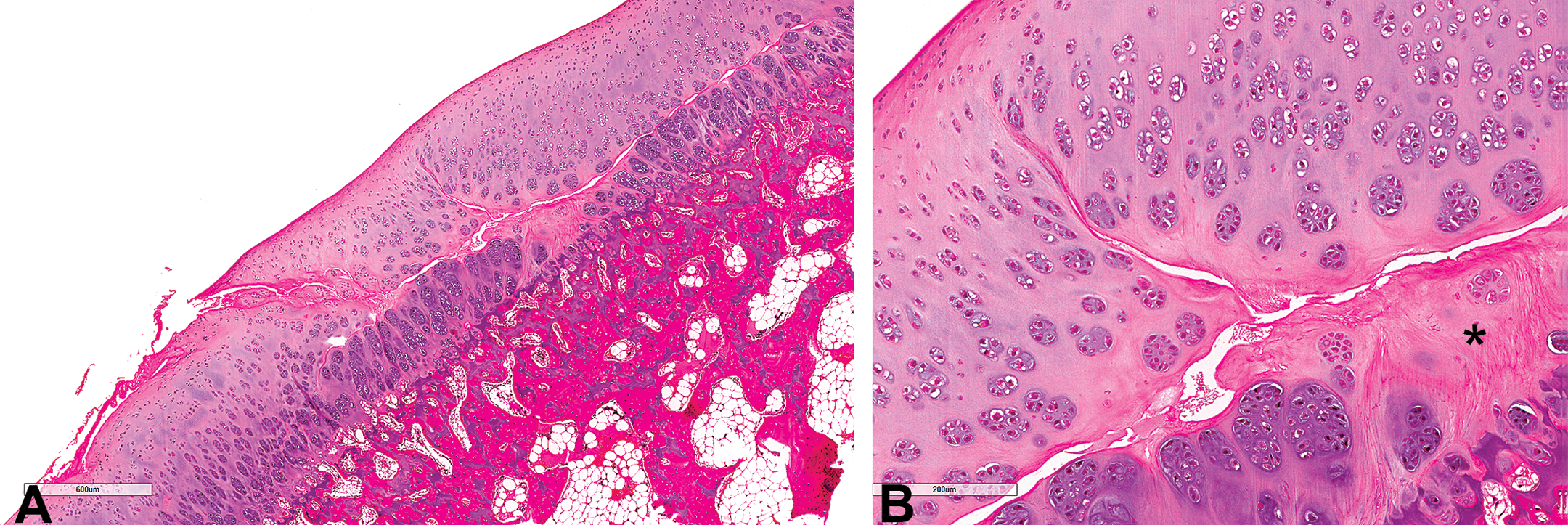
H&E-stained images of distal femoral articular cartilage from a rabbit treated with 30 mg/kg twice daily showing (A) microfracturing of the growth plate zone of hypertrophic cartilage with extension up to the articular surface and (B) matrix degeneration and chondrocyte necrosis (*).
In the femorotibial joint, minimal to moderate epiphyseal growth plate dysplasia was noted in animals treated with 15 and 30 mg/kg twice daily, but this was not accompanied by physeal fractures. Minimal to moderate reduced bone marrow cellularity was noted in the femoral head, femorotibial joints, and sternum in animals treated with 15 and 30 mg/kg twice daily. Finally, a minimal to mild increase in degenerate/necrotic follicles was noted in the ovaries of animals treated with 5–30 mg/kg twice daily.
Discussion
Fostamatinib forms an active metabolite, R406, which is a mixed tyrosine kinase inhibitor that potently inhibits SYK in in vitro isolated enzyme (IC50 41 nM) and cell-based assays (IC50 range 31–171 nM; Braselmann et al. 2006). SYK is a nonreceptor protein tyrosine kinase that is primarily expressed in immune cells (B cells, mast cells, macrophages, and neutrophils). It couples immune receptors such as the B-cell receptor on B cells (Kurosaki and Hikida 2009), IgE FcεR1 receptor on mast cells (Matsubara et al. 2006), and the FcγR receptor on macrophages (Crowley et al. 1997) to signal transduction pathways that ultimately results in cytokine production, proliferation, differentiation, survival, and phagocytosis (Kaur, Singh, and Silakari 2013).
In this study, we noted decreases in thymic and splenic organ weights together with decreases in circulating white blood cell counts and bone marrow cellularity in male and female animals. Although organ weight changes were not associated with any histological effects, reduced lymphoid tissue mass and decreased circulating white blood cells are consistent with SYK inhibition since knockout mice show deficiencies in both B cell and thymocyte development (Turner et al. 1995). In addition, drug-induced cytopenias have been reported in non-Hodgkin lymphoma, rheumatoid arthritis, and immune thrombocytopenic purpura patients treated with R778 (fostamatinib; Efremov and Laurenti 2011). The cause of the changes in hematology is unknown; but since VEGFR inhibitors have been associated with myelosuppression, it is possible that the increase in red cell distribution width reflects an off-target response to VEGFR inhibition (Gerber et al. 2002; Chen and Cleck 2009). Finally, a minimal to mild increase in degenerate/necrotic ovarian follicles was noted in treated animals. The significance of this change is unknown, as ovarian follicles, unlike corpora lutea, are avascular and therefore not expected to be affected by angiogenic inhibition.
R406 also has significant off-target inhibitor activity for the VEGF and FGF family of receptors, with in vitro isolated enzyme IC50 values similar to the SYK potency (IC50 4–60 nM; Rolf et al. 2015). In vivo potency, however, is likely to be much lower since elevation of blood pressure (a recognized biomarker of VEGFR activity [Curwen et al. 2008]) is much less compared to other more potent VEGFR inhibitors (fostamatinib < 5 mmHg vs. 25 mmHg for sunitinb; Rolf et al. 2015). VEGF and FGF family receptors deliver potent angiogenic signals that promote endothelial cell survival, growth, migration, and differentiation (Cross and Claesson-Welsh 2001; Shibuya and Claesson-Welsh 2006). Inhibition with bevacizumab, sunitinib, and recentin (which inhibit the VEGF pathway) as well as PD176067 (which inhibits the FGF pathway), all induce epiphyseal growth plate dysplasia in rats and monkeys (Ryan et al. 1999; Patyna et al. 2008; Wedge et al. 2005; Brown et al. 2005). Inhibition of angiogenesis by other classes of molecule, such as MMPs (Vu et al. 1998), and vascular targeting agents (Hall, Westwood, and Wadsworth 2006) also induces growth plate dysplasia, suggesting that this change is pathognomonic for antiangiogenic treatment.
The mechanism underlying growth plate dysplasia is through inhibition of endochondral ossification in the terminal zone of hypertrophic cartilage of the growth plate due to delayed vascular invasion which is a necessary prerequisite to signal chondrocyte apoptosis prior to conversion to bone (Gerber and Ferrara 2000). This results in accumulation of vertical columns (chondrons) of hypertrophic chondrocytes within an otherwise normal but thickened growth plate. In this study, we observed growth plate dysplasia (and in 1 animal articular cartilage dysplasia) in the rabbit that appeared morphologically identical to the same changes observed in rodents treated with a range of antiangiogenic compounds (Hall, Westwood, and Wadsworth 2006), and where the mechanism has been well elucidated (Gerber, Vu et al. 1999). Since the active metabolite of fostamatinib, R406, inhibits VEGFR and FGFR family members with a similar potency to the intended therapeutic target, SYK (IC504–60 nM; Braselmann et al. 2006; Rolf et al. 2015), and since the primary target, SYK, plays no known role in osteogenesis, the dysplastic changes noted in this study are believed to be due to angiogenic inhibition of VEGF and/or family FGF receptors leading to the retention of hypertrophic chondrocytes secondary to delayed vascular invasion. This observation therefore extends the notion that epiphyseal growth plate dysplasia is an invariable consequence of angiogenic inhibition in all preclinical species and that this change can be used as a reliable and quantitative biomarker of antiangiogenic effects in preclinical studies.
The rabbit skeleton is relatively fragile, comprising 7–8% of the total body weight, whereas the skeletal muscle constitutes 50% of the body weight (Percy and Barthold 2001) which makes the rabbit particularly susceptible to fractures of the hind legs and spine. In our study, we noted fracture of the proximal head through the epiphyseal growth plate in 6 male and 1 female animal, as well as a microfracture of the articular cartilage, suggesting that dysplasia conferred mechanical weakness to the growth plate. In children, the relatively high incidence of physeal fractures (approximately 15% of all bone fractures; Lipp 1998) supports this idea whereas in rats, the physis has been identified as a frequent point of failure under shear stress (van Leeuwen et al. 2004) with the zone of hypertrophic cartilage being the weakest zone (Splengler et al. 1980) and a common site for failure (van Leeuwen et al. 2004). This is because the increased size of hypertrophic chondrocytes reciprocally reduces matrix collagen content, and therefore the strength of the tethers which run between columns of chondrons (Alexander 1976). However, fractures can run through different layers of the growth plate with the transition between the zone of hypertrophic cartilage and the proliferating zone being identified as a frequent site of failure (Smith, Geist, and Cooperman 1985; Williams et al. 1999). Thickening of the zone of hypertrophic cartilage further weakens the growth plate (Salter 1992; Williams et al. 1999), and it is therefore likely that in our study dysplasia with expansion of the zone of hypertrophic cartilage predisposed to growth plate fracture. We noted that fractures ran parallel to the plane of the physis through the hypertrophic and bordering zones and are consistent with this notion. It is also likely that the rabbit growth plate models the potential adverse effects of antiangiogenic therapy in children more accurately than the mouse or rat (in which growth plate fracture has not been reported) since the magnitude of the sheer stresses are more likely to be comparable as a result of the increased size and muscle to skeletal mass ratio in the rabbit. We would therefore suggest that treatment of children with potent antiangiogenic molecules may be associated with the additional adverse risk of growth plate fracture and possibly articular cartilage microfracture.
Footnotes
Author Contributions
Authors contributed to conception or design (AH, TM, JS); data acquisition, analysis, or interpretation (AH, TM, MR); drafting the manuscript (AH); and critically revising the manuscript (AH, TM, JS, MR). All authors gave final approval and agreed to be accountable for all aspects of work in ensuring that questions relating to the accuracy or integrity of any part of the work are appropriately investigated and resolved.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.