
Abstract
Keywords
Introduction
Middle ear cholesteatoma is a non-neoplastic destructive lesion that is characterized by abnormal growth of the keratinizing squamous epithelium in the temporal bone. Congenital middle ear cholesteatoma is the white mass located medial to the intact tympanic membrane, with no previous history of otorrhea, perforation, or otologic procedures. 1 However, the European Academy of Otology and Neurotology and the Japanese Otological Society (EAONO/JOS) working group have stated that a history of previous otitis media or effusions does not exclude congenital middle ear cholesteatoma. 2 Conductive hearing loss is the most common presenting symptom. The Potsic staging system for congenital cholesteatoma has been widely used given its simplicity and predictability. Potsic and colleagues demonstrated a strong association between stage and residual disease, ranging from a 13% risk in stage I to 67% in stage IV. 3 The strong correlation between the stage of congenital cholesteatoma and the residual or recurrent rate was also supported by 2 follow-up studies.4,5 In the current study, we report a case series of 3 patients managed in our department between June and August 2023, and present a summary of the recent literature.
Case Report
Case 1
A 14-year-old boy was admitted to our department with hearing loss in the left ear for 5 years and nasal obstruction with open-mouth breathing for 3 years. He was diagnosed with adenoid hypertrophy and secretory otitis media at a local hospital. He was administered oral antibiotics and nasal hormone spray, but his hearing was not improved. He had no history of otorrhea, tinnitus, vertigo, or previous ear surgery. A computed tomography (CT) demonstrated 2 masses of soft tissue density that occupied the mesotympanum with ossicular destruction (Figure 1). Otoscopy revealed 2 white masses located behind the posterior-inferior quadrant (PIQ) and anterior-superior quadrant (ASQ) of the left tympanic membrane. Pure tone audiometry demonstrated moderate conductive hearing loss in the left ear (61.25 dB HL; 0.5, 1, 2, 4 kHz). The tympanogram showed binaural A type. The patient was diagnosed with a left congenital cholesteatoma and underwent adenoidectomy and transcanal endoscopic ear surgery under general anesthesia. The tympanomeatal flap was elevated from the malleus short process, the handle, and the umbo, and the disrupted long process of the incus was found. The range of cholesteatoma was more extensive than that was shown on CT images, and an open type cholesteatoma was revealed at the PIQ, ASQ, and the eustachian tube tympanic orifice. The cholesteatoma was completely removed and ossiculoplasty with partial ossicular replacement prosthesis was undertaken, and the substantial air-bone gap decreased.
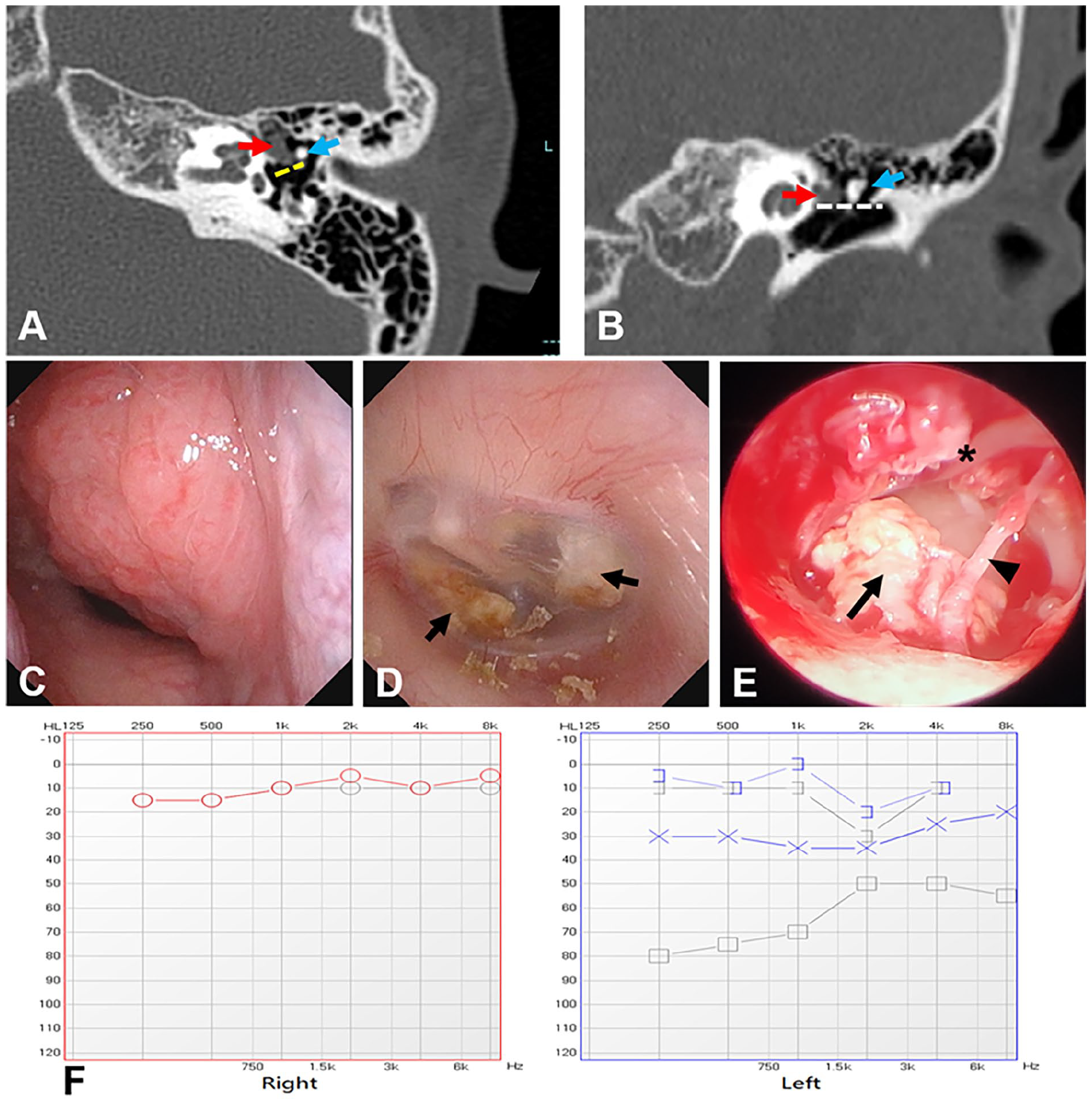
Preoperative, operative, and postoperative findings in case 1. The axial (A) and coronal views (B) of the computed tomography scan show soft tissue density in the left ear cavity. Red arrow, cholesteatoma in the anterior-superior quadrant (ASQ); blue arrow, the malleus; yellow dashed line, the erosion of the long process of the incus; white dashed line, the level at the handle of the malleus dividing the middle ear into superior and inferior quadrants. Nasopharyngoscopy shows adenoid hypertrophy (C). Preoperative otoscopy shows 2 white masses (black arrow) behind the posterior-inferior quadrant (PIQ) and ASQ of the left tympanic membrane (D). Operative findings of the left ear (E): the cholesteatoma was located in the PIQ and ASQ, and the long process of incus was disrupted (black arrow: cholesteatoma; arrowhead: chorda tympani nerve; asterisk: malleus handle). Pre- (gray lines) and postoperative (blue lines) pure tone audiometry of the left ear (F) shows the substantial air-bone gap is decreased, and normal hearing is found in the right ear.
Case 2
A 4-year-old girl presented to our department with otalgia and hearing loss in the left ear. Following treatment with oral antibiotics, her ear pain went away. However, her hearing ability was not improved. She had no history of otitis media or previous otologic surgery. She was in good health and had no cleft palate. A CT scan showed a large number of soft tissue densities occupied the middle ear and mastoid cavity (Figure 2). Otoscopy revealed that the tympanic membrane was intact and the tympanic cavity was filled with white mass. Pure tone audiometry demonstrated mild conductive hearing loss in the left ear (28.75 dB HL; 0.5, 1, 2, 4 kHz). The tympanogram showed a B type of the left ear. We chose a postauricular incision and performed a canal wall-up mastoidectomy. During the operation, we identified and preserved the chorda, and found a closed-type congenial cholesteatoma spreading to the whole tympanic cavity and the sinus tympani, but the ossicular chain was intact. The cholesteatoma was too large to remove en bloc, thereby we opened it and reduced its size by removing inner keratin debris. To remove the cholesteatoma around the ossicular chain, we removed the incus and the head of the malleus. After resecting cholesteatoma, we carefully checked the tympanum and confirmed that there was no residual cholesteatoma matrix in the tympanum using a 30° endoscope, and implanted partial ossicular replacement prosthesis.
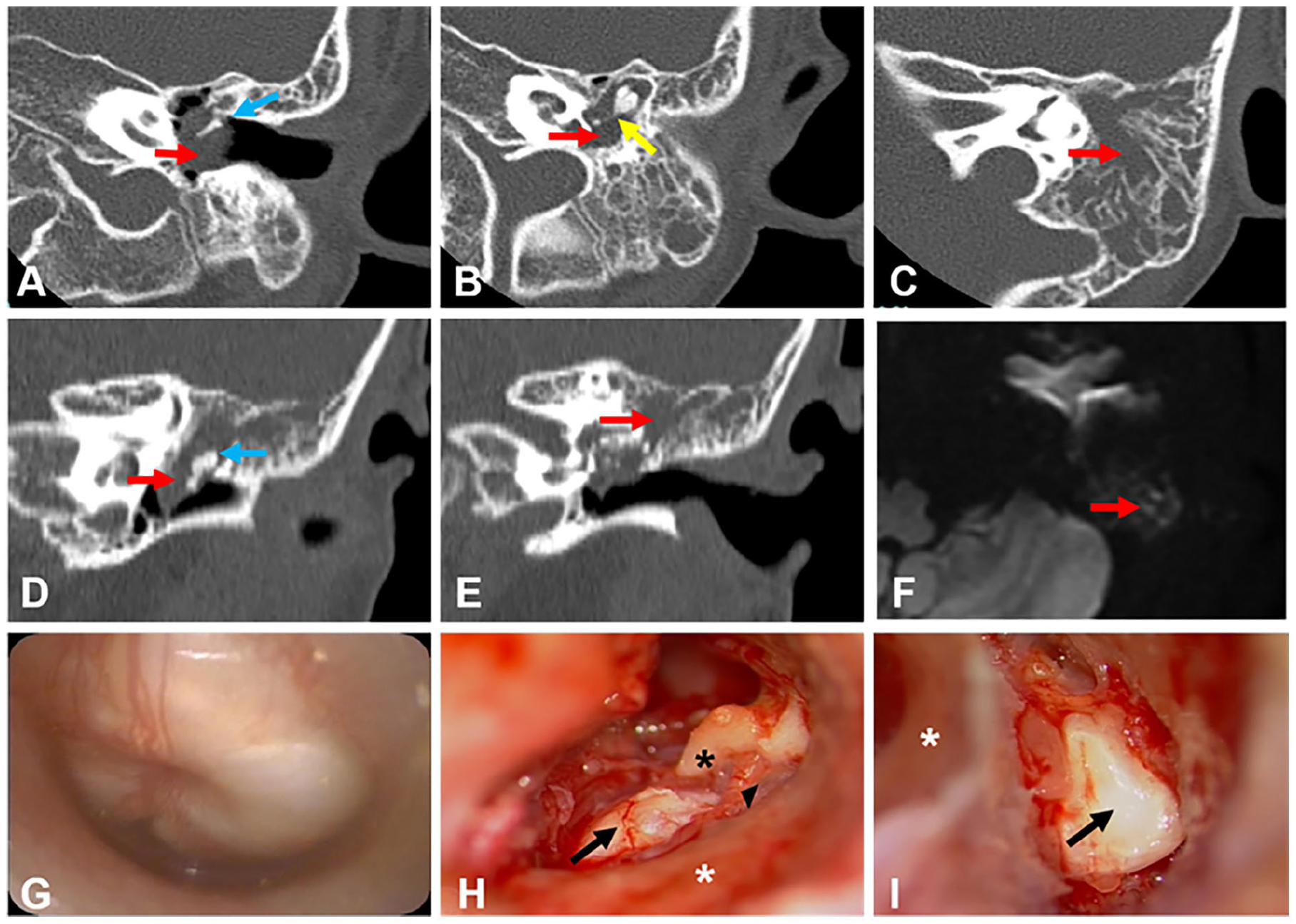
Preoperative and operative findings in case 2. The axial (A-C) and coronal views (D, E) of the computed tomography scan show a large cholesteatoma in the left epitympanum, mesotympanum, hypotympanum, and mastoid antrum, with no bony erosion of the scutum or ossicles. Red arrow, cholesteatoma; blue arrow, malleus; yellow arrow, incus. Axial diffusion-weighted imaging MRI shows a high signal on a high b-value image (red arrow; F). Preoperative otoscopy revealed that the tympanic membrane was intact and the tympanic cavity was filled with white mass (G). Operative findings of the left ear (H, I): the closed type cholesteatoma occupied the tympanic cavity and mastoid antrum (black arrows). Black arrowhead, chorda tympani nerve; black asterisk, malleus handle; white asterisk, posterior bony external canal wall.
Case 3
A 23-year-old man presented to our department with a long history of hearing loss in the left ear. He had no prior history of otorrhea, otalgia, tinnitus, vertigo, or previous ear surgery. CT showed a tiny soft tissue density in the middle ear cavity and no obvious abnormality in the mastoid cavity (Figure 3). Otoscopy was normal, with no whitish mass behind an intact tympanic membrane. Pure tone audiometry demonstrated moderate conductive hearing loss in the left ear (63.75 dB HL; 0.5, 1, 2, 4 kHz). The tympanogram showed binaural A type. The patient underwent transcanal endoscopic ear surgery under general anesthesia. During the operation, we found that the long process of the incus was destructed and that there was an open-type congenital cholesteatoma located medial to the disrupted long process of the incus. We resected the cholesteatoma and the destroyed incus and implanted a partial ossicular replacement prosthesis.
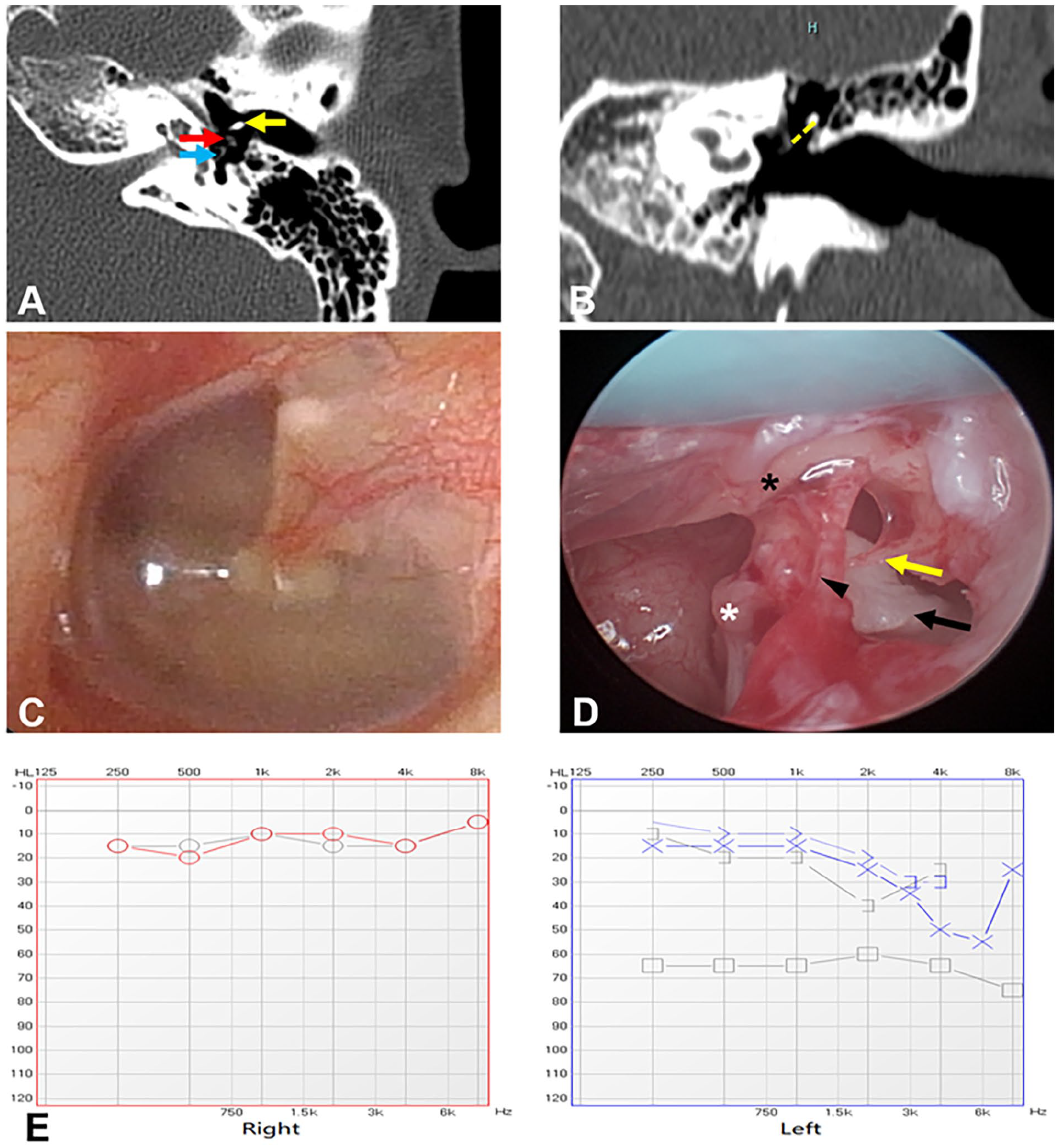
Preoperative, operative, and postoperative findings in case 3. The axial (A) and coronal views (B) of the computed tomography scan show small soft tissue density in the left tympanic cavity and with bony erosion of the incus long process. Red arrow, small soft tissue; yellow arrow, malleus; blue arrow, stapes; yellow dashed line, the damaged long process of the incus. Preoperative otoscopy revealed that the tympanic membrane was intact and no white mass was found (C). Operative findings of the left ear (D): the cholesteatoma behind the destructed incus long process. Black arrow, cholesteatoma; yellow arrow, the damaged incus long process; black arrowhead, chorda tympani nerve; black asterisk, malleus handle; white asterisk, stapes. Pre- (gray lines) and postoperative (blue lines) pure tone audiometry of the left ear (E) showed that the substantial air-bone gap was decreased, and the right hearing was normal.
Discussion
The annual incidence of cholesteatoma was reported to be 3 per 100,000 in children and 9.2 per 100,000 in adults. 4 Congenital middle ear cholesteatoma was reported to account for 2% to 5% of all middle ear cholesteatomas.6-8 Thanks to the increased awareness and the popularity of ear endoscopy, the incidence of congenital middle ear cholesteatoma is on the rise. This disease is more common in the young than in the adult population. A previous study showed that the median age of congenital middle ear cholesteatoma patients at presentation was 5 years, and male patients outnumbered females. 9
The components of middle ear cholesteatoma are characterized by 3 layers: the perimatrix, matrix, and cystic contents. Based on histopathology, the congenital cholesteatoma can be categorized into closed type and open type, 10 and the recurrence rate was higher in open type than in closed type cases.11-13 The open-type cholesteatoma is more common in older children (>5 years of age) or adults and is more invasive as a flat keratinized epithelium spreading along the surface of the mucous membrane rather than forming epithelial masses and sacs. The ossicular chain is often severely eroded, mainly at the incudostapedial joint, leading to severe conductive deafness and it is difficult to be removed by surgery.11,14 The closed-type cholesteatoma is localized and cystic, and the ossicular chain is usually unaffected. This type of cholesteatoma is more commonly reported in the literature, is relatively easy to remove, and is often diagnosed in young children (mean age 4 years).11,15 Our findings demonstrate that 2 cases (case 1, 14-year-old male and case 3, 23-year-old male) had open-type congenital cholesteatoma, and 1 case (case 2, 4-year-old female) had closed-type congenital cholesteatoma. According to the EAONO/JOS working group, cholesteatomas can be divided into 3 categories: the congenital type, which is specific to childhood; the acquired type, which affects both children and adults; and unclassifiable cholesteatomas whose origin cannot be precisely traced. 3 The most popular hypothesis for congenital middle ear cholesteatoma is the epidermoid cell rest theory. 16 Congenital cholesteatoma can be located anywhere in the temporal bone 17 : petrous apex, mastoid, middle ear, middle ear and mastoid, and external auditory canal; therefore, it is difficult to explain with a single theory. Unlike primary acquired cholesteatoma, congenital middle ear cholesteatoma often occurs in healthy ears with no prior history of disease and with normal eustachian tube function. However, as congenital cholesteatoma develops, it blocks the eustachian tube. When otitis media manifests, the tympanic membrane can be perforated, and cholesteatoma can be the consequence of secondary infection. In this case, it is difficult to be distinguished from congenital cholesteatoma.
The Potsic’s staging system is frequently utilized to define the stage of congenital cholesteatoma, as follows: stage I, disease confined to a single quadrant; stage II, cholesteatoma in multiple quadrants, but without ossicular involvement or mastoid extension; stage III, ossicular involvement without mastoid extension; and stage IV, mastoid disease. 3 Stage III was the most common, comprising more than 60% of cases. 18 According to the Potsic stage, 2 of our cases were classified as stage III and the remaining 1 as stage IV. Potsic stage is positively correlated with residual disease risk. The mean threshold of air conduction in the preoperative hearing test was also correlated with stage, higher thresholds were recorded with higher stages. 12 The most frequent anatomic location of congenital middle ear cholesteatoma is the ASQ, followed by the PSQ.10,18 A recent study showed that there was a site bias of congenital cholesteatoma between patients from Asia and those from Europe and America. Indeed, cholesteatomas in patients from Asia or Western countries tended to be localized to the PSQ 19 or ASQ, 18 respectively. The disease progression of congenital middle ear cholesteatoma is relatively slow and unilateral hearing loss in children is often overlooked, so larger lesions remain undetected until incidentally diagnosed during routine physical examination. Previous studies showed that 66% of congenital middle ear cholesteatoma cases were detected during the examination of hearing loss or the follow-up of otitis media with effusion or acute otitis media, 34% of cases were asymptomatic and the diagnosis was done by routine otoscopy and otologic screening. 20 Other studies have reported that 80% of congenital middle ear cholesteatoma diagnoses are asymptomatic. 21 Hearing loss may be noticeable when cholesteatoma is large enough to occupy and block the middle ear cavity or when the ossicles have eroded. The ossicular chain is the first middle ear structure affected, and the most commonly eroded ossicular is the incus, where damage occurs nearly 100% of the time.
High-resolution CT (HRCT) of the temporal bone has been the primary imaging modality to determine the extent of middle ear cholesteatoma and the anatomic abnormalities.22-24 Thanks to its excellent spatial resolution, HRCT has a high sensitivity when it shows a free middle ear cavity or mastoid, but it is not specific enough to differentiate cholesteatoma from fluid, cholesterol granuloma, or granulation tissue. Typical findings associated with cholesteatoma on HRCT include a soft tissue lesion in the middle ear, with associated bone erosion, most often in the ossicles. 25 Small closed-type congenital cholesteatomas are typically observed as round-shaped lesions, while the open-type congenital cholesteatomas display irregular shapes. 26 MRI has better tissue differentiation; however, the signal-intensity characteristics of cholesteatomas are not specific. MRI sequences show a cholesteatoma hypointense/isointense on T1-weighted images and hyperintense on T2-weighted images compared with brain tissue. Diffusion-weighted imaging MRI (DWI-MRI) has been a valuable tool to detect cholesteatoma. A meta-analysis to assess the utility of DWI-MRI in the preoperative evaluation of cholesteatoma showed that the overall sensitivity and specificity were 0.94. 27 The superiority of DWI-MRI in detecting cholesteatoma was also confirmed in extra studies,28-30 but it should be noted that its capability in detecting small size cholesteatomas (<3 mm) was limited. 28 As recommended in the latest review, 25 in the initial diagnostic setting, HRCT is the main imaging modality for patients referred for conductive hearing loss with suspected middle ear lesions behind the intact tympanic membrane, and MRI with DWI is another useful technique for the detection of cholesteatoma when there is equivocal soft tissue or fluid in the middle ear. In postoperative settings, MRI with DWI is the primary modality for differentiation of recurrent cholesteatoma versus granulation tissue and scar, and HRCT is useful for evaluating postoperative changes in the mastoid cavity and ossicular prosthesis.
Currently, surgery is the primary management to treat cholesteatoma. The main goal of surgical treatment is to completely remove cholesteatoma, to keep a dry ear, and to avoid recurrence. Two types of surgical approaches are commonly applied: the conventional microscopic approach and the endoscopic approach. The conventional microscopic approach consists of 2 main surgical procedures, canal wall down (CWD) and canal wall up (CWU) mastoidectomy. The CWD approach can expose the entire lesion and achieve the purpose of complete resection. However, CWD removes the posterior wall of the external ear canal, making the external ear canal and mastoid cavity a common cavity, and there are postoperative wound healing and cavity problems, including the accumulation of keratin debris and ear wax, requiring long-term ear cleaning. 31 Because the temporal bone is still developing, CWD is not recommended in pediatric patients. 32 Currently, CWU is more frequently applied, with the benefit of less structural damage during surgery, preserving the integrity of the posterior extraosseous auditory canal and the contour of the ear canal, which can mitigate healing symptoms. 31 In addition to facilitating the postoperative fitting of hearing aids, CWU surgery can also avoid cavity problems such as the accumulation of debris and the need to avoid exposure to water. Notably, given that CWU does not adequately expose the portion of the middle ear cleft, the recurrence rate is higher than CWD. This caveat necessitates a planned second-look procedure 6 to 12 months after the initial CWU surgery. According to a recent study, the CWU approach to cholesteatoma treatment has a nearly 3 times greater likelihood of recurrence than CWD surgery. 33
Recently, endoscopic ear surgery has been widely employed as a promising treatment for removing congenital cholesteatoma in children. The utilization of a high-definition visualization endoscope offers a better view of hidden areas that are difficult to observe when using microscopic vision, facilitating middle ear manipulation and reducing recurrence and residual of middle ear cholesteatoma. 34 Opinions vary about the appropriate surgical treatment for congenital middle ear cholesteatoma. Several studies have shown that endoscopic ear surgery can effectively and safely remove congenital cholesteatoma in children, and its effectiveness is comparable to that of conventional microscopic surgery.35-38 Congenital cholesteatomas in Potsic stage I and II are good indicators of transcanal endoscopic ear surgery, and congenital cholesteatomas in stage III can also be treated by transcanal endoscopic ear surgery with an atticotomy. Our cases comprised 2 stage III and 1 stage IV congenital middle ear cholesteatomas, and we performed transcanal endoscopic ear surgery for the 2 stage III cases and endoscopic-assisted conventional microscopic approaches including mastoidectomy for the stage IV case. Previous studies have reported lower rates of residual lesions when using the microscope combined with the endoscope for rechecking and monitoring cholesteatoma.39-41 Notably, surgery should be personalized according to the patient’s disease extent and location.
We have followed up on these cases for half a month, 1 month, 3, and 6 months after surgery. The wound healed well and the patients were satisfied with the improvement of hearing. However, our study also has some limitations, such as short follow-up time and the absence of 1-year postoperative imaging. We plan to monitor these cases for an extended period.
Conclusions
Congenital cholesteatoma of the middle ear is not exclusive to children but may also occur in adults. The presence of congenital cholesteatoma should be considered in patients with intact eardrum, white soft tissue behind the eardrum, and unilateral conductive hearing loss. Also, in children with unilateral secretory otitis media, when the white soft tissue shadow inside the tympanic membrane remains intact after treatment, the possibility of congenital cholesteatoma should be considered. The accurate diagnosis of congenital cholesteatoma requires a combination of audiology, otoscopy, and imaging. The surgical treatment should not be limited to one modality but should be appropriate according to the location and range of the lesions, and the combination of endoscopy and microscope should be considered whenever appropriate.
Footnotes
Author Contributions
L.X. collected and analyzed data. L.Z. conceived the study, analyzed data, and wrote the manuscript.
Data Availability Statement
All data generated or analyzed during this study are included in this published article.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: this work was supported by a grant from Tongji Hospital.
Ethical Statement
This study was approved by the Institutional Review Board of Tongji Hospital. The authors have obtained written informed consent from the patient or the patient’s legally authorized guardian.