
Abstract
Alveolar rhabdomyosarcoma is a rare pediatric malignant tumor with a poor prognosis, and it is exceedingly rare for this tumor to manifest on the skin of the nasal dorsum. Therefore, timely and accurate treatment can improve the survival rate of patients. We reported a case of a 4 year-old child with acinar rhabdomyosarcoma of the nasal dorsum, and the patient was cured by surgery and postoperative chemotherapy without recurrence. This case report contributes to the understanding of this rare tumor.
Keywords
Introduction
Alveolar rhabdomyosarcoma (ARMS) is a subtype of rhabdomyosarcoma (RMS) that is distinguished by its acinar pattern. Typically, it afflicts individuals aged 10 to 25 years and commonly appears in the limbs and trunk, but its occurrence on the skin of the nasal dorsum is extremely rare. In this article, we present data on a single pediatric patient with ARMS located on the nasal dorsum who was admitted to our hospital.
Case Report
A 4-year-old male was admitted to the hospital with a painless mass on the right side of the nasal dorsum. More than 1 year ago, the child’s parents had discovered a rice-sized tumor on the right side of the nasal dorsum, which did not cause pain or itching. The child had been examined at our hospital’s dermatology department, where a dermoscopy suggested a myxoid cyst, but no treatment was administered. As the mass continued to grow, the child was referred to our department for further evaluation and management.
On clinical examination, a semicircular, smooth-surfaced, 1.5 × 1.5 cm translucent mass was identified on the right side of the nasal dorsum, which elicited slight pressing pain but no noticeable fluctuation. After admission, routine examinations were conducted, but no abnormalities were detected. Given the likelihood of a benign tumor, imaging studies were not ordered. Subsequently, we performed a tumor resection of the nasal dorsum, and intraoperative rapid pathology confirmed a diagnosis of a small cell malignant tumor. With parental consent, we excised the tumor along a safe margin of no less than 0.5 cm from its edge. The surgical wound’s base was located on the periosteum’s surface, and we confirmed the absence of obvious malignancy in the marked margins through a second rapid pathology. Therefore, we consider that the tumor is limited to the nasal skin. Following the tumor’s removal, the skin defect measured approximately 3 × 3 cm. We collaborated with the plastic surgery department to procure full-thickness skin from the upper arm to perform a nasal dorsum skin graft. Postoperative pathology and immunohistochemical analyses indicated that the mass was a small cell malignant tumor with features consistent with RMS. Immunohistochemistry showed Vim (+), CD56 (+), Desmin (+), Myogenin (+), MYOD1 (+), CK (−), NSE (−), CgA (−), Syn (−), S-100 (−), CK5/6 (−), P63 (−), LCA (−), CD3 (−), CD20 (−), CD99 (−), ALK (+), ki67 (60%+), and fusion PAX-FOXO1(+). Reticular fiber staining revealed a nest-like distribution of tumor cells, in line with ARMS (Figure 1).
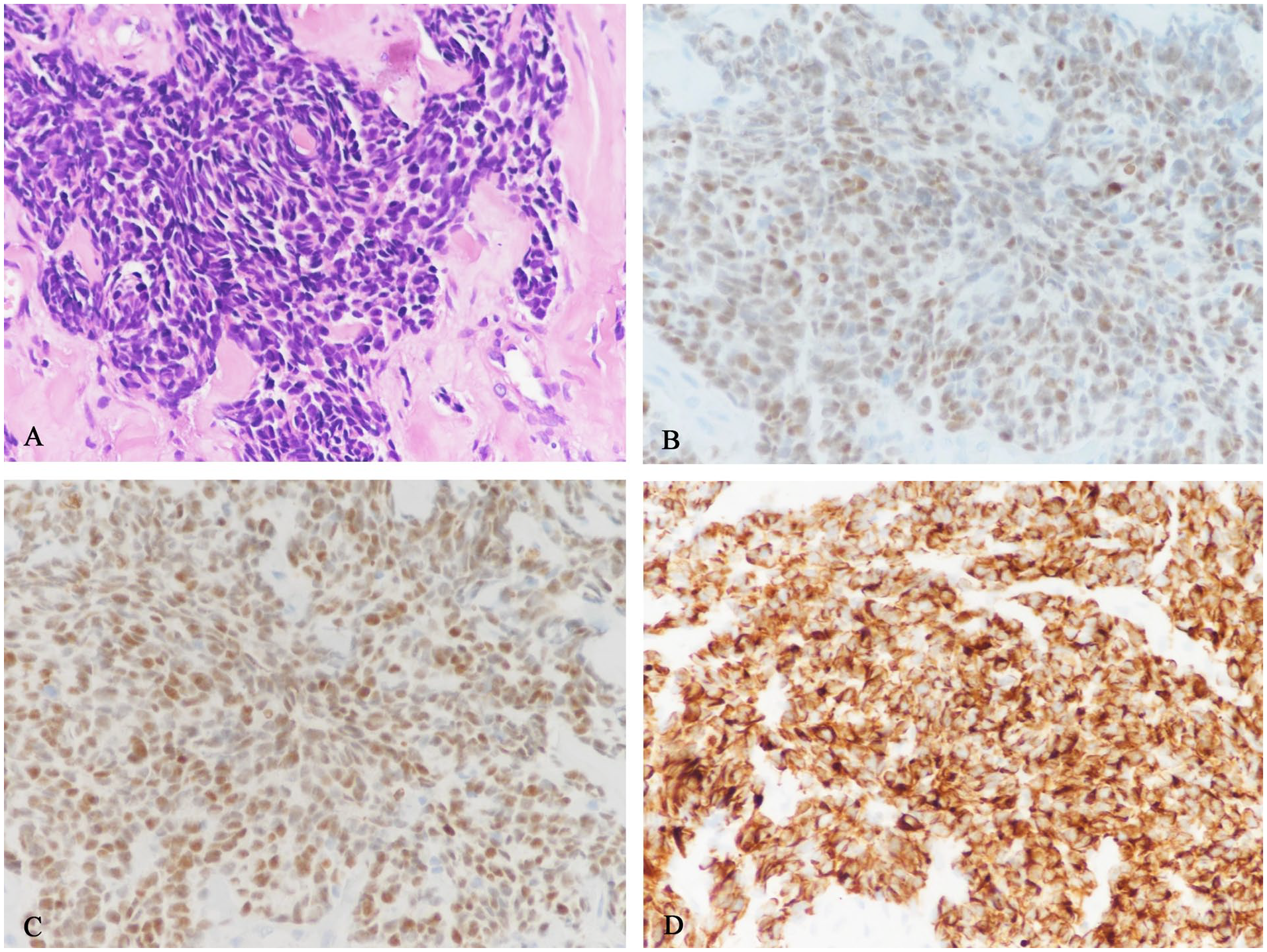
(A) Specific tumor cells with nest-like distribution were seen under hematoxylin-eosin (HE) staining (×400), (B) it shows Myogenin (+) under immunohistochemistry, (C) MYOD1 (+) can be seen under immunohistochemistry, and (D) it shows Desmin (+).
On the 11th day after surgery, the patient was transferred to the pediatrics department for further management and examination. Cervical computed tomography (CT) revealed lymph nodes in the II and III areas of the neck on both sides, with the left measuring approximately 2.3 × 0.8 cm and the right measuring approximately 2.6 × 1.0 cm. Craniocerebral magnetic resonance imaging (MRI) indicated postoperative changes in the nasal region, while other related examinations did not reveal any notable abnormalities (Figure 2).
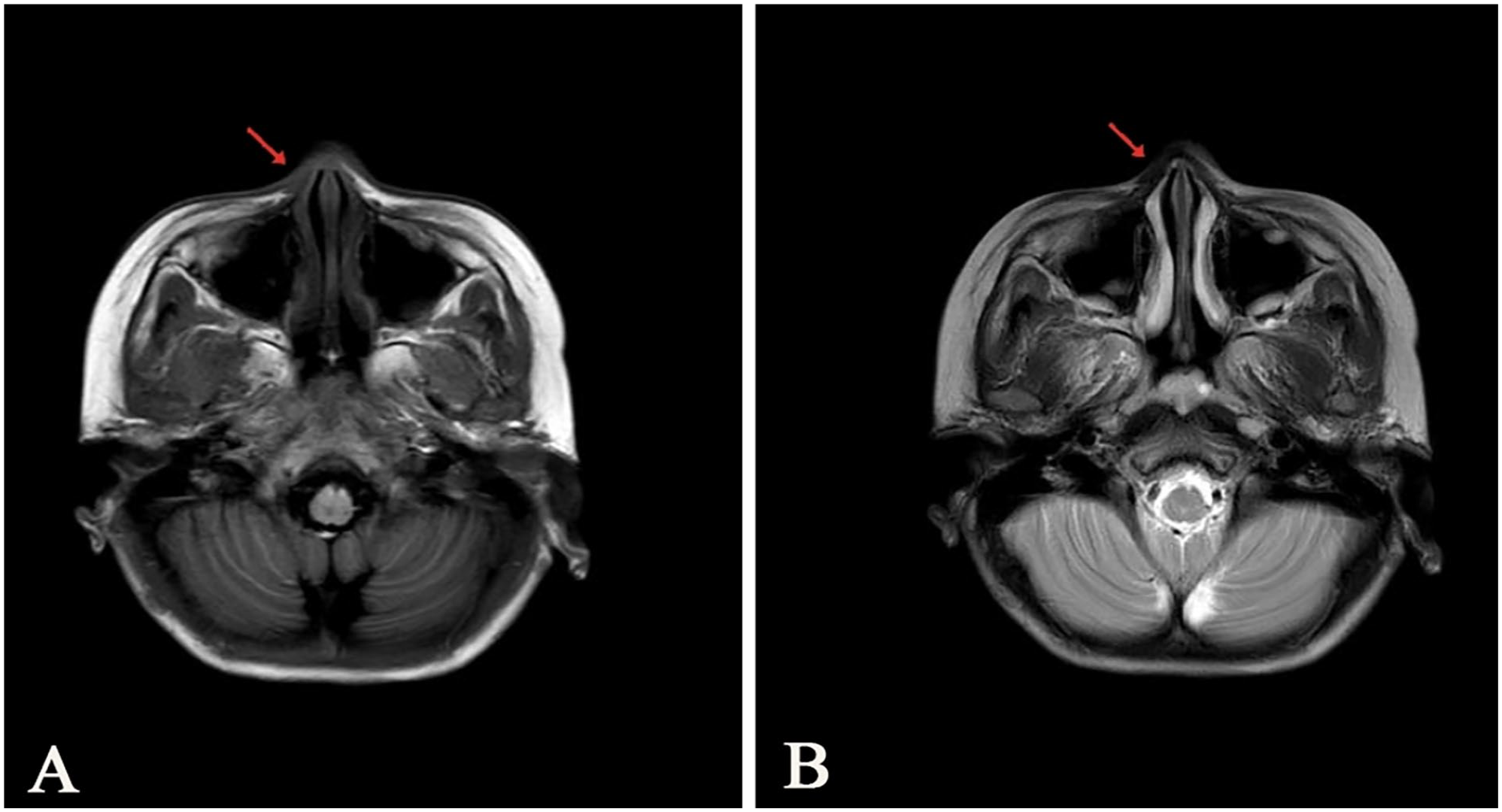
(A) Half a month after the operation, the brain-enhanced MRI in T1WI indicated that the right nose presented postoperative changes, and (B) there was no obvious abnormality in T2WI of brain-enhanced MRI 1 year after operation.
The patient’s parents declined postoperative radiotherapy but consented to chemotherapy. We employed the actinomycin D, adriamycin, vindesine, cyclophosphamide, and IEV (ifosfamide, etoposide, vincristine) regimens for alternating chemotherapy with alkalization and antiemetic supplements. One year following the operation, the patient’s neck lymph nodes had decreased in size compared to their previous dimensions, and there was no evidence of tumor recurrence on the nasal dorsum (Figure 3).
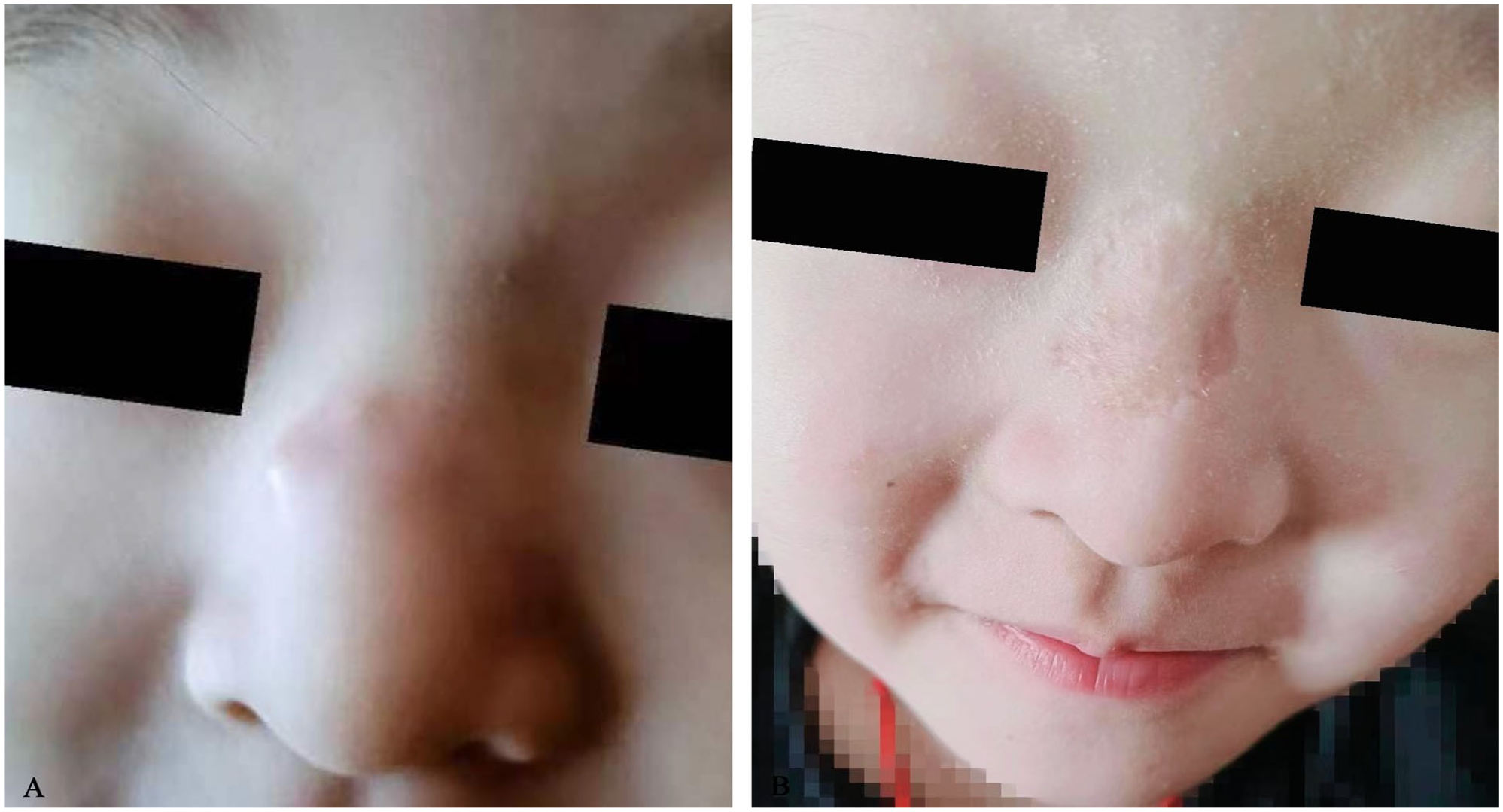
(A) The patient’s nose condition at admission and (B) the patient’s nasal condition 1 year after surgery.
Discussion
RMS is a malignant tumor that originates from the striated muscle tissue or mesenchymal tissue that differentiates into striated muscle, which is more common in children. 1 The etiology of RMS is unknown, which may be related to chromosomal abnormalities and gene fusion. According to WHO’s pathological classification in 2020,2,3 RMS is classified into embryonal, alveolar, spindle cell/sclerosing, and pleomorphic subtypes. In the head and neck area of children, the embryonic type is more common and the alveolar type is rare. 4 ARMS mainly occurs in adolescents aged 10 to 25 years, mostly in deep soft tissues of limbs, however is rare in nasal dorsum skin.
RMS typically manifests as a mass, which may or may not cause pain and may compress adjacent structures. Imaging is a crucial diagnostic tool for this condition, with MRI being the preferred modality. MRI can reveal the lesion’s size, location, and relationship with surrounding tissue structures, as well as the presence of pelvic lymph node metastasis. On T1-weighted images (T1WI), RMS is usually low to equal signal intensity, while on T2-weighted images (T2WI), it is equal to higher than signal intensity. ARMS presents with marked heterogeneity and extremely high-signal intensity on T2WI.5,6 Additional imaging studies, such as CT, positron emission tomography-CT, bone scans, and lumbar puncture, may be necessary to identify distant metastases. However, a definitive diagnosis of RMS depends on postoperative pathology and immunohistochemistry.
Pathologically, ARMS is characterized by small round cells arranged in sheets or nests, separated by a fibrous vascular stroma. Some cells may undergo degeneration and necrosis, losing adhesion and floating in the acinar cavity to form a typical vesicular structure. 7 Unlike embryonal rhabdomyosarcoma (ERMS), some of the tumor nuclei in ARMS are situated around eosinophilic cytoplasm, which can create characteristic multinucleated giant cells. Myogenic tumor markers, Desmin (+), and MyoD1 (+), aid in diagnosing ARMS, and Myogenin (+) is a crucial reference index for identifying RMS from other small cell tumors. ARMS may have chromosomal t (2; 13) or t (1; 13) translocations that result in PAX3-FOXO1 and PAX7-FOXO1 gene fusions. These fusions often correlate with recurrence and bone metastasis and have a poor prognosis.2,8
The main treatments for RMS are surgical excision and chemoradiotherapy. The Intergroup Rhabdomyosarcoma Study Group and the European Pediatric Soft Tissue Sarcoma Study Group have classified RMS into low, intermediate, high, and central aggressive groups based on risk. 9 According to the definition of moderate-risk RMS in Children’s Oncology Group (COG), 10 nonmetastatic ARMS in children is classified as a moderate-risk group, indicating the need for postoperative multidrug combination chemotherapy, regardless of whether the tumor is completely removed by surgery. The vincristine, actinomycin D, and cyclophosphamide (VAC) regimen is the gold standard chemotherapy regimen for treating ARMS internationally. In recent years, the VAC and IEV regimens have been alternately employed for ARMS treatment in the intermediate-risk group, following the Shanghai Children’s Medical Center (SCMC)-RMS-99 11 scheme, If preoperative imaging evaluation indicates that complete tumor removal may be difficult, preoperative chemotherapy and radiotherapy can be administered for 3 to 4 courses before elective surgery. The total duration of chemotherapy is typically no more than 12 courses. The chemotherapy process should be based on its effectiveness and the specific treatment timeline and should monitor liver and kidney function and hearing while reducing bone marrow suppression. ARMS have an invasive growth pattern with high malignancy, and lymph node metastasis or hematogenous spread may occur early. Therefore, the prognosis for ARMS is poorer than that for ERMS, with a 5 year survival rate of less than 50%.12,13
Conclusion
ARMS is a rare disease in children that can be easily overlooked at the initial diagnosis. Early detection, diagnosis, and treatment are crucial for improving the survival rate of children with ARMS, but it remains a clinical challenge. In this case, the possibility of a malignant tumor was not considered before the operation. There were several reasons for this, including the fact that dermoscopy indicated a myxoid cyst, the tumor did not exhibit any malignant appearance or clinical biological behavior, the patient was young, and no specific imaging was performed. However, the rapid pathological examination was conducted promptly, and the appropriate treatment was implemented based on the pathological results. The patient is currently recovering well but requires ongoing follow-up.
Footnotes
Authors’ Note
Jingyu Chen and Fan Zhang contributed equally to this work. Verbal informed consent was obtained from the patient(s) for their anonymized information to be published in this article.
Data Availability Statement
All data generated or analyzed in this study are included in this article and its online supplemental material. Further inquiries can be directed to the corresponding author.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.
Ethics Statement
Written informed consent for publication of this case report and any accompanying images was obtained from the patient’s parents.