
Abstract
Purpose
There has been a lack of evidence-based management strategies on the nasal presentations of Rosai–Dorfman disease (RDD). We aim to investigate the clinical manifestation, treatment, and outcomes in patients with nasal RDD.
Methods
We retrospectively reviewed available medical records of patients diagnosed with nasal RDD from 2014 to 2021 at our department.
Results
A total of 26 patients were included with a marked female preponderance (2.25:1). The most common symptom and affected sites were nasal congestion (31%) and nasal cavity (73%), respectively. The average times of biopsies was 1.5 times (range: 1–3). The histiocytes were positive about S100 and CD68 and negative for CD1a with common emperipolesis. The mean duration of follow-up was 34 months (range, 3–87). One patient with concomitant nasal small B-cell lymphoma achieved complete remission after chemoradiotherapy. Recommended treatments were endoscopic resection (92%) and oral corticosteroids (21%). Surgery was performed to remove the resectable lesion as completely as possible. Corticosteroids induced almost 100% overall remission. Of the relapses, two patients achieved an overall response and one remained in progressive stage after subsequent excision. Two patients only received dissection biopsy that responded to oral corticosteroid administration and combined therapies of lenalidomide and dexamethasone, respectively.
Conclusions
Diffuse lesions in nasal cavity and sinuses, and even widely affected nasal skull base, laryngopharynx, orbit, and cavernous sinus, should be considered the possibility of Rosai–Dorfman disease. Characteristic immunohistochemical staining is helpful for the diagnosis. Endoscopic surgical therapy remains the mainstream treatment for patients enduring an unbearable course. Oral corticosteroid administration serves as an adjuvant therapy for first-line treatments.
Introduction
Rosai–Dorfman disease (RDD) is an idiopathic, benign histiocytic condition of unknown origin with marked nonmalignant proliferation of distinctive histiocytes within lymph node sinuses and extranodal sites. The histiocytes were positive about S100 and CD68 and negative for CD1a with ubiquitous emperipolesis. The prevalence rate of RDD has been reported to be 1:200,000, with approximately 100 new cases being diagnosed per year in the United States. 1 Classically, Rosai–Dorfman disease clinically manifests as chronic painless cervical lymphadenopathy with pyrexia, leucocytosis, increased erythrocyte sedimentation rate, and hypergammaglobulinemia. The population sites of these abnormal histiocytes are not confined to lymph node sinuses, and 43% of RDD patients have at least one site of extranodal lesions, including the skin (20.8%), oral cavity (27.3%), nasal cavity/paranasal sinuses (22.9%), lower respiratory tract (22.3%), eyelid/orbit (22.3%), and larynx (16.7%). 2 Of this large-scale cohort, Foucar et al. highlighted that involvement in the nasal cavity and/or paranasal sinuses may appear as the first symptom. 2 Additionally, the registry indicated that head and neck manifestations were more common in patients of Asian descent, most universally affecting the nasal cavity. 3 Importantly, we obtained pathological data from our medical records, which indicated that nasal presentations of Rosai–Dorfman disease recorded up to 54.2% of total confirmed cases within the last seven years, while it may be a bias towards noted RDD patients in a specialized medical centre. However, optimal therapeutic guidelines on the management of nasal RDD have not yet been established given the lack of systematic data. Herein, we analyzed the clinical traits, therapies, and prognosis of 26 retrospective histopathologically proven cases of nasal presentations of Rosai–Dorfman disease in a specialized tertiary institute.
Methods
The medical data of patients diagnosed with nasal RDD at our specialized referral centre of August 2014 to September 2021 were reviewed. All patients that had definitive histopathological diagnosis were considered for inclusion. Data extracted from the medical data included demographic features, clinical manifestations, endoscopic and radiologic findings, histopathological traits, treatment strategy, and prognosis evaluations. Enrolled patients were followed up by a review of the medical records or telephone interview for acquiring additional information. Two independent residents were responsible for the collection and review of medical records to reduce errors and bias. A large percent of patients did not routinely receive positron emission tomography–computed tomography (PET-CT) scans for the treatment. Hence, we mainly utilized available imaging data, including contrast-enhanced CT scans and magnetic resonance imaging (MRI) scans. The involved sites and scope of disease was assessed based on endoscopic records and radiographic findings, and also included those found intraoperatively or during the follow-up. According to whether involving the lymph nodes, nasal RDD was divided into 'classical' and 'extranodal'. Based on consistent definitions of comorbidity, nasal RDD was classified as 'neoplasia-associated' RDD, 'immune-related' RDD, and 'IgG4-related' RDD. As there is lack of consensus treatment recommendation of nasal RDD, these patients received multiple therapeutic modalities. We evaluated therapeutic response by reviewing the clinical outcomes clinically and radiologically. Existing studies indicated that RDD is a chronic relapsing/remitting disease. Thus, we chose to record the overall response rate (ORR) as assessment criteria, which included complete response and partial response (partial resolution of symptoms, residual endoscopic or imaging findings). Data were analyzed by descriptive statistics. This retrospective study was approved by our institutional review board.
Results
Clinical Profiles and Presenting Features
Clinical data and baseline features of patients with nasal RDD.

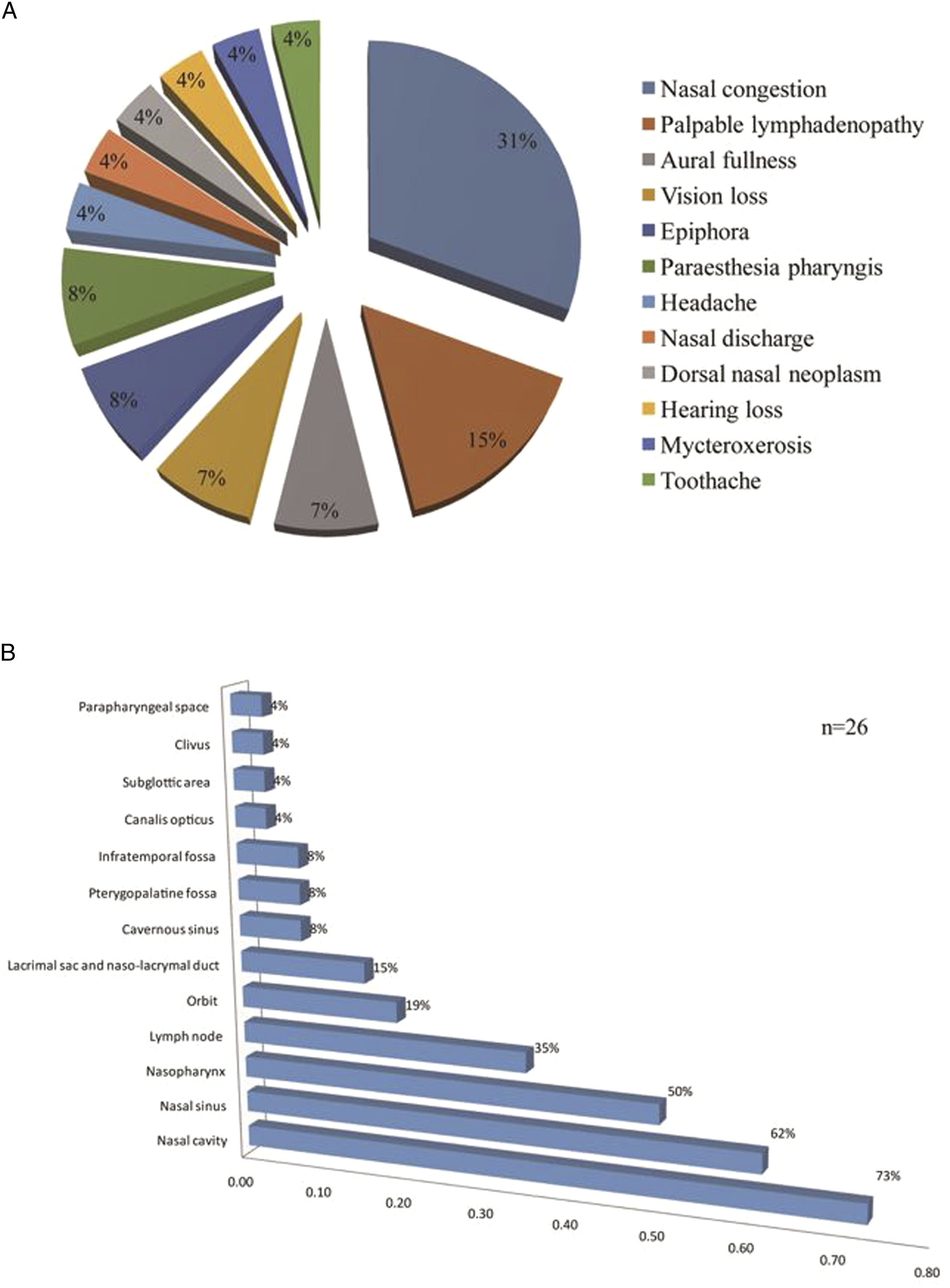
Clinical manifestations and location involvement among patients with nasal RDD (A) presenting features and (B) site involvement.
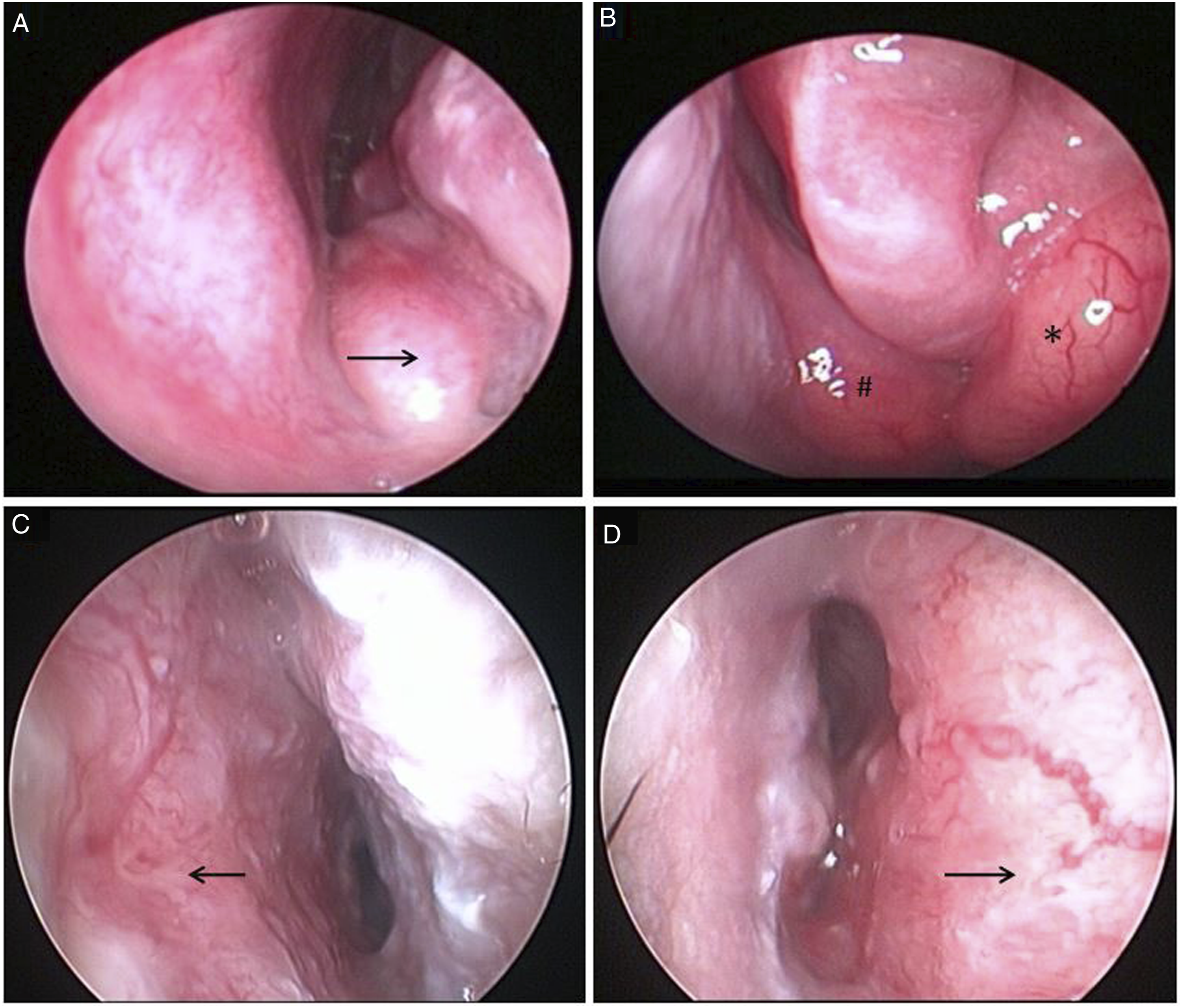
Endoscopic results of nasal RDD. (A and B) Endoscopic signs of multisite lesions located in the left inferior nasal meatus (arrow), nasal septum (#), and middle nasal meatus swelling from the maxillary sinus (*). (C and D) Endoscopic findings of diffuse thickening of septum mucosa (arrow).
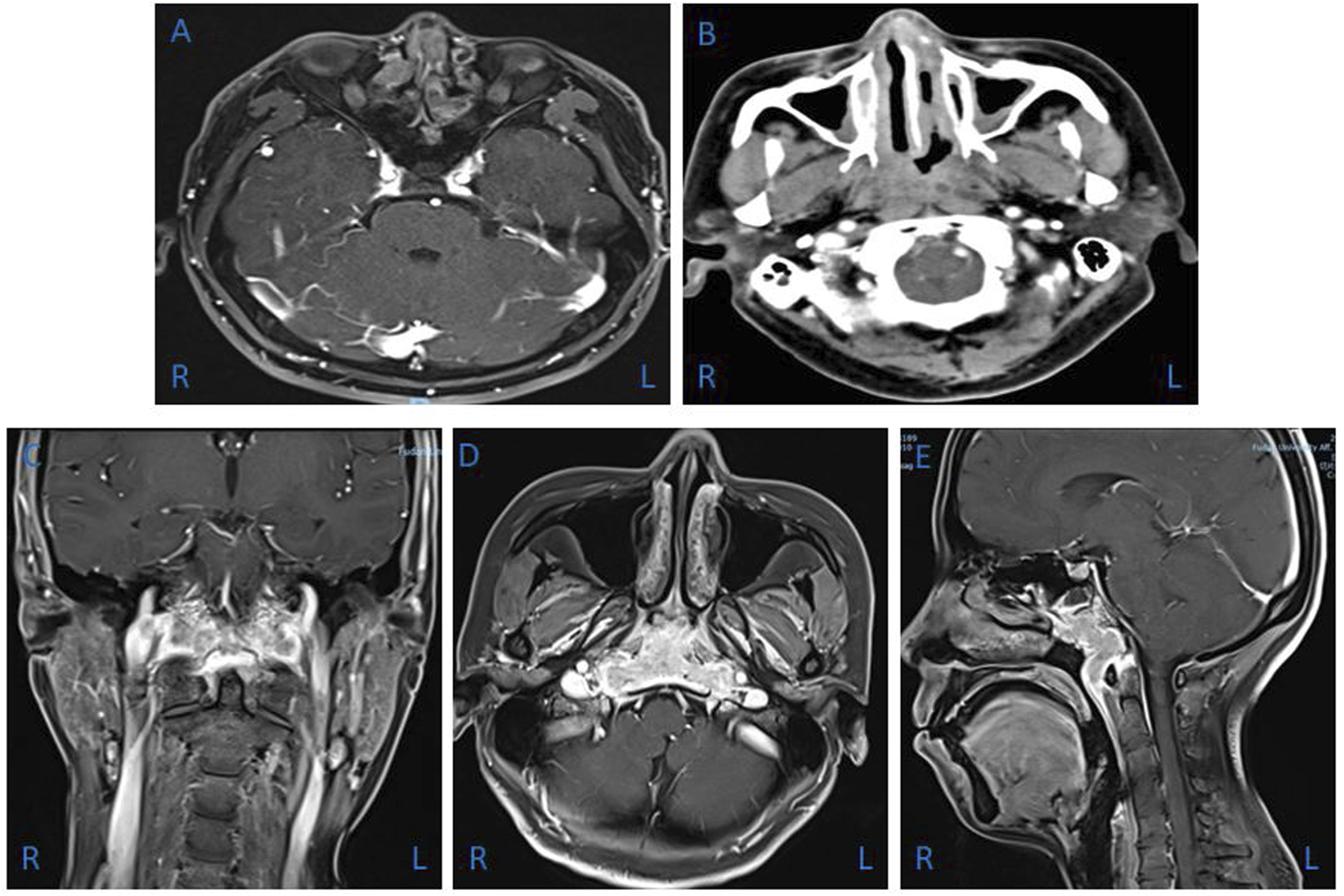
Imaging findings of nasal RDD. (A) Axial contrast-enhanced MRI depicted a hyperplastic soft tissue mass in the bilateral nasal cavity. (B) Enhanced axial CT of the sinonasal cavity demonstrated septal and nasopharyngeal diffuse soft tissue mass. (C, D, and E) Enhanced coronal, axial, and sagittal MRI showed an extensive soft tissue lesion in the nasopharynx and middle skull base, involving the bilateral longus cephalus, clivus, and jugular foramen. MRI: magnetic resonance imaging.
Laboratory examination of patients with nasal RDD.

“-” = not mentioned.
Nasal RDD Subtype
Overall, 7 patients had high expression of IgG4 and IgG levels in diseased lymphoplasmacytic cells (e.g. >100 cells per high-powered field in lymph nodes with an IgG4-positive to IgG-positive plasma cell ratio ˂40%). Three had elevated serum IgG levels, but without serum IgG4 levels examined. Storiform fibrosis was not observed, whereas emperipolesis of plasma cells and lymphocytes penetrating through large S100-positive histiocytes was common. Corresponding serologic evaluation was not performed due to the lack of clinical symptoms of accompanying autoimmune diseases. Immune-related nasal RDD was not found. Only one patient had neoplasia-associated nasal RDD. She underwent endoscopic surgery, and postoperative pathological findings indicated small B-cell lymphoma and nasal RDD concomitantly. None of the patients had a familial predisposition.
Histological Examination
The average times of biopsies was 1.5 times (range: 1–3). All lesions had similar histopathologic features. Under the microscope, there was a polymorphic group of histiocytes, plasma cells, and mature lymphocytes isolated by fibrous septum. Some lymphatic follicles were seen with germinal centres. In addition, there were prominent histiocytes that contained abundant pale to vacuolated cytoplasm and a large nucleus with prominent nucleoli. Most of these cells showed lymphophagocytosis or emperipolesis. These histiocytes were positive about S100 and the CD68 antibody but negative for CD1a (Figure 4). Histopathological results of nasal RDD. (A and B) Nasal RDD histiocytes are S100+ and CD68+ by immunohistochemistry, respectively (×100). (C) Histopathologic section showing a polymorphous population of cells consisting of mature lymphocytes and plasma cells interspersed with histiocytes. Histiocytes are large with vesicular nuclei and abundant cytoplasm with engulfed lymphocytes and plasma cells, a phenomenon called lymphophagocytosis or emperipolesis, a hallmark of RDD (arrow) (×200, haematoxylin and eosin).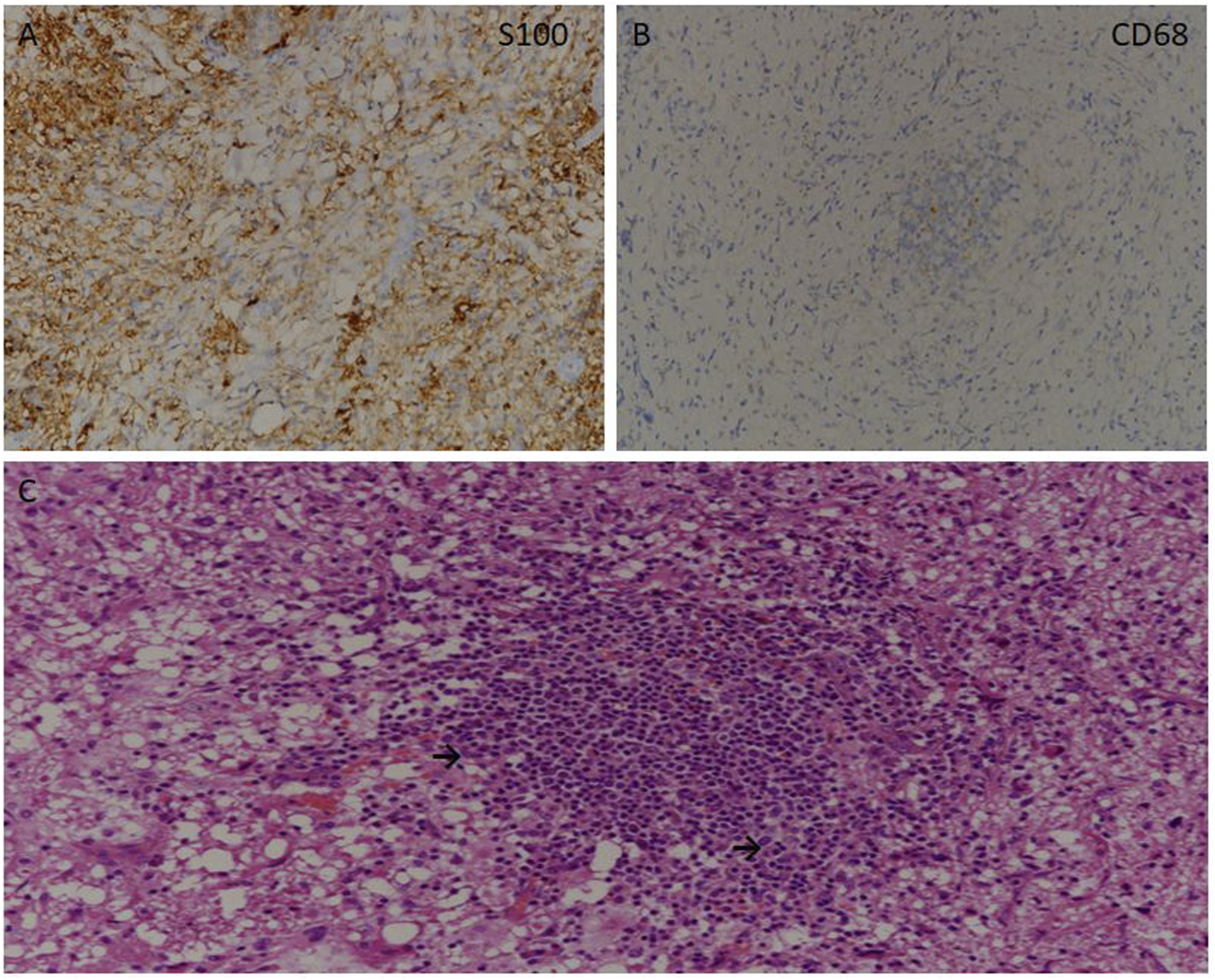
Treatments and Prognosis
Treatments and overall response rates (ORRs) in patients with nasal RDD.
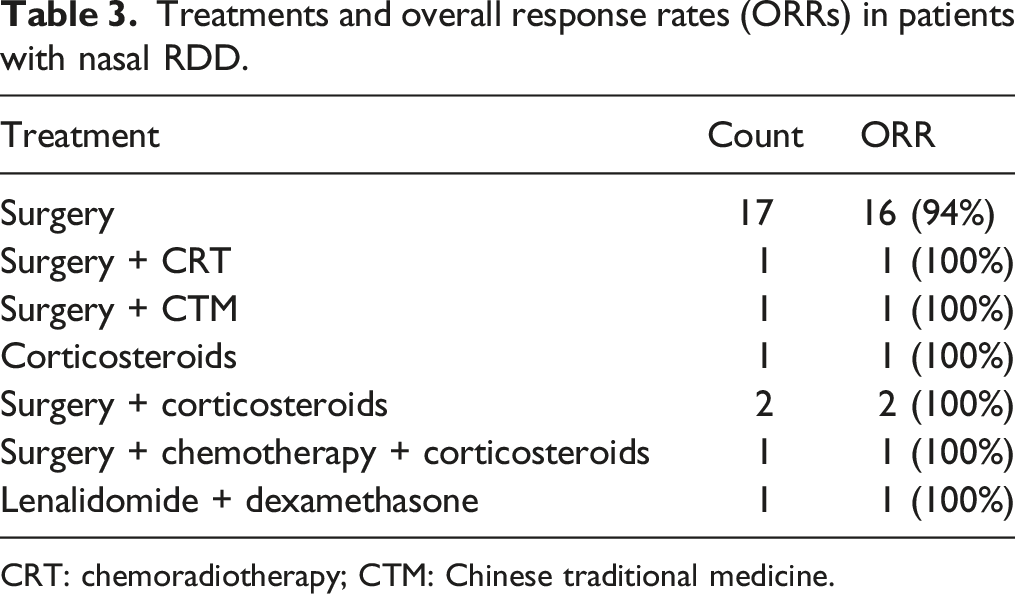
CRT: chemoradiotherapy; CTM: Chinese traditional medicine.
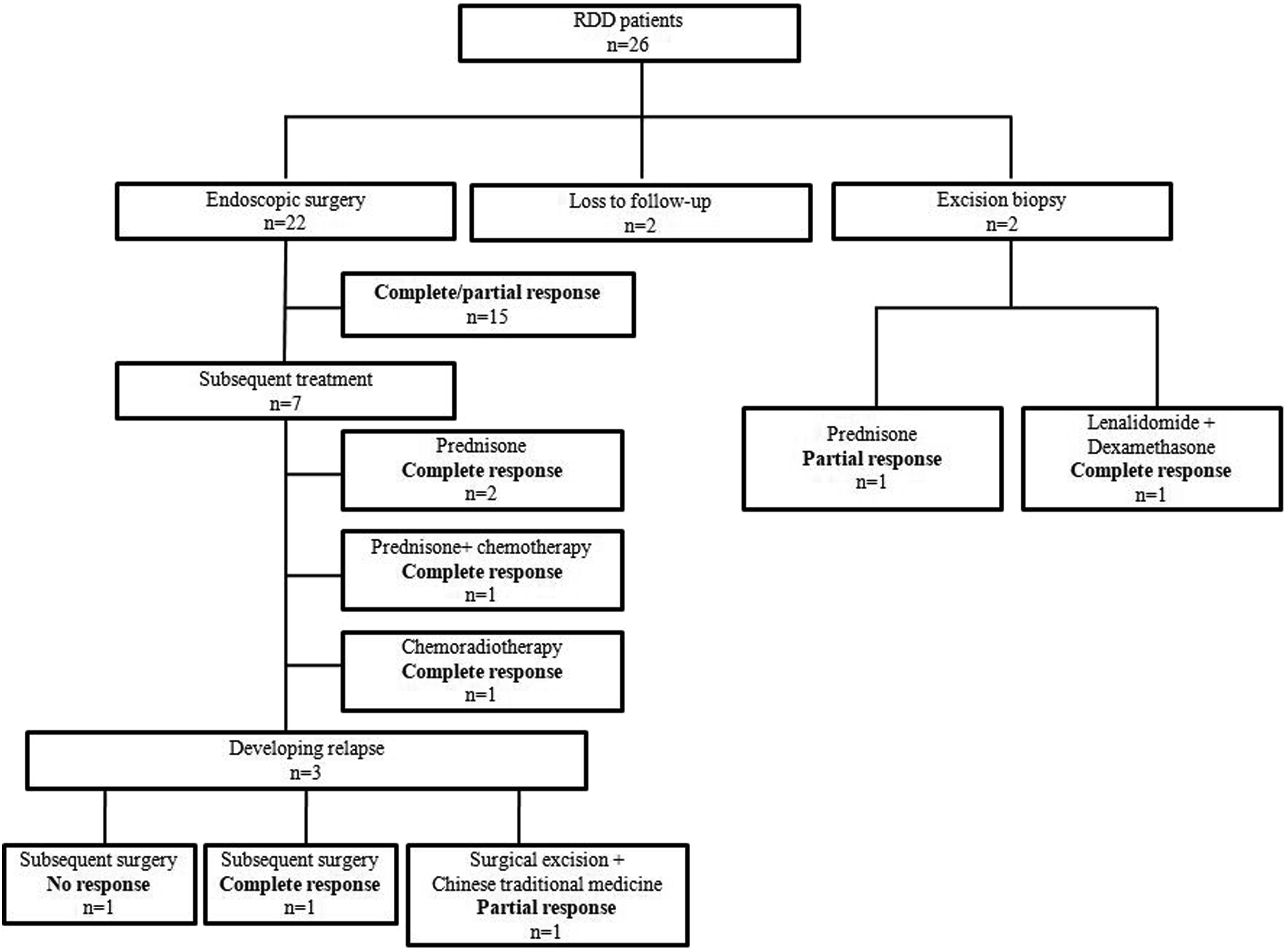
Treatments and outcomes of patients with nasal RDD from diagnosis until response.
Discussion
Since RDD was first described in 1969 as a rare histiocytosis, there has been a paucity of systematic studies. In addition, most studies of RDD involving the nose merely reported case reports or small-scale reports.4,5 Hence, we shared empirically summarized series of patients with nasal RDD for clinical features, treatment approaches, and outcomes. Over the past decades, the recognition of this disease appears to be increasing, and especially within the last three years, our tertiary referral centre saw patients with nasal RDD at an average rate of 5 cases. Compared to the historical cohort reported by Foucar et al., our cohort had more patients who were older (mean age 47 versus 20 years). 2 Female preponderance of our study was more significant than that in a previous cohort of sinonasal RDD patients (female:male 2.25 versus 1). 6 Contrary to the historically reported RDD cohorts with massive lymphadenopathy, we found that the most cases presented with nasal lesions. Lymphadenopathy was the second most common manifestation. In addition, the involvement sites of enrolled cases were not limited to bilateral cervical lymph nodes, as described by Foucar et al. in the initial landmark series of RDD. 2 The reason for this difference in site involvement is unknown but may be related to a difference in the population of the 2 studies or excluding some classic cases of nasal RDD treated in the community in our study. It may also be biased towards incidentally found nrRDD when being extensively evaluated by means of nasal endoscopy and imaging for primary nrRDD diagnosis and received endoscopic treatments. Our findings emphasize that nasal RDD is inherently syndromic with a wide range of manifestations, and our experience may be more representative of some patients who develop specific symptoms as initial manifestations are diagnosed in our specialized centre.
Based on the available evidence, the causes of Rosai–Dorfman disease remain unknown. However, proposed causes include viral infection, immunologic disorders, and activation disorders of histiocytes and may be associated with familial and/or malignant processes.5,7 In our series, no patients had familial history. Histopathological re-review of previous biopsy slides by institutional expert pathologists did not indicate IgG4-related disease. Interestingly, one patient was concomitantly diagnosed with nasal small B-cell lymphoma, consistent with prior reports.4,5,8 Some patients had abnormal changes in routine blood tests, serum biochemistry, and immunologic examinations, but combined with their medical history and other presentations, they did not support the diagnosis of viral infection or immunologic disorders. Abla O et al. suggested that laboratory evaluation should include complete blood count, comprehensive metabolic panel, coagulation studies, and an evaluation of C-reactive protein, uric acid, lactate dehydrogenase, and serum immunoglobulins in the diagnosis of RDD. 5 All patients in our study underwent preoperative contrast-enhanced MRI or CT, while only 2 underwent PET-CT scans. A single-centre retrospective study showed that PET-CT detected lesions were not recognized by anatomic cross-sectional imaging in 30% of RDD patients with available prior CT or MRI. 9 Ronald S. Go also proposed that full-body FDG-PET-CT is recommended as part of the baseline evaluation of RDD. 10 However, MRI of the head tends to be superior for evaluation of the sinuses and nasal skull base compared with PET-CT. 11 Hence, we suggest routinely performing contrast-enhanced MRI combined with nasal endoscopy to evaluate suspected lesions. It is worth further exploration whether all patients should receive PET-CT scans, even if there is value in excluding systemic RDD involvement. Diagnosis is made only on the basis of histopathology and immunohistochemistry. However, the histopathological diagnosis of RDD can be challenging due to its rarity and nonspecific histologic findings, especially in extranodal forms. 8 Abla O et al. proposed that RDD tissue may contain very few lesional cells and often shows a pronounced inflammatory background with plasma cells or lymphoid follicle formation and neutrophilic infiltration. 5 The difficulty in diagnosing RDD histopathologically is manifested by the possibility of requiring multiple biopsies. As described in our data, some patients underwent even the 3 biopsies. Diagnostic features include mixed, florid infiltrate of large pale histiocytes, abundant plasma cells, and lymphocytes with strong reactivity to CD68 and S100 protein on immunohistochemistry. Emperipolesis is a commonly reported histologic finding but is not specific to this entity, which is basically consistent with the immunohistochemical results of our cases. 12 Therefore, the diagnosis of RDD should include a comprehensive clinical and radiologic examination, as well as histopathologic analysis. Neurologic and psychological assessments are also recommended. A history of inherited conditions predisposing to RDD (e.g., ALPS), malignancies and other neoplasia associated with RDD, and other autoimmune disorders should be evaluated based on clinical symptoms and family history.
Systematic studies analyzing first-line treatments and outcomes in RDD are lacking. Unknown causes suggest that current treatments are usually symptomatic. Watch and wait is a reasonable treatment strategy for patients with asymptomatic and mild disease, as spontaneous remission has been reported to occur in 40% of these patients.5,8 In our series, all patients suffered unbearable symptoms and required treatment. Surgery is also a reasonable curative option for those with isolated disease or for debulking symptomatic disease. Hence, 24 (92%) received radical endoscopic surgery. During the follow-up period, 3 patients developed recurrence, and no recurrent lesions underwent spontaneous regression. Despite the significantly lower relapse rate compared with previous studies, all patients underwent a second operation and subsequent multiple therapies.8,13,14 Of these, 2 remain in sustained clinical remission, but another young patient had progressive disease, and whether to perform genetic sequencing in molecular alteration should be considered. Other empirically used therapies also led to sustained clinical responses. Our study suggests that 2 patients undergoing excision biopsy benefit of oral prednisone treatment and combined therapy with lenalidomide and dexamethasone. Another 3 were given oral prednisone therapy as part of the surgical treatment and led to complete symptomatic remission. No side effects were found from corticosteroid therapy. The optimal doses and duration of steroid treatment is unknown at this time; treating with optimal response, followed by a slow taper, is a reasonable strategy. 5 Adverse effects of steroids should be carefully monitored, although these are generally well tolerated. 8 Chemoradiotherapy was performed for the patient with small B-cell lymphoma-associated RDD, and no signs of relapse were found. Aradhana K et al. concluded that low-dose radiation either in combination with surgery or corticosteroids in nodal or extranodal lesions showed better local control through a five-year retrospective analysis. 15 However, Abla Oet al. suggested that radiation therapy (RT) should only be used for palliative purposes of patients with multifocal symptomatic disease. 5 In addition, chemotherapy is recommended for patients with disseminated and multifocal RDD or for those for whom both surgery and radiotherapy have failed.16-18 We believe that it is beneficial to carry out randomized controlled clinical trials and provide more valuable evidence for treatment options.
Conclusion
The clinical course of nasal RDD is unpredictable, with episodes of exacerbation and remission that could last many years. The disease is often self-limited with a positive outcome; nevertheless, death occurs to 5–11% of patients. 19 Overall, our novel findings highlight that endoscopic surgical excision is the mainstay of treatment given that complete excision affords symptom reversal, low recurrence rates, and positive long-term prognosis. In our series, systematic involvement in nasal RDD was uncommon; nevertheless, corticosteroid therapy in universal response was indicated as the preferred adjuvant therapy after aggressive surgery. In addition, despite progress made in the understanding of curative options of alternative strategies, such as intralesional administration, radiotherapy, chemotherapy, immunomodulators, and targeted agents, we should further make efforts to study ontogeny and pathogenesis of nasal RDD. While watching and waiting could be considered for asymptomatic and mild patients, close follow-up remains necessary to indicate early relapse.
Footnotes
Acknowledgements
We thank all the participants for their contribution and participation.
Authors’ Contributions
Xu HY and Li WP conceived and designed the study. Zhang HK and Wang H acquired the data. Xu HY and Zhang C drafted the manuscript. Li WP performed the statistical analysis. Wang DH supervised the study.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This study was financially supported by the National Natural Science Foundation of China (No. 81870703); Shanghai Shen Kang Hospital Development Center (SHDC12018118); Science and Technology Commission of Shanghai Municipality (23YF1404600); and Clinical Research Plan of the SHDC (SHDC2020CR2005A).
Ethics Approval
This study was approved by the Institutional Review Board of the Affiliated Eye Ear Nose and Throat Hospital (AEENTH) at Fudan University (Shanghai, China).
Consent for Publication
Written informed consent for publication of anonymized case details, which included accompanying images, was obtained from the patients.
Availability of Data and Materials
The datasets used and/or analyzed during the present study are available from the corresponding author on reasonable request.