
Abstract

Keywords
Significant Statement
Phosphaturic mesenchymal tumors (PMTs) are rare tumors commonly arising in soft tissues and bones. They are the main cause of tumor-induced osteomalacia (TIO), a paraneoplastic syndrome caused by production of FGF23 and other phosphatonins. “Non-phosphaturic” variant of PMTs (i.e., not associated with TIO) have been also reported. We describe a patient with a PMT of the middle ear, a very rare site for PMTs, in which TIO failed to develop although three phosphatonins were expressed.
Main Document
Phosphaturic Mesenchymal Tumors (PMTs) are defined by WHO as “morphologically distinctive neoplasms that cause tumor-induced osteomalacia (TIO) in most affected patients, usually through production of FGF23.” 1 They are rare, usually benign and commonly located in soft tissues and bones.1-3 Patients with TIO present hypophosphatemia, renal phosphate wasting, inappropriately normal/low serum levels of 1,25-dihydroxyvitamin D and clinical features related to systemic osteomalacia including pain, weakness, gait disturbances and fractures. 3 In addition to FGF23, PMT-tumor cells are known to produce other phosphatonins [e.g., Matrix Extracellular Phosphoglycoprotein (MEPE) and Fibroblast Growth Factor-7 (FGF7)] involved in the regulation of bone and mineral metabolism and thought to play synergistic/additive actions with FGF23 in the development of TIO.2-4 Identification and complete resection of the tumor is imperative for treatment. Indeed, TIO is definitely cured upon complete removal of the tumor.1-3 For not completely excised or inoperable TIO-associated PMTs, medical treatment with phosphate supplements and calcitriol is needed, radiotherapy has been employed and humanized anti-FGF23 monoclonal antibodies have been recently approved.3,5 Nearly half of TIO-associated PMTs show either FN1-FGFR1 or, rarely, FN1-FGF1 fusion gene.2,3,6,7 Interestingly, tumors with morphological features and molecular signature overlapping those of PMTs and with expression of FGF23 have been also reported in the absence of TIO and, for these reasons, defined “non-phosphaturic” variant of PMTs.1-3,8
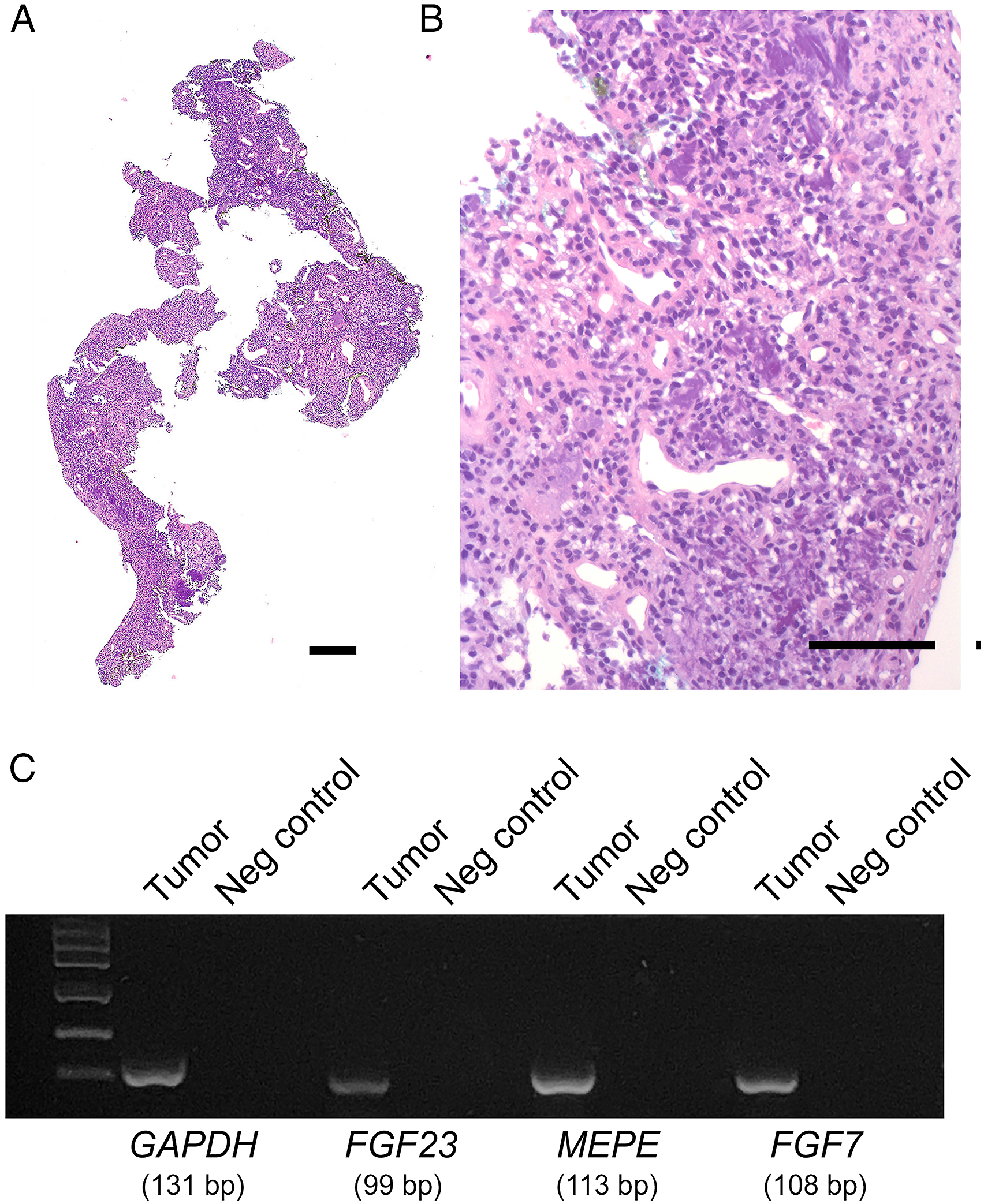
We report here a 70-year-old woman presenting with progressive right hearing impairment. Oto-microscopy showed intact tympanic membrane. Pure-tone audiometry revealed right mixed hearing loss with an air-bone gap of 25 db on high frequencies (1 kHz, 2 kHz, and 4 kHz). Computed Tomography (CT) and Magnetic Resonance Imaging (MRI) revealed a 1-cm tumor involving the right middle ear (Figure 1). The patient underwent surgery with en bloc removal of the tumor through endaural approach under general anesthesia. Histologically (Figure 2A-B), the tumor consisted of bland, round-ovoidal to spindle cells arranged around variable sized blood vessels. Focal matrix calcifications were observed while atypia, mitoses, necrosis, and multinucleated giant cells were not. Immunohistochemical analysis, performed as described previously,4,8-10 showed immunoreactivity of the neoplastic cells for Vimentin and CD56, but not for pan-cytokeratin, CD31, CD34, Smooth Muscle Actin, S100, STAT6, HMB45, and nuclear β-Catenin. The Ki-67 proliferation index was less than 5%. Based on these findings, the diagnosis of PMT was made.1-3 To confirm the diagnosis, total RNA was extracted from the formalin-fixed paraffin-embedded (FFPE) tumor block and used for Reverse Transcriptase Polymerase Chain Reaction (RT-PCR). The analysis demonstrated the expression of FGF23, MEPE, and FGF7 (Figure 2C). As all available clinical data, blood tests and bone mineral density failed to reveal changes consistent with TIO, the diagnosis of “non-phosphaturic” variant of PMT was eventually made. Axial (A) and coronal (B) CT and coronal MRI (C) images showing the tumor (arrow) englobing the ossicular chain and extending from the epithympan to the tympanic membrane. Whole mount section of the FFPE block prepared from the tumor tissue sample obtained at surgery is shown in A. Details of the neoplastic cells and matrix calcifications (arrows) are shown in B. Agarose gel electrophoresis showing the expression of FGF23, MEPE, and FGF7 in the tumor is illustrated in C. For RNA isolation, FFPE sample was treated with xylene to remove paraffin and then incubated with Proteinase K at 56°C for 1 hour. Total RNA was isolated as described previously.
11
cDNA was obtained by reverse transcription of 1 μg and used as template for PCR analysis using the following primer sets: FGF23 (NM_020638.3): 5’-TCTGCATGGATTTCAGAGGCA-3’ (forward) and 5’-AGACGTCGTACCCGTTTTCC-3’ (reverse), product size 99 bp; MEPE (NM_020203.6): 5’-GCAAAGCTGTGTGGAAGAGCA-3’G (forward) and 5’-GGACAATATTTTCTTTAGATGATAGC-3’ (reverse), product size 113 bp; FGF7 (NM_002009.4): 5’-CTGTCGAACACAGTGGTACCTG-3’ (forward) and 5’-CCAACTGCCACTGTCCTGATTTC-3’ (reverse), product size 108 bp. GAPDH (product size 131 bp) was used as housekeeping gene and amplified using the primers 5’-GTCTCCTCTGACTTCAACAGC-3’ (forward) and 5’-ACCACCCTGTTGCTGTAGCCAA-3’ (reverse). The identity of the amplification products was verified by DNA sequencing. Negative controls included the specific primers in the absence of cDNA. A and B: hematoxylin–eosin. Bars: 200 μm in A and 100 μm in B.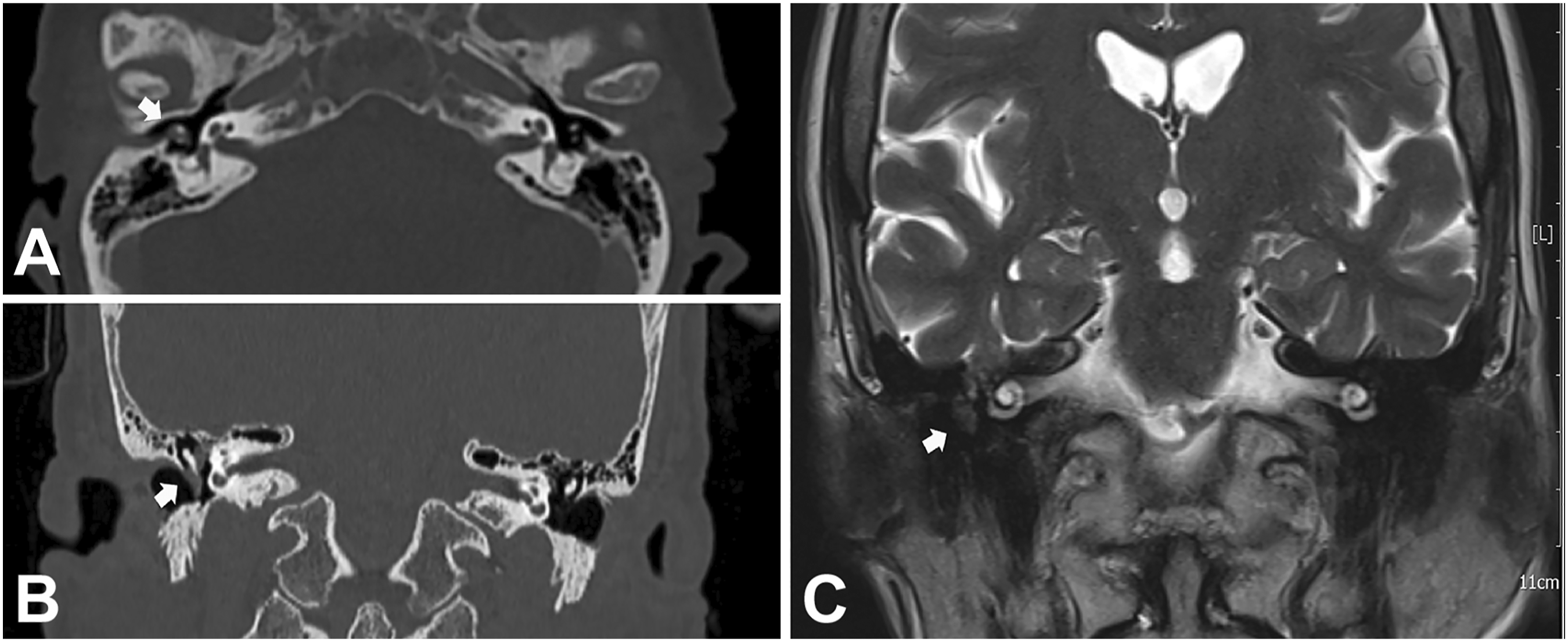
Otolaryngologists might not be familiar with PMTs because development of this tumor in the middle ear/temporal bone is extremely rare.12-18 In our case, as in few others involving this site,16,17 PMT was not associated with TIO. Production of FGF23 by the neoplastic cells is the usual mechanism by which PMTs usually cause TIO.1-3 For this reason, FGF23 needs to be evaluated in the serum when TIO is clinically suspected.2,3,19 Moreover, demonstration of its expression in the tumor tissue, at mRNA (either by RT-PCR, as in our case, or by chromogenic in situ hybridization) or protein (by immunohistochemistry) level, is valuable to differentiate PMTs from the wide spectrum of mesenchymal tumors with which they may be confused. 2 In our case, in addition to the expression of FGF23, we also demonstrated the expression of MEPE and FGF7, two other phosphatonins known to be involved in the pathogenesis of TIO.2,3 To the best of our knowledge, expression of three phosphatonins in a “non-phosphaturic” variant of PMT has never been described. It is likely that the absence of TIO in our patient reflected an early detection of the tumor (i.e., before any manifestation of TIO) as a result of local symptoms (hearing impairment) that led to timely imaging identification and surgical treatment of the tumor itself. However, other mechanisms (e.g., secretion/expression of non-functional forms or low levels of phosphatonins, undefined compensatory mechanisms) cannot be excluded.2,3,8,17
As recurrence (and metastasis as well) of PMTs, including the “non-phosphaturic” variant,16,20 have been reported also years after surgery, long-term follow-up for these patients is mandatory. In our case, follow-up based on clinical evaluation, monitoring of phosphate serum levels and MRI failed to reveal recurrence of the tumor and development of TIO for 2 years.
Footnotes
Authors’ Note
The data sets used and/or analyzed during the current study are available from the corresponding author on reasonable request.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.
Ethics Statement
All the clinico-pathologic investigations detailed in the manuscript have been conducted in accordance with the Declaration of Helsinki and its later amendments or comparable ethical standards.
Informed Consent
Written informed consent for publication of data and images was obtained from the patient.