
Abstract

Main Document
A 39-year-old woman presented with 3-year history of muscular weakness, back pain, and fractures of vertebrae and hip. She had been walking on crutches for several months and was under treatment with phosphate and calcitriol for hypophosphatemia. A sense of nasal obstruction and headache were also referred. Routine analyses revealed hypophosphatemia (1.6 mg/dL, normal range 2.6-4.5), renal phosphate wasting (TMP/GFR 1.07 mg/dL, normal range for age and gender 2.9-4.4), increased total (190 U/L, normal range 39-111) and bone-specific (77.9 U/L, normal range 11-30) alkaline phosphatase, low 1,25-(OH)2-Vitamin D (13.3 pg/ml, normal range 19.9-67) and normal values of serum calcium, and 25-(OH)-Vitamin D and parathyroid hormone. Magnetic resonance imaging and computed tomography revealed a 2-cm tumor involving the right nasal cavity, ethmoid, and anterior skull base (Figure 1). As Tumor Induced Osteomalacia (TIO) was suspected, 111In-Octreotide Scintigraphy was performed but it was negative. The tumor was radically excised by ethmoidectomy together with the endonasal portion.
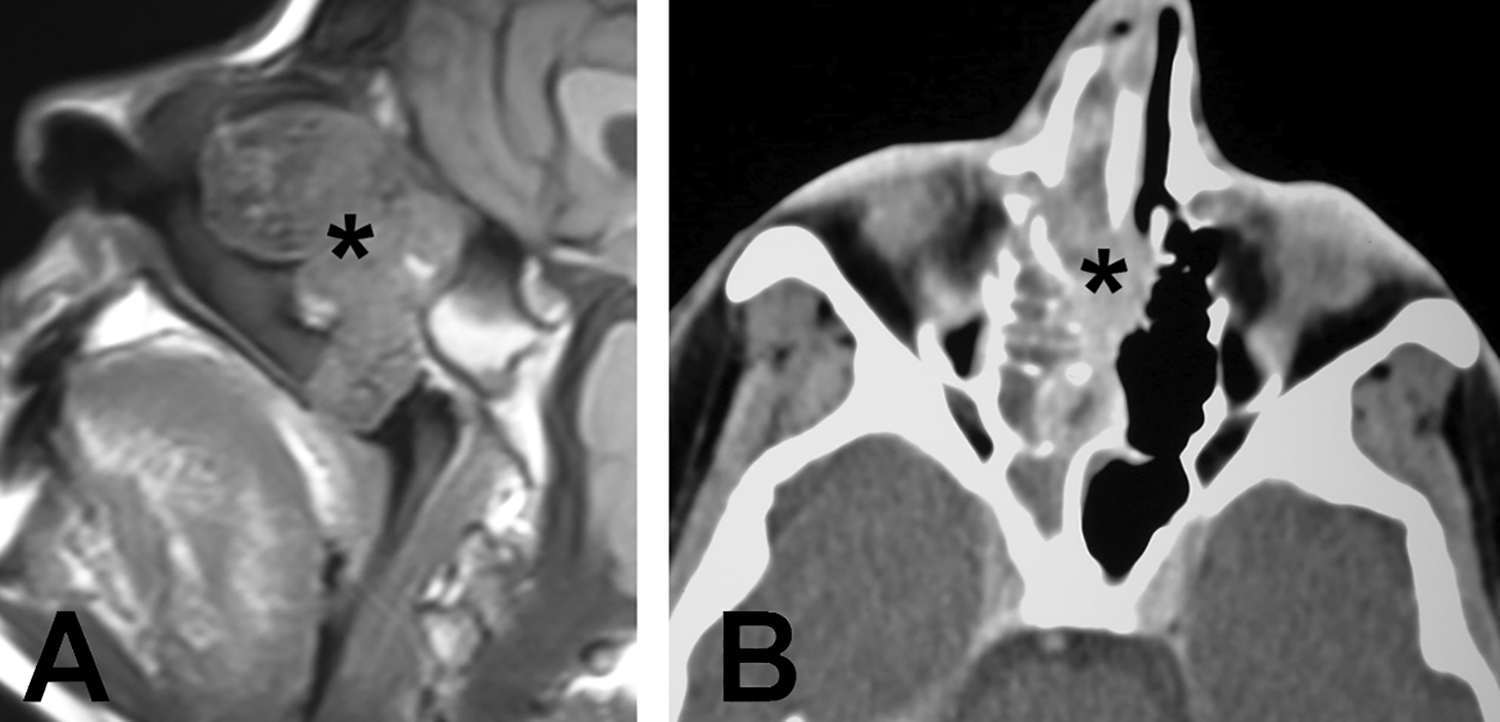
Sagittal magnetic resonance imaging (A) and axial computed tomography (B) demonstrate a tumor (asterisks) involving the right nasal cavity, ethmoid, and anterior skull base.
Histological examination revealed a tumor consisting of bland, round-ovoidal to spindle cells arranged around variable sized blood vessels ranging from small capillaries to cavernous vascular spaces (Figure 2A-D). Hypercellular areas, hemorrhages, siderophages, and clusters of mature adipocytes were also observed. Atypia, mitoses, necrosis, multinucleated giant cells and matrix mineralization were absent. Immunohistochemical analysis, performed as described previously, 1 showed immunoreactivity of the neoplastic cells for Vimentin and CD56 (Figure 2E), but not for pan-cytokeratin, CD31, CD34, STAT6, HMB45, Melan-A, nuclear β-catenin, and smooth muscle actin. The Ki-67 proliferation index was less than 5%. A small portion of fresh tumor tissue was used for reverse transcriptase polymerase chain reaction (RT-PCR). The analysis, performed as described previously,2,3 demonstrated expression of fibroblast growth factor-23 (FGF23) and matrix extracellular phosphoglycoprotein (MEPE; Figure 3). Based on these findings, the diagnosis of phosphaturic mesenchymal tumor (PMT) was made.
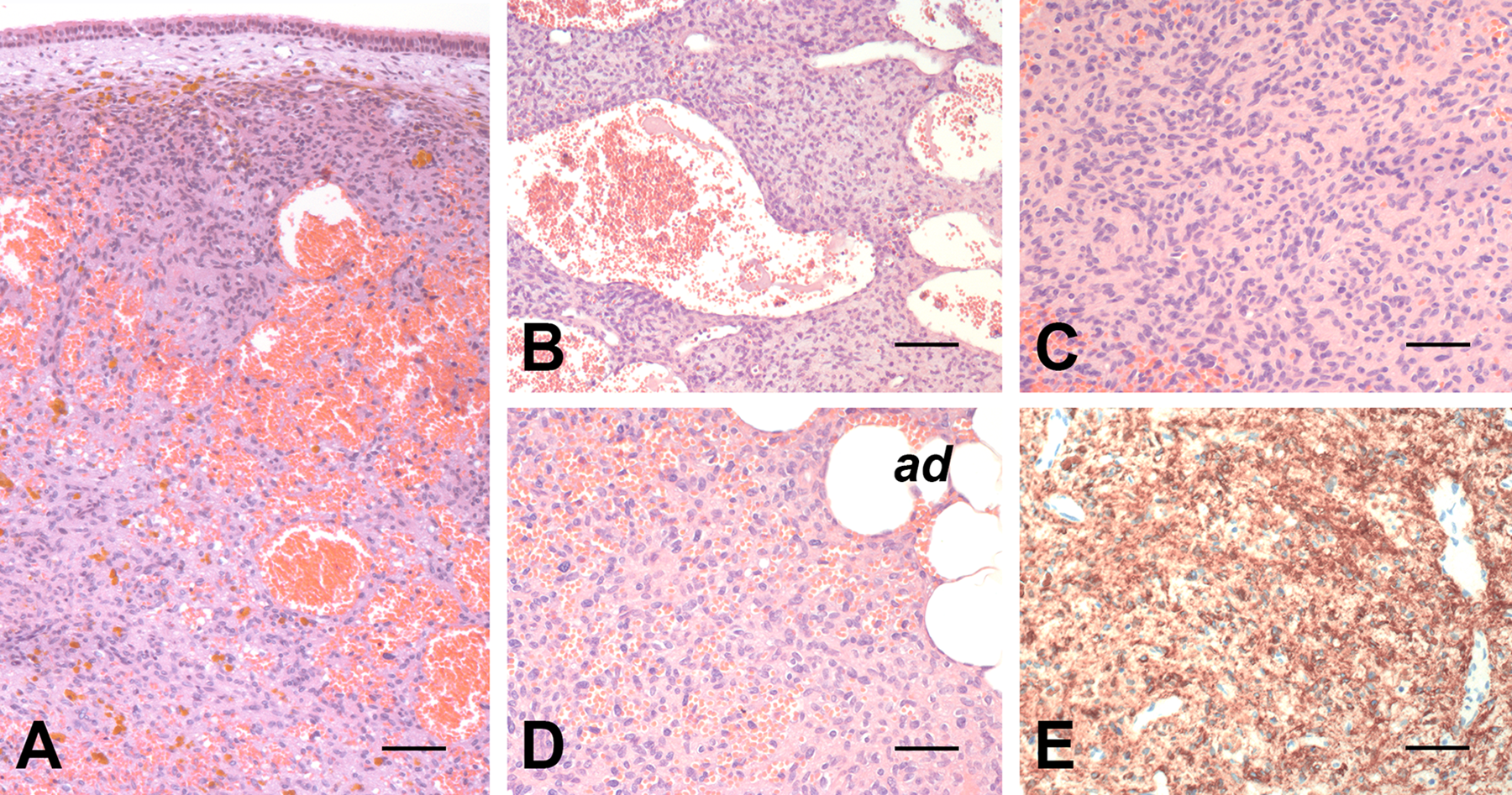
Histology of the tumor. The tumor growths beneath the surface respiratory epithelium (A) and consists of bland, round-ovoidal to spindle cells intercalated among vascular spaces of different size (B). Panels C and D illustrate a more solid area of growth of the neoplastic cells and intra-tumoral adipocytes (ad), respectively. Immunoreactivity of the neoplastic cells for CD56 is illustrated in E. A-D, hematoxylin–eosin. Bars: 150 µm in A, 100 µm in B and E, and 80 µm in C and D.
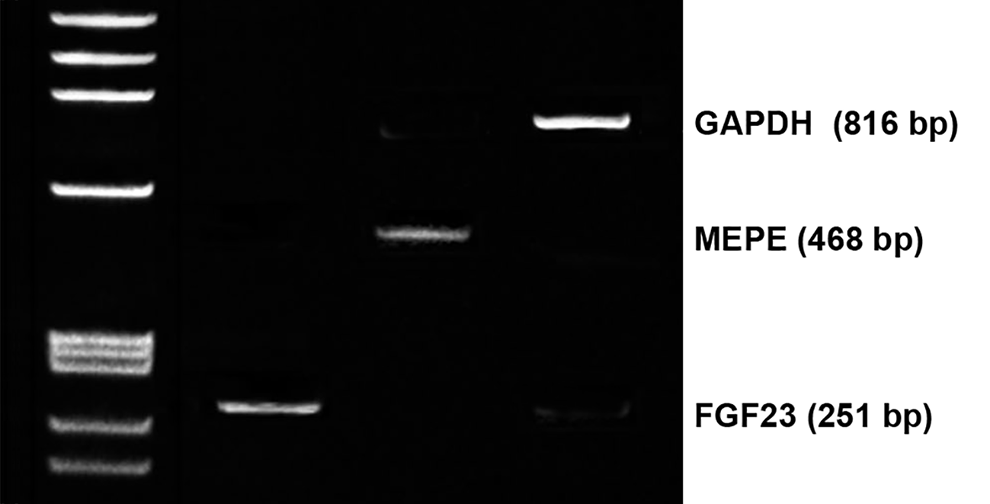
Agarose gel electrophoresis shows the expression of FGF23 and matrix extracellular phosphoglycoprotein (MEPE) in the tumor. Target cDNA sequence of FGF23 and MEPE were amplified as described previously.2,3 The identity of the amplification products was verified by DNA sequencing. GAPDH was used as housekeeping gene.
Two months after surgery, the patient was able to walk without crutches, pain was resolved, and biochemical parameters (serum phosphate, TMP/GFR, and 1,25-(OH)2-Vitamin D) were all normalized.
Phosphaturic mesenchymal tumors are rare, typically benign, tumors commonly arising in soft tissues and bones.4,5 They are the main cause of TIO, a paraneoplastic syndrome caused by tumor cell secretion of hormones (phosphatonins) involved in the regulation of phosphate metabolism and bone mineralization. 1 Patients with TIO present hypophosphatemia, renal phosphate wasting and low serum levels of 1,25-(OH)2-vitamin D, and clinical features related to systemic osteomalacia (bone pain, weakness, gait disturbances, fractures) for which a timely and accurate diagnosis and prompt surgical treatment are mandatory.4,5
Otolaryngologists and maxillofacial surgeons might not be familiar with PMTs because they are uncommon in the head and neck and rarely involve the naso-ethmoidal region.6,7 At this level, PMTs often lack the distinctive (grungy) calcified matrix and multinucleated giant cells that characterize PMTs of soft tissues and bones and commonly present clusters of mature adipocytes and florid hemangiopericytoma-like vasculature associated with fascicles of myoid-appearing cells that may mimic other vascular and perivascular tumor.5,8 To this regard, even though not specific, the immunoreactivity of the neoplastic cells for CD56 may be helpful because its expression in mesenchymal tumors is limited. 9
Production of the “phosphatonin” FGF23 by the neoplastic cells is the mechanism by which PMTs cause TIO.4,5 For this reason, it is commonly used for diagnosis and follow-up of patients with TIO.4-6,10 It can be measured in the serum and demonstrated in the tumor by RT-PCR (as in our case), chromogenic in situ hybridization (CISH), and immunohistochemistry.4-6,10 To date, evaluation of FGF23 expression, in particular by CISH, is the most specific tool for differentiation of PMTs from the wide spectrum of benign and malignant tumors with which they may be confused (for review see Folpe5). In our case, we also demonstrated the expression of MEPE, another “phosphatonin” known to be involved in the pathogenesis of TIO.4,5 The co-expression of two “phosphatonins” suggests their synergistic/additive effect in the development of TIO in our patient.
Identification and complete resection of the tumor is imperative for treatment. Indeed, as in our case, TIO is definitely cured upon its complete removal. However, when small, PMTs may be difficult to locate and remain occult also after particularly sensitive radionuclide scans.4,5 In addition, they can either recur if incompletely resected or be inoperable or, in a minority of cases, show a malignant behavior with development of metastasis that cannot be always predicted by histology. 6 In these cases, medical treatment with phosphate supplements and calcitriol is needed and humanized anti-FGF23 monoclonal antibodies represent a promising procedure.4,5 The recent identification of FN1-FGFR1 or FN1-FGF1 fusions in most of PMTs4,5 will also favor the development of targeted therapy approaches for these cases.
Footnotes
Authors’ Note
The data sets used and/or analyzed during the current study are available from the corresponding author on reasonable request. All the clinicopathologic investigations detailed in the manuscript have been conducted in accordance with the Declaration of Helsinki and its later amendments or comparable ethical standards. Written informed consent for publication of data and images was obtained from the patient.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.