
Abstract
Hyaline fibromatosis syndrome (HFS) is a rare, autosomally-recesfvsive disease characterized by papulonodular skin lesions, soft tissue masses, joint contractures, gingival overgrowth, and osteolytic bone lesions. Mutations in capillary morphogenesis gene 2 are responsible for both these conditions. Generally, an autosomal recessive pattern is assumed to be the most common mode of inheritance. Here, we report an unusual case of a twenty-three-year-old female patient with HFS who reported with a chief complaint of growing nasal mass for three months. There was no history of pain or bleeding associated with the nasal mass. Due to the growing mass, she experienced right nasal obstruction, which compromised her quality of life. There was an unremarkable family history. Her physical examination revealed multiple asymptomatic pinkish-white papulonodular lesions located at multiple sites. Intra orally, she had generalized gingival enlargement. Her nasal examination revealed a right sided nasal mass, bright red in color. The lesion was soft on palpation. All the results of hematological and biochemical tests were normal. However, skeletal radiographic examination showed the joint contractures on her knees and elbows without the presence of osteolytic bone lesions. The nasal lesion was surgically excised and histopathological examination revealed features suggestive of HFS.
Keywords
Introduction
The term hyaline fibromatosis syndrome (HFS) first described by Nofal et al. entails 2 variants of a disfiguring and disabling degenerative disease, the juvenile hyaline fibromatosis (JHF) and the infantile systemic hyalinosis (ISH). 1 These 2 conditions represent different degrees of severity of the same disease. The first case of JHF was diagnosed in 1873 and was named molloscum fibrosum by Murray. 2 It is characterized by widespread accumulation of hyaline amorphous deposits in the skin and other organs of patients leading to an abnormal growth of hyalinized fibrous tissue with mucosal, cutaneous, and osteoarticular involvement and occasional systemic involvement. 3 The onset of clinical manifestations of JHF is normally in the first three to four months of life, while ISH typically manifests in the first weeks to months of life. In both the disorders, mental development is normal. It is more prevalent in the Middle East due to higher rates of consanguineous marriages and till date, less than a hundred cases have been reported worldwide. 4
Hyaline fibromatosis syndrome is caused by mutations in toxin receptor 2 gene (ANTXR2), also known as capillary morphogenesis gene 2 (CMG2) at 4q21 which leads to loss of function of the transmembrane protein anthrax toxin receptor 2. 5 Lesions on the skin may be disfiguring. Two types of skin lesions are generally present. Pink pearly papules and plaques are commonly located on the nasolabial folds, chin, forehead, ears, back of the neck, and perianal region, whereas large subcutaneous tumors are found on the scalp and less frequently on the trunk, extremities, and eyelids. 6 A common intraoral finding is gingival enlargement that may interfere with feeding and may result in poor oral hygiene, infection, and dental caries. A painful flexion contracture results in a severe limitation of mobility, particularly in the large joints. Bone involvement may present in the form of osteoporosis, pathologic fractures, and osteolytic lesions of the long bones. 1
We report one such extremely unusual case. To our knowledge, this is a first reported case in the literature wherein patient with HFS presented with a growing nasal mass. Ethical approval to report this case was obtained from. We also obtained written informed consent from the patient for their anonymized information to be published in this case.
Case Presentation
A 23-year-old female patient, who was a known case of HFS, reported with a chief complaint of growing nasal mass for three months. The mass caused right nasal obstruction, which adversely affected the patient’s quality of life and therefore the patient reported to Otorhinolaryngology, Head and Neck Surgery clinic.
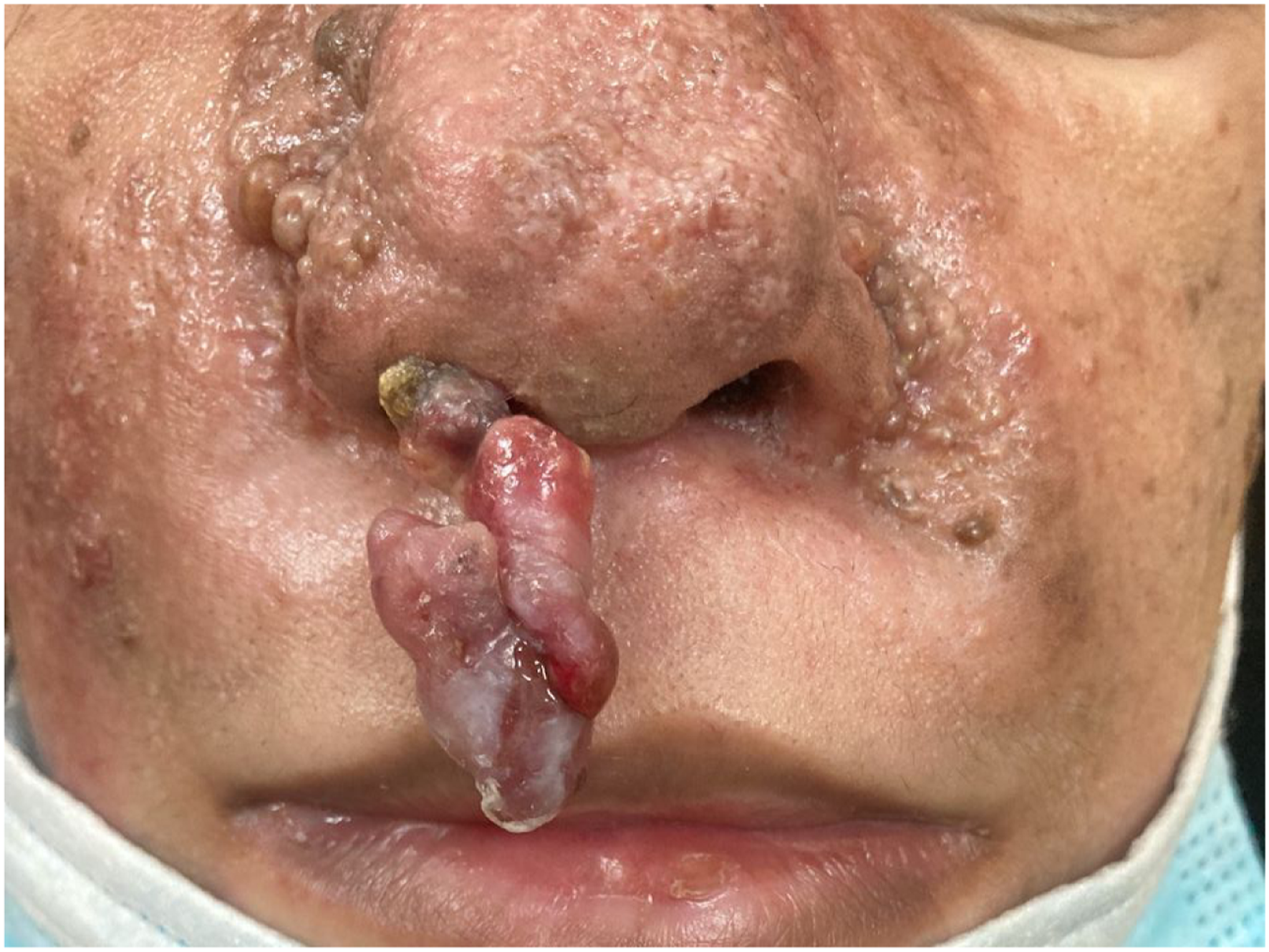
The mass started increasing in size gradually. There was no history of pain, bleeding or discharge from the mass. She denied any past history of nasal trauma, surgery, or illicit drug intake. Family history was negative for HFS. Her nasal examination revealed a right sided nasal mass, bright red in color, arising from the alar rim, around 3x1.4cm in dimension. The lesion was soft on palpation. Figure 1 shows nasal mass protruding from right side of nasal cavity. Microscopic description: Sections show a polyp with its surface lined by keratinized squamous epithelium with foci of ulcerations and underlying granulation tissue formation and marked inflammatory infiltrate consisting mainly of plasma cells. No significant eosinophils seen. The polyp core shows multiple dilated blood vessels with intervening edematous stroma mixed with foci of hyalinization.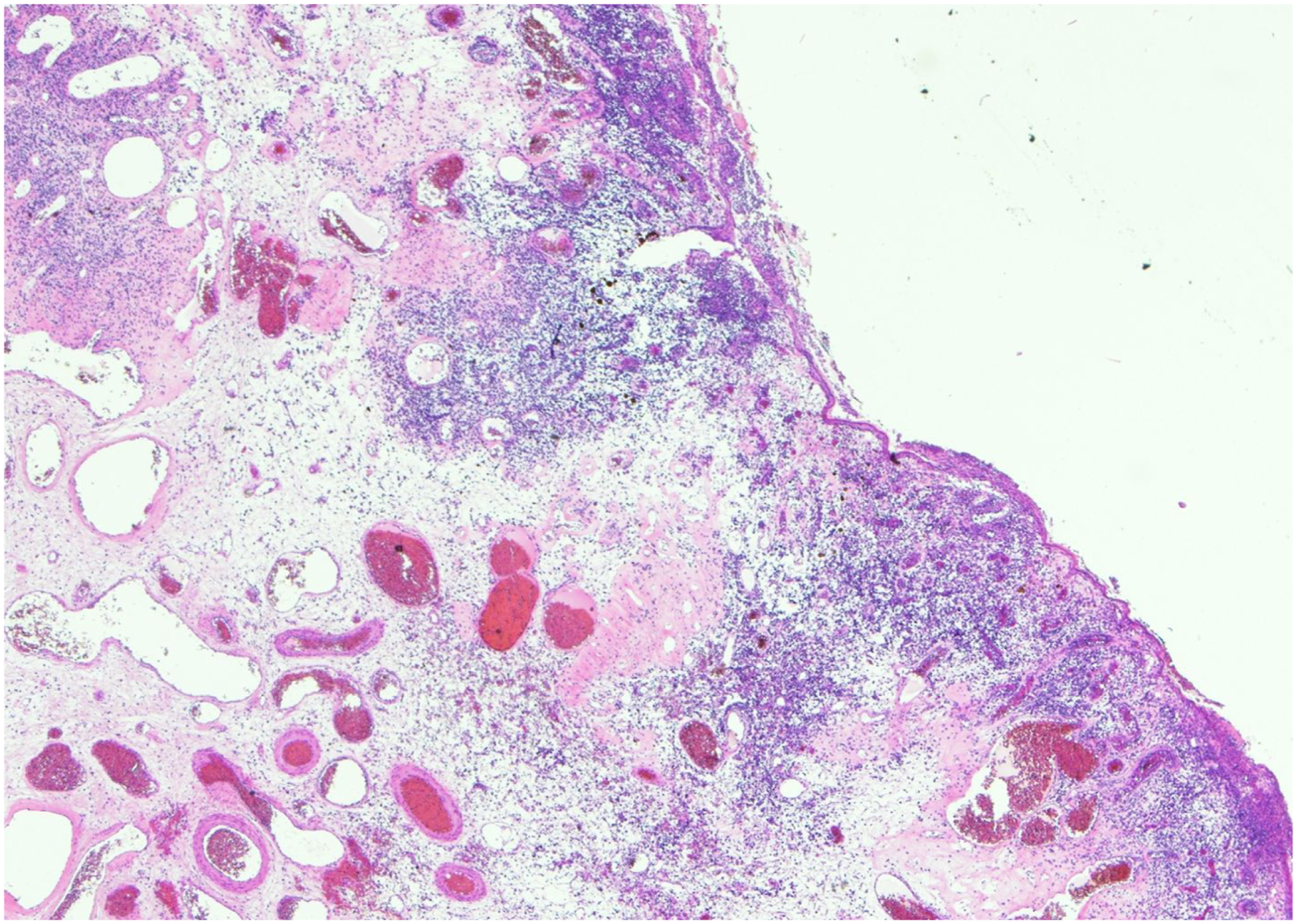
On physical examination, she had multiple asymptomatic skin lesions at different body sites. Pinkish-white papulonodular lesions were present on her digits, back, chin, and gluteal sulcus. Her eyesight and hearing were normal. Intraoral examination revealed generalized gingival enlargement covering more than two-third surface of her teeth.
All the results of hematological and biochemical tests were normal. Skeletal radiographic examination showed the joint contractures on her knees and elbows without the presence of osteolytic bone lesions. Histopathological examination of nasal mass revealed marked vascularity along with ulceration, edematous changes, focal epithelial atrophy, and foci of hyalinization suggestive of JHF. Figure 2 and 3 show sections of nasal polyp with surface lined by keratinized squamous epithelium along with foci of ulceration, granulation tissue formation and marked inflammatory infiltrate consisting mainly of plasma cells. Eosinophils are not predominantly seen. The polyp core shows multiple dilated blood vessels with intervening edematous stroma mixed with foci of hyalinization. Microscopic description: Sections show a polyp with its surface lined by keratinized squamous epithelium with foci of ulcerations and underlying granulation tissue formation and marked inflammatory infiltrate consisting mainly of plasma cells. No significant eosinophils seen. The polyp core shows multiple dilated blood vessels with intervening edematous stroma mixed with foci of hyalinization. image showing protruding nasal mass.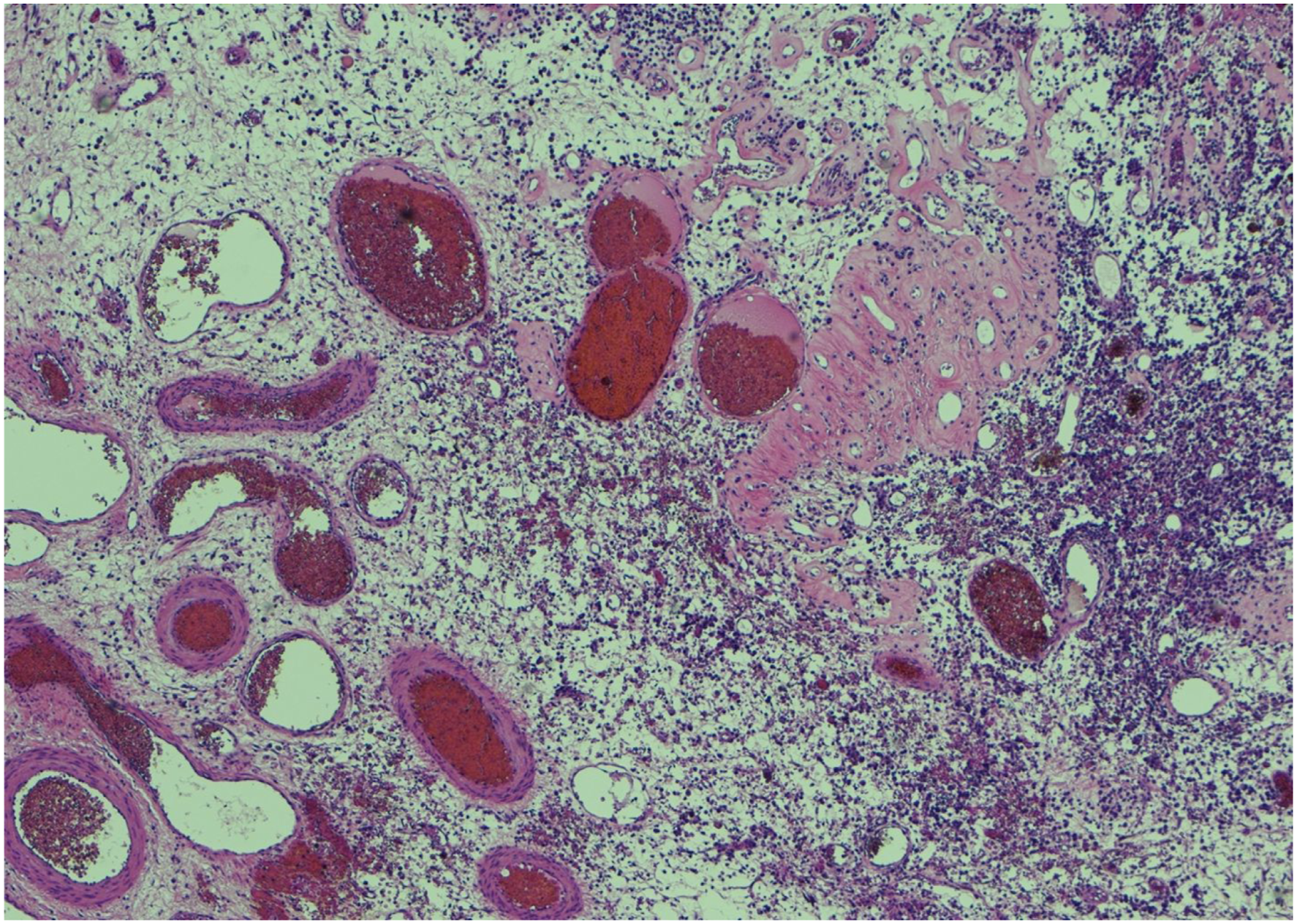
Discussion
Although JHF's pathogenesis is unknown, several mechanisms have been proposed to explain its abnormal production which include accumulation of glycosaminoglycans and glycoproteins, along with impaired synthesis of procollagen or tropocollagen. 7 Deuquet et al. classified the ANTXR2 mutations into 4 major classes: class I missense mutations in the vWA domain; class II other missense mutations in exons 1–11; class III mutations leading to premature stop codon (frameshift and splicing mutations); and class IV missense mutations in the cytosolic tail. 8 Class III mutations are the most frequently reported, followed by class I mutations. 3
JHF follows an autosomally recessive mode of inheritance but sporadic cases occur as well. 9 In our case, neither parents nor the siblings of the patient had JHF.The disease is defined by a constellation of clinical findings including pearly skin papules or subcutaneous firm nodules, joint contractures, acral osteolytic lesions, gingival hypertrophy, and normal intelligence. 9 A tumoral infiltration of the joint capsule has been proposed as the major cause of contractures. 10
Other findings reported in the literature are muscle weakness, scoliosis, generalized osteoporosis, and reduced height and weight.9,10 It is rare to have cognitive impairment, which is in line with the fact that ANTXR2 protein is minimally expressed or not at all in the brain. 11 Gingival enlargement is a significant finding and can interfere with feeding in severe cases. 12 In our case, skin lesions were present on digits, back, chin, and gluteal sulcus of the patient. The joint contractures were localized to knees and elbows. Generalized gingival enlargement was also present in our patient. However, osteolytic lesions and subcutaneous nodules were not present in our case.
So far, no treatment has been discovered that can cure or stop the progression of HFS. Certain therapeutic strategies have been used, including penicillamine, methotrexate, and steroids (both systemic and intra-lesional), with limited rate of success. 13 Capsulotomy of contractures has shown some temporary benefits. Surgical excision is the preferred method for the treatment of subcutaneous nodules. 14 Gingival enlargement is treated by carrying out gingivectomy. 14 Oral hygiene maintenance is important to prevent recurrence. Supportive care is the cornerstone of treatment. Maintaining range of motion and preventing contractures require regular physiotherapy. Pain is often treated with nonsteroidal anti-inflammatory drugs and opiates, though gabapentin has also been advised in some cases. 15 In our case, the nasal mass was treated with surgical excision. Patient showed improvement of right nasal obstruction in subsequent follow up.
To our knowledge this is the first reported case of patient with HFS and nasal mass along with histopathological features of the disease. HFS usually has a fatal course. Intractable diarrhea, recurrent infections, and organ failure are the main causes of death. 16 The differential diagnosis of HFS includes Winchester syndrome, congenital generalized fibromatosis, hereditary gingival fibromatosis, and nodular amyloidosis. Winchester syndrome is characterized by generalized osteoporosis, contractures of small joints, short stature, and corneal opacities. 9 In our case, patient did not have any ocular manifestation. Congenital generalized fibromatosis is characterized by multiple subcutaneous and dermal nodules, and visceral involvement. 12 In our case there was no visceral involvement. Hereditary gingival fibromatosis has only gingival manifestations. 14 In our case, there were extraoral manifestations also.
To summarize, HFS is an autosomal recessive disease caused by mutations in ANTXR2 leading to dysfunction of transmembrane protein anthrax toxin receptor 2. The most common mode of presentation in newborns is extreme pain when being handled minimally.
Conclusion
The prognosis of HFS is poor, and most treatment approaches have not proven to be beneficial. The disfiguring, disabling, and immobilizing influences of HFS have an adverse effect on patients’ lives. Considering the multisystemic nature of the disease, a multidisciplinary team approach is recommended.
Footnotes
Authors Contributions
Alkholaiwi, Feras: Study design and conception, writing the draft, and approving the final manuscript. Abdulmalik Alsheikh: Histopathological diagnosis and approving the final manuscript.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.