
Abstract
A 12-year-old female with a history of multicentric infantile myofibromatosis (IM) presented with a tender, enlarging cheek mass and trismus. Imaging identified an intramasseteric tumor. Given the unknown etiology of the tumor and her bothersome symptoms, the mass was excised using a transoral approach with concurrent facial nerve monitoring. Her pathology report confirmed the diagnosis of a myofibromatosis lesion embedded within the masseter muscle. While IM can often present with lesions in the head and neck region, the intramasseteric location is rare and presents unique considerations for surgical approach. Myofibromatosis lesions typically occur before two years of age, although there are some rare documented cases of multicentric myofibromatosis lesions presenting at older ages. Furthermore, this patient’s family history of similar lesions suggests a familial variant, which may have implications for disease behavior and need for further work-up, monitoring, and management. Overall, this was an unusual presentation of IM given the patient's age, prevalent family history, and the location of the mass. This case report adds to the literature and discusses the clinical differential of a pediatric cheek mass, the surgical considerations for an intramasseteric tumor, and the natural history of infantile myofibromatosis.
Introduction
The differential diagnosis for a growing cheek mass in a pediatric patient is broad and includes neoplastic, congenital, inflammatory, and developmental entities. Lesions of the parotid salivary gland represent the most frequently encountered cheek masses, however, masses of the masseter should be included on the differential. A variety of pediatric masseteric masses have been described, including intramuscular hemangioma, schwannoma, granular cell tumor, solitary fibrous tumor, myxoma, rhabdomyosarcoma, odontogenic keratocyst, nodular fasciitis, and benign muscle hypertrophy. 1 Imaging findings and histopathology can help confirm the diagnosis. The treatment for these lesions can vary depending on location and extent of involvement and can range from observation alone to surgery with adjuvant chemotherapy. We discuss the case of a pediatric patient who presented with an enlarging cheek mass who was found to have an infantile myofibromatosis lesion within her masseter muscle.
Case Report
A 12-year-old female patient presented to the pediatric otolaryngology clinic with a palpable mass that had been rapidly growing on her left face over the past three months. The mass was associated with intermittent pain and moderate trismus. Examination revealed a firm, fixed 2 cm mass on the left cheek near her jawline with no associated left facial weakness or palpable lymphadenopathy. Of note, as an infant she had a history of a forehead mass that was found to be an infantile myofibromatosis tumor. Furthermore, her family history was significant for 2 brothers and her mother with myofibromatosis. One brother had a facial mass requiring surgical excision, and the other brother passed away from complications of visceral myofibromatosis disease. To further complicate her presentation, the patient had a remote history of acute idiopathic facial nerve weakness on her right side (contra-lateral to the palpable mass) which returned to near-normal function (grade 2 House-Brackmann scale) over time.
The location of the patient’s mass initially raised suspicion for a parotid tumor, and—given the history of rapid growth, trismus, and discomfort—prompt additional work-up was recommended. MRI of the head and neck with contrast demonstrated an isolated expansile mass (approximately 2 × 1.5 × 1.3 cm) within the left masseter muscle with T1 signal isointense to the adjacent muscle, T2 hyperintensity, and postcontrast peripheral enhancement. There was no adjacent fat stranding, extension outside of the masseter muscle, or lymphadenopathy (Figure 1).
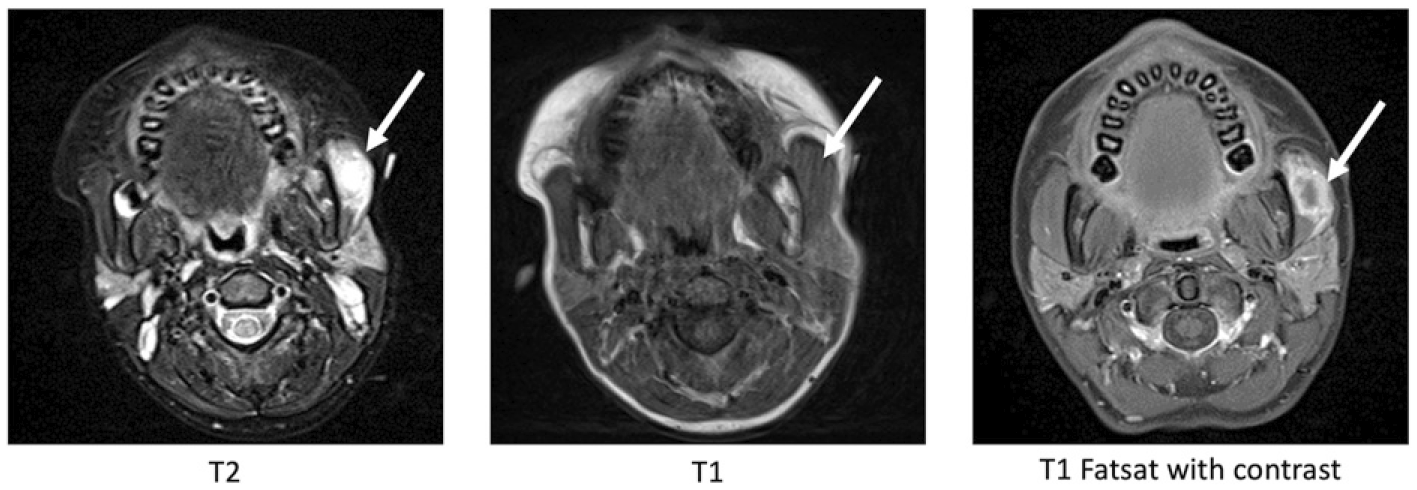
MRI of the head and neck with and without contrast.
Imaging appeared consistent with a myofibromatosis lesion and confirmed the location in the masseter. Surgical excision of the mass was recommended given rapid growth, associated symptoms, and the unknown definitive nature of the mass. FNA was deferred due to the deep location of the mass, patient age, and plan for complete excisional biopsy regardless of FNA results. The location of this mass prompted consideration of different surgical approaches for excision, including parotidectomy approach versus transoral approach, with decision-making based on mass size, cosmesis, oncologic safety, and risk to the facial nerve (Figure 2).
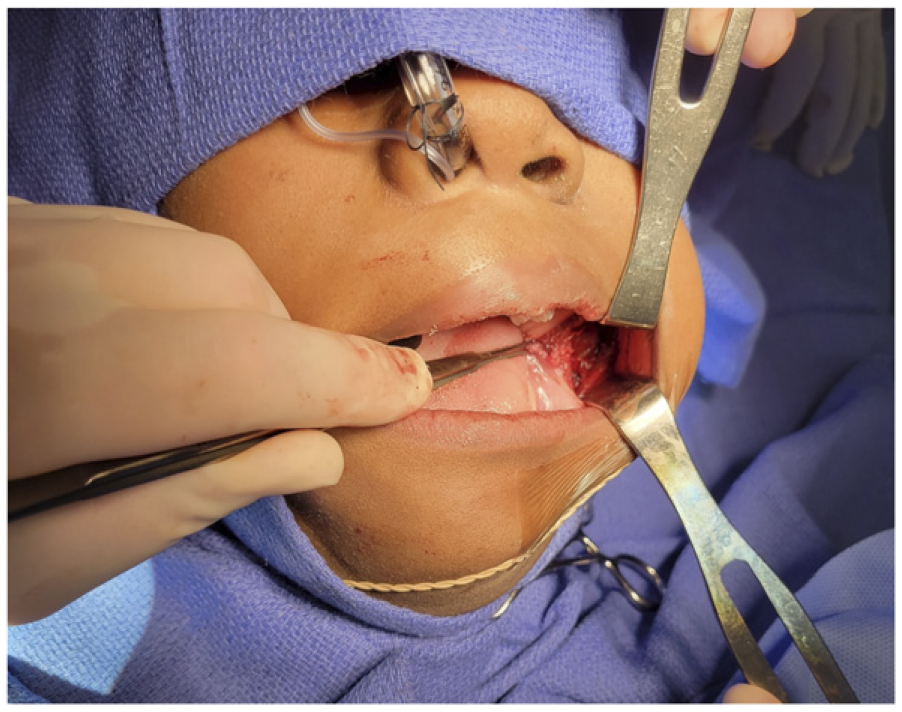
Excision of left masseter lesion using a transoral approach.
Our patient was prepped for both open parotidectomy and the transoral approach, however, with the patient under anesthesia, a more in-depth bimanual examination revealed the feasibility of the transoral approach which was thus undertaken. This involved a mucosal incision inferior to the course of the parotid duct. Dissection was continued through the buccinator to the masseter muscle. The mass was identified buried within the masseter muscle and was excised using a combination of blunt and sharp dissection. The soft tissue defect was not dramatic, and thus a fat graft was deferred. A gross total resection was achieved, and the intraoral incision was closed primarily (Figure 3).
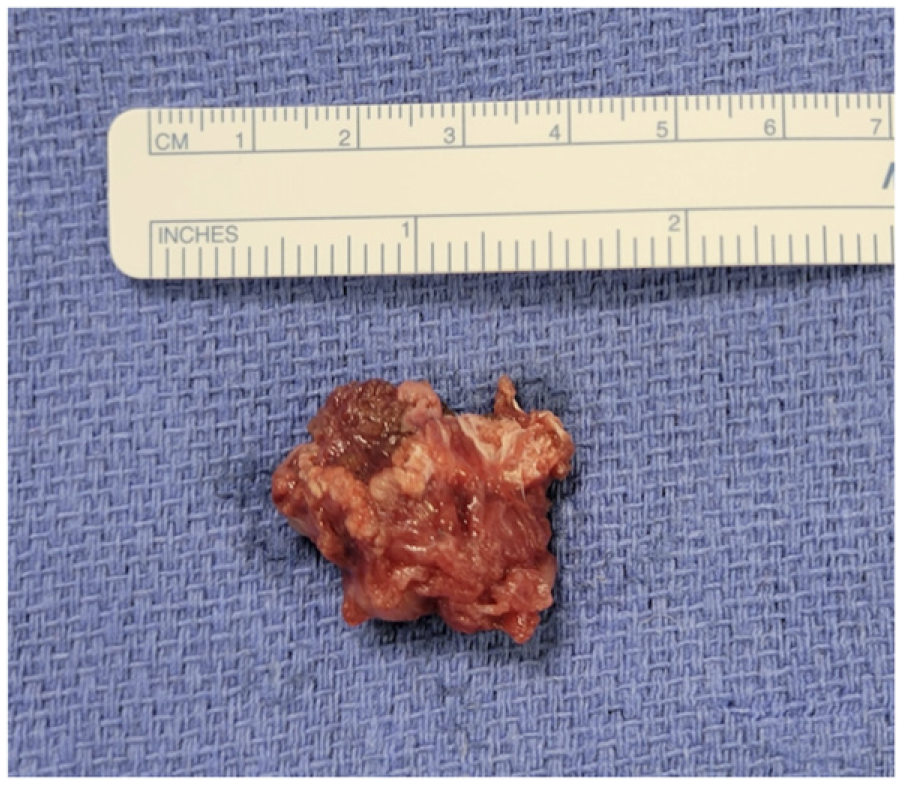
Gross image of intramasseteric lesion following removal.
Pathology revealed infantile myofibromatosis infiltrating into surrounding skeletal muscle and soft tissue. Histopathologic findings of IM classically include spindle shaped cells involving the dermis, subcutaneous, or soft tissues. 1 Following surgery, our patient recovered well and had improved trismus and swelling. Pediatric hematology was consulted for further management of her IM.
Discussion
There is a broad differential for masses in the cheek region which can involve salivary glands, neural, lymphatic, bony, and vascular structures. 1 Lesions of the parotid gland, such as pleomorphic adenoma or mucoepidermoid carcinoma, would be near the top of the differential list. Other considerations included myofibromatosis and other tumors such as a hemangioma or rhabdomyosarcoma. Developmental or inflammatory etiologies were on the differential for this patient, albeit less likely. One retrospective study found that the most common pediatric cheek masses were lymphatic malformations, followed by venous and arterial malformations.(2) Various case reports also describe hemangioma presenting as a check mass. Although intramuscular location of hemangioma is overall uncommon, when located in the head and neck region the masseter is the most common site. Other common etiologies presenting as pediatric cheek masses include lipomas and neurofibromas. Despite the wide variety of etiologies, the most common presentation for all pediatric cheek lesions is an asymptomatic enlarging cheek mass, making physical exam and history alone insufficient for diagnosis.
Imaging was essential for discerning between possible etiologies. Differential diagnostic considerations after MRI included conditions such as rhabdomyosarcoma, lymphoma, sarcoid, abscess, and infantile myofibromatosis. This case demonstrated typical MRI signal characteristics of myofibromatosis in which the mass is isointense to muscle on precontrast T1 weighted imaging. On T2 or STIR imaging infantile myofibromatosis lesions are typically T2 hyperintense, although some can also demonstrate central T2 hypointensity. Peripheral postcontrast enhancement is typical for this lesion and is perhaps the most distinctive imaging characteristic as many masses are T1 isointense and T2 hyperintense to muscle. The lack of inflammatory fat stranding and clinical exam findings suggested against an abscess or other inflammatory etiology.
After further characterizing the mass through imaging, excisional biopsy was planned. Several surgical approaches have been published regarding pediatric cheek masses including direct incision over the mass, standard parotidectomy approach, and transoral approaches. The most common of these techniques is the standard parotidectomy. 1 A parotidectomy approach for mid-cheek masses has been shown to be effective oncologically and safe for facial nerve preservation. However, potential drawbacks include negative cosmetic effects, such as a facial scar and potential for asymmetric facial contours. Although infrequent, facial nerve injury and Frey syndrome are also potential morbid consequences that must be considered with this approach.(2–4)
Transoral approaches for removal of accessory parotid gland tumors have been shown to be successful with potential for less disruption to facial nerve branches. 5 Additionally, a transoral approach has less cosmetic morbidity with no visible scars or depressions, and also avoids the risk of Frey syndrome. The biggest risks still include facial nerve damage and potentially less exposure leading to decreased oncologic safety and inadvertent capsular dissection. 5 In addition, transoral approach may have increased risk of injury to the parotid duct. When suspicion for malignancy is high, the transoral approach can be combined with intraoperative frozen samples to optimize both cosmesis and safety.1,5 This type of approach has been described as a reasonable approach to complete gross dissection of intramassteric fibrous tumors. 6 Removal of a tumor from the cheek area, whether achieved through the parotidectomy or transoral approach, has the potential to leave a soft tissue defect or concavity, thus the use of autologous fat graft should be considered. 7
This case report describes a rare presentation of infantile myofibromatosis (IM) in the masseter muscle of a pediatric patient. IM is a benign, fibrous tumor that can be found in skin, muscles, bones, and sometimes visceral organs. The most common location for the tumor is the head and neck region, although documentation of intramassteric IM lesions is rare.8,9 Most cases occur within the first 2 years of life, although families with associated mutations may have recurrent myofibromatosis at later ages. 10
Infantile myofibromatosis has a reported incidence of 1:150,000 live births, but it should be kept in mind that there are minor forms that often go unnoticed, so the incidence is likely higher. 11 IM is most commonly sporadic although there are familial cases which are typically inherited in an autosomal dominant fashion.11,12 Within families the phenotype may vary from asymptomatic carriers to lethal generalized IM. 13 In familial cases, the most common identified mutation is within the platelet-derived growth factor beta (PDGFRB) gene, however, Wu et al have also identified a mutation in NOTCH3 that results in upregulation of PDGFRB expression in fibroblasts.11,14 Antonescu et al have also identified a mutation within somatic serum-response factor (SEF) genes that result in IM. 15 Our patient has a significant family history suggesting a familial case of infantile myofibromatosis; however, she has not yet had comprehensive genetic testing.
Treatment of these IM lesions vary by location, extent of involvement, and corresponding symptoms. Initial treatment of infantile myofibromatosis is close observation as most lesions regress spontaneously by about 2 years of age, likely as a result of massive apoptosis. 16 Solitary lesions that have not regressed can be surgically excised, as seen in our patient. 13 Systemic therapy is recommended for multicentric visceral disease due to the significant mortality associated (73–93%) with cardiopulmonary or gastrointestinal involvement.13,17 Possible chemotherapeutic regimens include vincristine/daptomycin and vinblastine/methotrexate.14,18 Imatinib is also effective for known PDGFRB mutations. 19
While there are currently no widely used guidelines for screening for visceral disease in patients with infantile myofibromatosis some researchers suggest beginning screening in any patient with multiple lesions or in those that have a proven PDGFRB variant or first-degree relative with IM.13,20 Initial screening should include genetic counseling, comprehensive physical exam, an abdominal ultrasound, and a cardiac ultrasound. Furthermore, Hettmer and colleagues recommend continuing abdominal ultrasounds every 3 months until age 2, then every 1–2 years until the age of 12. 13 Any suspicious exam findings including new onset heart murmur, new/rapidly enlarging lesion, bowel obstruction, or respiratory difficulties should prompt further imaging with a whole-body MRI and repeat cardiac ultrasound even in the setting of a previously negative evaluation.13,20
In conclusion, we report a case of multicentric infantile myofibromatosis in the masseter muscle of a 12-year-old female. Although rare, it is important to consider myofibromatosis on the differential when evaluating pediatric patients with unidentified enlarging tumors in the head and neck region. Parotid tumors are the more likely location for a cheek mass of unknown etiology; however, an intramasseteric tumor should be considered. Although myofibromatosis lesions are benign tumors, surgical excision can be valuable in treating symptomatic growth, confirming the underlying etiology, and excluding more sinister oncologic etiologies. Depending on adequate intraoperative exposure, a transoral technique is a good option to treat these tumors as it improves cosmetic outcomes and potentially decreases likelihood of nerve injury. Following surgical excision, patients should be referred to hematology-oncology for further genetic work-up and possible adjuvant therapy.
Footnotes
Declarations of Conflicting Interests
The authors have no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors received no grant or other financial support for the research, authorship, and/or publication of this article.
Ethical Approval
Our institution does not require ethical approval for reporting individual cases or case series.
Human and Animal Rights
This study does not contain any studies with human or animal subjects.
Informed Consent
Written informed consent was obtained from the patient's legally authorized representative (mother) for anonymized patient information to be published in this article. In addition, written assent was obtained from the pediatric patient.