
Abstract
Objectives:
To describe the management of a 5-year old female with a painless, mobile cheek mass.
Methods:
A retrospective chart review of presentation, imaging, pathology and management.
Results:
Magnetic resonance imaging showed a heterogenous mass with solid and lipomatous components. The mass was a lipoblastoma on histopathology and was excised completely with no evidence of recurrence.
Conclusions:
The diagnosis and management of a cheek mass in a child is challenging. Imaging is important but not diagnostic. Surgical excision is the primary management of a lipoblastoma.
Keywords
A 5-year-old female presented with a slowly enlarging left cheek mass. The mass was firm, mobile, and painless. There were no systemic symptoms such as fevers, malaise, weight loss, or night sweats. The child was born full-term with a past medical history of transient erythroblastopenia of childhood that resolved. She had no previous hospitalizations or surgeries. The remainder of the head and neck examination was normal.
A computed tomography (CT) scan with IV contrast demonstrated a soft tissue mass in the left masticator space extending posteriorly into the retromaxillary fat pad and superiorly into the pterygopalatine fossa. As part of the mass contained fat density, the initial diagnosis was of a lipoma with an atypical solid component. Magnetic resonance imaging (MRI) showed a heterogenous solid T2 hyperintense mass that measured 2.6 cm × 1.8 cm. The non-lipomatous component abutted the alveolar ridge and had minimal heterogeneous increased T1 signal as well as increased T2 signal on fat suppressed images. Post gadolinium T1-weighted images with fat suppression demonstrated peripheral enhancement (Figure 1). The lipomatous component extended superiorly and medially into the pterygopalatine and infratemporal fossa. The mass did not have the classic appearance of a dermoid due to the absence of intrinsic diffusion restriction.
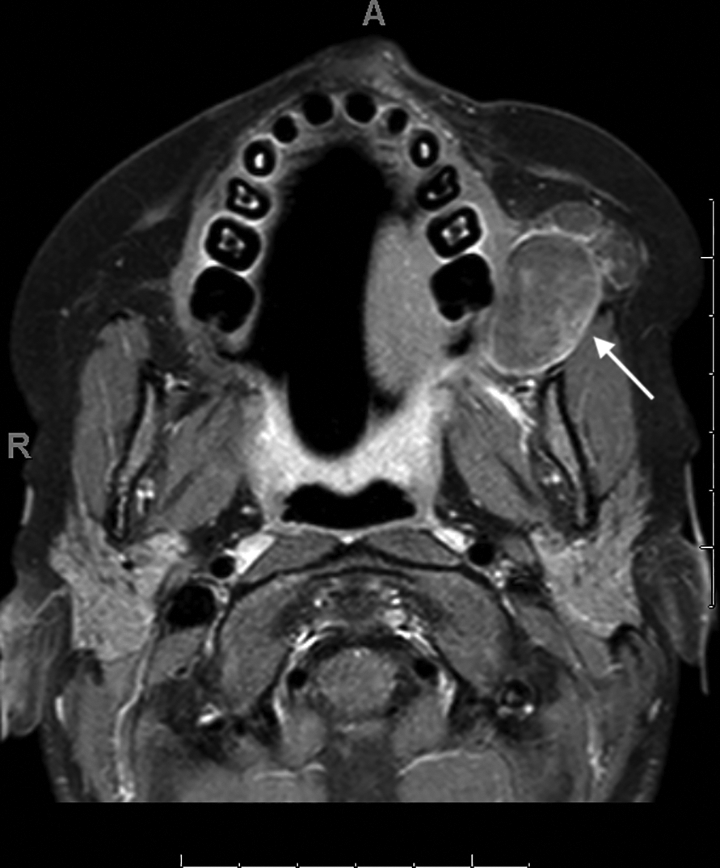
Axial post gadolinium T1-weighted image with fat saturation shows a masticator space mass with peripheral enhancement. There is incomplete suppression of fat signal in the medial component (arrow).
The child underwent excision of the mass through a transverse incision in the buccal mucosa just anterior to the opening of Stensen duct. The buccinator muscle was divided to expose the mass. It was nondescript and consisted of several enlarged lobules of fatty tissue that felt firmer than the surrounding buccal fat. Blunt finger dissection was used to remove the portion extending behind the maxillary sinus. Care was taken to avoid removal of native buccal fat to avoid a sunken cheek deformity. The incision was closed primarily. The child had no postoperative complications and was discharged the following day. At 3-month follow-up, the patient did not have trismus and had no evidence of recurrence.
Histopathologic examination demonstrated a lipoblastoma with complete excision (Figure 2). The solid component was composed of irregular lobules of myxoid tissue and adipocytes in various stages of maturity, ranging from lipoblasts to mature adipocytes. A delicate branching vasculature was noted throughout the mass. The fatty component contained some lobules of mostly mature adipocytes with few lipoblasts, while other lobules had extravasated mucin. No increase in cellularity or nuclear atypia was observed in the solid and fatty components. Fluorescence in situ hybridization for the DDIT3 C/EBP homologous protein (CHOP) rearrangement, a common marker for myxoid liposarcoma, was negative. 1 The karyotyping results for the solid component revealed a cell population with a loss of chromosome 8, an extra chromosome 6, and additional genetic material on chromosomes 4, 6, 16, and 25. The karyotype of the fatty component was normal.
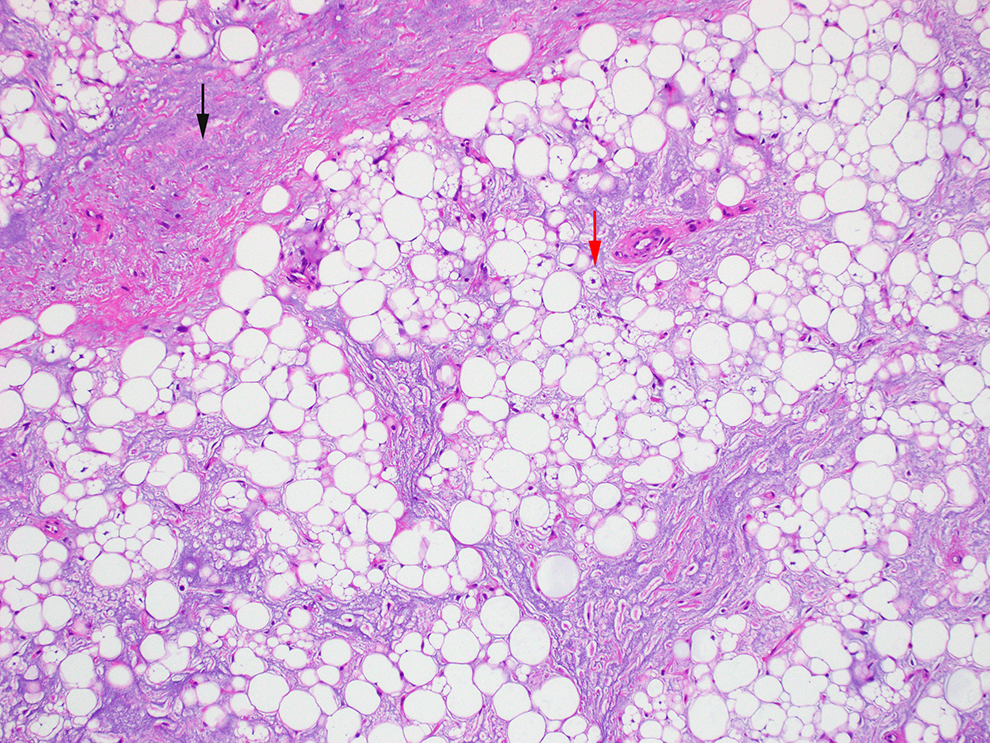
Fibromyxoid stroma and adipocytes: Histology showed lipoblasts (red arrow) and adipocytes in various stages of maturation in the background of fibromyxoid stroma (black arrow), without nuclear atypia (Hematoxylin and eosin (H&E), 100× magnification).
Discussion
Lipoblastoma is a rare, benign adipocytic tumor that presents in children under 10 years old and has a preponderance for boys. 2 These tumors are typically reported in the limbs, trunk, and retroperitoneum. Lipoblastoma is rarely diagnosed in the head and neck, with the neck being the most common site in this region. 3 These are rapidly growing tumors that can lead to location-dependent problems such as nerve compression or airway obstruction. 4 Definitive diagnosis involves histopathology. Lipoblastoma has been associated with the chromosomal breakpoint 8q11-q13 that mutates the PLAG1 gene. 5
The differential diagnosis for a cheek mass in a child includes adipocytic tumors, vascular malformations (lymphatic, venous, arteriovenous, infantile hemangioma), neurofibroma, and teratomas. 6 Lymphatic malformations, also known as lymphangiomas, frequently occur in the head and neck. 7 On MRI, lymphatic malformations are usually T1 hypointense and T2 hyperintense cysts, and fluid levels sometimes present due to hemorrhage.4,7 Due to the location and similar appearance, a lymphatic malformation can be mistaken for a lipoblastoma.4,6
Among adipocytic tumors, the differential diagnosis includes lipoma, myxoid liposarcoma, and lipoblastoma. Lipoma is a benign tumor composed entirely of fat. Lipomas typically show homogenous, hyperintense T1 and T2 signal with complete suppression of signal using fat suppression techniques and no enhancement on post gadolinium sequences. 7 It is important to distinguish myxoid liposarcomas and lipoblastomas as the management is different. Myxoid liposarcomas are sensitive to radiotherapy and chemotherapy unlike lipoblastomas where the management is surgical excision. Myxoid liposarcomas rarely occur in patients younger than 20 years old. 2 They are diagnosed with histopathology and identification of the distinct t(12;16) translocation, which results in the FUS-DDT3 (CHOP) gene fusion.1,2
A variety of scans may be part of the preoperative surgical planning for a cheek mass in a child including CT, MRI, MR angiography, and venography. Differentiating among lipomatous tumors with imaging alone is difficult. Lipoblastomas are well-defined, lobular, soft tissue masses that are heterogenous due to variable ratios of fat to myxogenous stroma. 7 As seen with this child, MRI T2-weighted images are hyperintense. 7 T1-weighted imaging can be either hypointense if the tumor contains more immature lipoblasts or hyperintense if the lesion contains a greater proportion of mature adipocytes.4,8 Magnetic resonance angiography and venography is helpful in excluding vascular anomalies.
Treatment of lipoblastoma is complete surgical excision without adjuvant therapies. Since this is a benign tumor, subtotal resection can be considered to avoid damaging important structures such as Stensen duct or branches of the facial nerve. For incomplete resections, a staged approach is recommended with subsequent surgeries utilizing a different incision. 9 The recurrence rate for lipoblastomas of the head and neck is 27%, while the recurrence rate in other locations is lower and ranges from 14% to 24%. 4 For recurrent lipoblastoma, re-excision is recommended. Adjuvant chemotherapy and radiotherapy have a limited role but are options for recurrent tumors in the cervical and retroperitoneal regions.10,11 There are reports of spontaneous resolution of lipoblastomas and maturation into lipomas, but progressive rapid growth requires intervention and precludes watchful waiting.12,13 Metastasis has not been reported. 13
Lipoblastomas are rare adipocytic tumors of the head and neck that should be considered in rapidly growing masses in children less than 10 years old. Imaging is important for surgical planning, but the definitive diagnosis is made with histopathology. Non-mutilating, complete surgical excision is the treatment of choice.
Footnotes
Declaration of Conflicting Interests
The author declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author received no financial support for the research, authorship, and/or publication of this article.