
Abstract
Isolated congenital anosmia (ICA) is a rare entity worldwide with poorly understood genetic variation. The diagnosis of ICA is made by exclusion of acquired causes of anosmia. Additionally, magnetic resonance imaging in ICA is essential for diagnosis, as it shows reduced or absent development of olfactory bulbs and shallow olfactory sulci. Here, we present the case of a 21-year-old man who presented to our clinic with complete anosmia since birth. The patient's history was negative for acquired causes of anosmia, and the physical examinations of the ears, nose, throat, head, and neck were all not remarkable. Smell testing revealed complete anosmia. The CT imaging was unremarkable; however, magnetic resonance imaging of the anterior brain and olfactory region showed bilaterally absent olfactory bulbs and olfactory tracts, with a shallow olfactory groove. The patient was then subjected to whole exome sequencing. Bioinformatics analysis was performed on the 37 genes associated with olfactory dysfunction, in which a missense variant was identified in the HS6ST1(NM_004807.3) gene was identified, which insilico tools predicted to be likely pathogenic. The results of this patient’s genetic analysis add to the possible genetic culprits reported in ICA cases. Additional genetic analyses are required to validate mutations and understand the heterogeneity of disease representation.
Introduction and Background
Anosmia is defined as the absence of smell due to complete loss of olfactory function. It is considered frequent as it affects 5% of the general population. 1 However, congenital anosmia in which an individual does not recall smelling since birth is rare, accounting for 1% of anosmic cases. 2 Reduction in the sense of smell has a great impact on human function and can be dangerous at times. Olfaction plays a role in detecting environmental or microbial hazards, tasting food, and improving social interactions. 1 Causes of anosmia include inflammatory, infectious, traumatic, congenital, autoimmune, neurodegenerative, and toxin-related.1,3 Congenital anosmia is more commonly syndromic, as seen in Kallmann syndrome (KS), the most studied cause of syndromic congenital anosmia. 4 Other syndromes in which anosmia is reported as a symptom include cystic fibrosis, CHARGE syndrome, 22q11 deletion syndrome, and congenital insensitivity to pain.5,6
However, congenital anosmia can rarely occur as an isolated deficit, which is then termed isolated congenital anosmia (ICA). Most ICA cases are sporadic and few are familial. 7 The genetic basis of ICA remains poorly understood. 8 The diagnosis of ICA begins with the exclusion of acquired causes of anosmia, followed by clinical testing and imaging. 9 Computed tomography (CT) is commonly employed to rule out known etiologies of anosmia, while magnetic resonance imaging (MRI) in ICA is essential for diagnosis. MRI in ICA cases shows reduced or absent development of olfactory bulbs and shallow olfactory sulci. 10 Although the diagnosis of ICA is relatively well reported, its genetic background and understanding remain obscure. With the help of advancements in genetic testing, we could begin to outline the genetic basis of such a rare and crucial disease. This article aims to report an ICA case, investigate the genetic basis of this case, and compare the findings with international reports on ICA.
Ethical Consideration
The study received approval from the Institutional Review Board Committee of King Faisal Specialist Hospital and Research Center (KFSHRC) Riyadh. Written informed consent was obtained from the patient according to the IRB regulations.
Research Design and Methods
This is a collaborative work between KFSH&RC, King Saud University, and King Abdulaziz City for Science and Technology. The patient underwent a full clinical examination and an anosmia scoring protocol followed by CT and MRI images. Complete family history was obtained from the patient followed by blood collection for DNA extraction and complete exome sequencing according to the standard protocol. Genetic data and bioinformatics analysis was performed to shortlist genetic variants and identify pathogenic changes responsible for the diseases.
Case Presentation
In this paper, we present the case of a 21-year-old male who came to our clinic with a complaint of complete loss of smell since birth. He did not recall smelling any scent to date; however, tasting remains intact. He denied having an upper respiratory tract infection (URTI) at the time of presentation or having chronic or frequent URTI. Furthermore, he denied having a history of facial or nasal trauma, delayed puberty, undescended testis, or surgical procedures involving the brain, ear, nose, or throat areas. He was not taking any medication at home, except salbutamol as needed for his bronchial asthma. His family history showed an affected uncle with loss of smell. Otherwise, his medical history is unremarkable. Upon examination, the patient was afebrile and the external examination of the nose was unremarkable. Anterior rhinoscopy and nasal endoscopy showed healthy mucosa without nasal inflammation, mucosal edema, nasal polyps, crustations, or secretions. Additionally, the oral cavity examination, as well as the complete examination of the ears, head, and neck, were unremarkable. Smell testing (via Smell diskettes
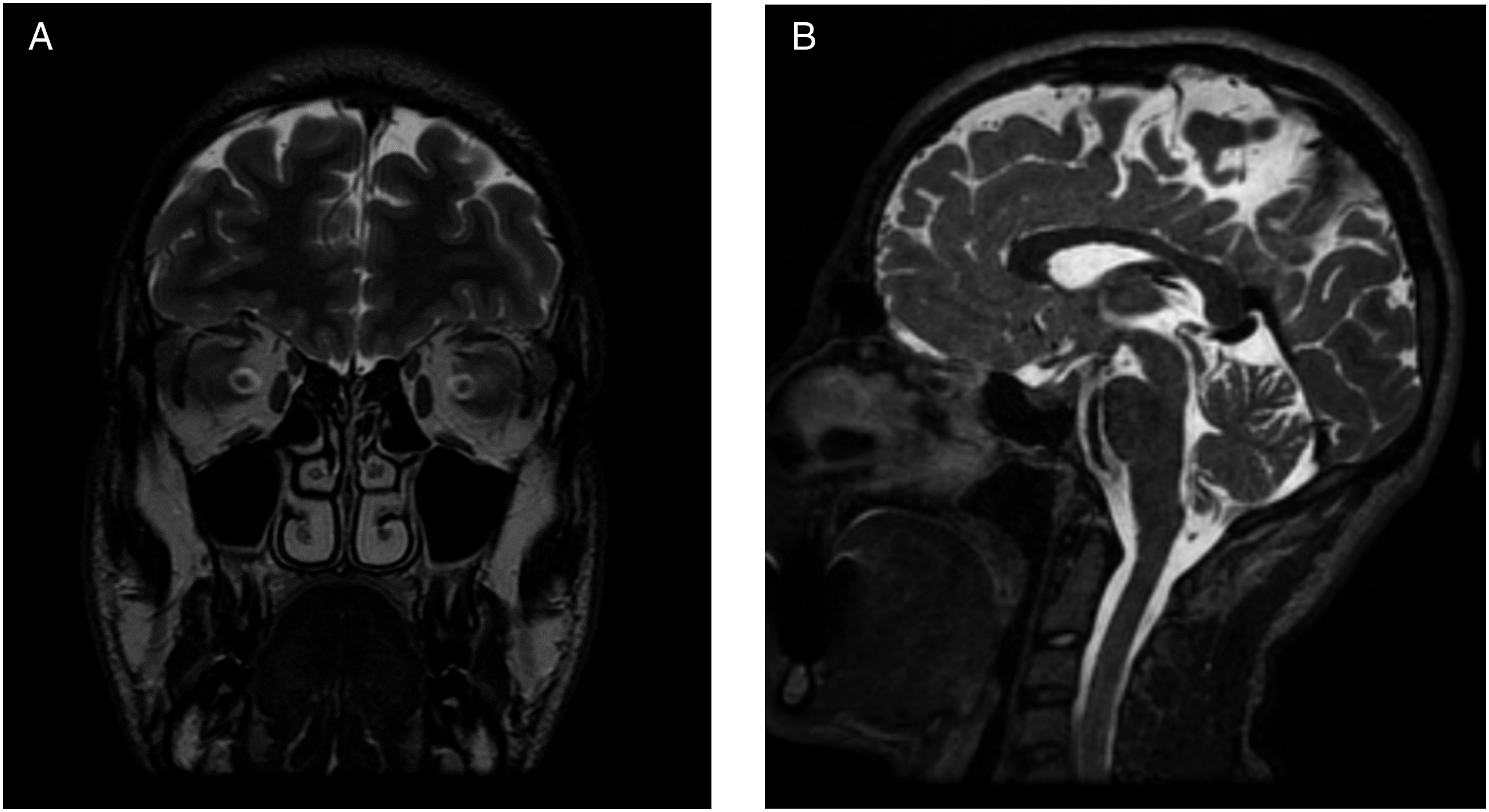
11
) revealed complete anosmia with a score of 0 out of 8. Investigations of the patient’s serum revealed no organ or endocrine dysfunction, normal inflammatory markers, and no abnormalities in electrolytes or minerals. The imaging by CT (Figure 1) showed well aerated paranasal sinuses without evidence of mucosal thickening or fluid density. Furthermore, the frontoethmoidal recesses, the sphenoethmoidal recesses, and the osteomeatal complexes were bilaterally patent, and the cribriform plate was symmetric without signs of inflammatory mucosal changes or active secretions. Furthermore, MRI of the anterior brain and olfactory region (Figure 2) showed bilateral absent olfactory bulbs and olfactory tracts, with shallow olfactory grooves. MRI did not show evidence of mass lesions, abnormal enhancement of the olfactory nerve area, cribriform plate or adjacent air cells, or any gross intracranial abnormalities. The patient was then subjected to complete exome sequencing. Bioinformatic analysis was performed as hypothesis-free analysis followed by additional filtration criteria focused on the 37 genes associated with olfactory dysfunction. A missense variant in the HS6ST1(NM_004807.3) gene which was predicted by insilico tools to be likely pathogenic. Coronal cut, bone window CT scan of the paranasal sinuses. In addition to minimal deviation of the nasal septum to the left, this is a normal scan that shows no evidence of inflammatory changes or bony lesions. (A) Coronal T2-weighted image of the anterior cranial fossa demonstrates the agenesis of the bilateral olfactory tracts and bulbs with shallow olfactory grooves. Preserved bilateral olfactory sulci. Small flow void structure in the left olfactory groove representing a branch of the left anterior cerebral artery. (B) Sagittal T2-weighted image of the midline brain showing normal midline structures and posterior fossa, indicating the lack of additional brain abnormalities.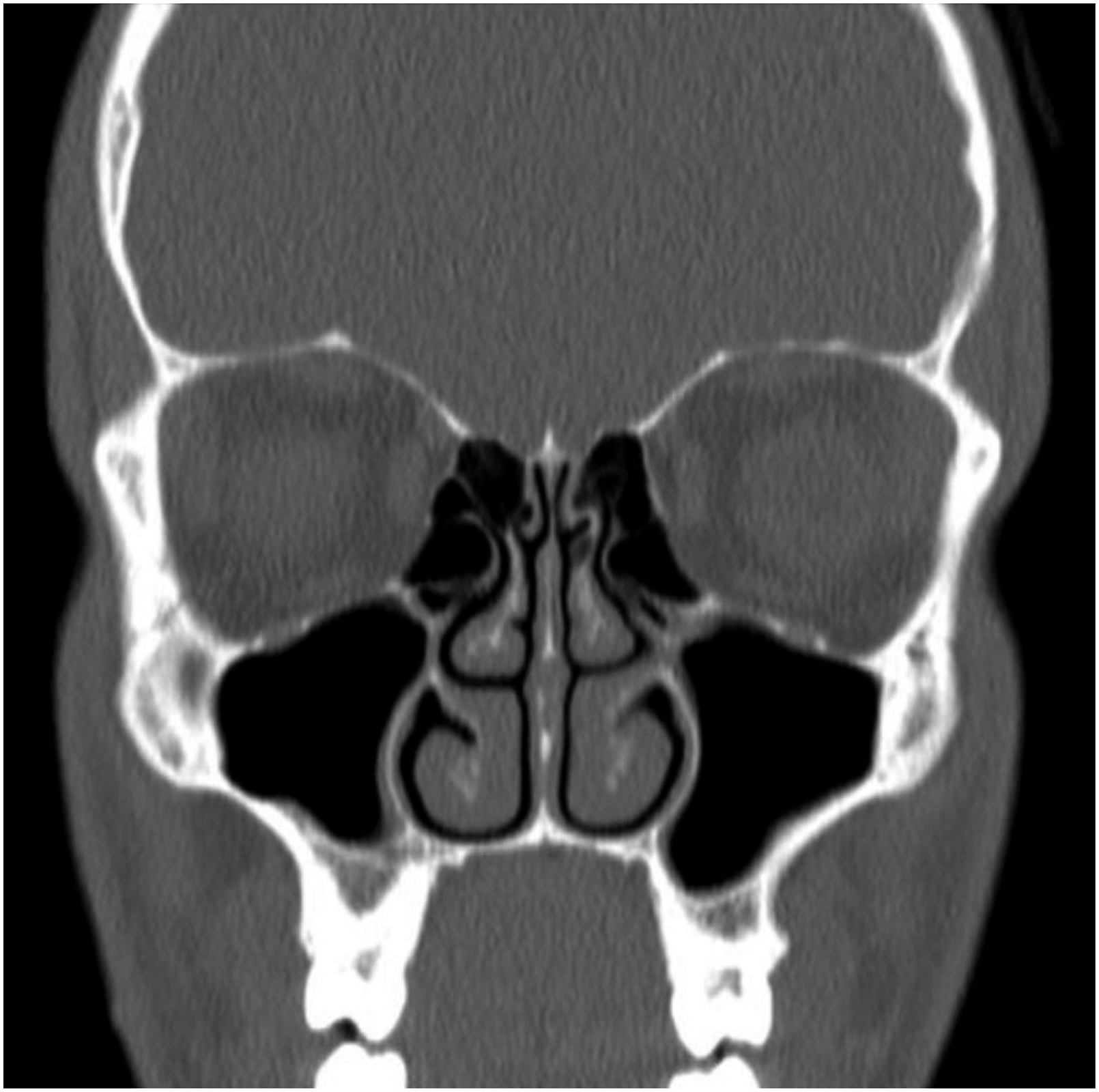
Discussion
Isolated congenital anosmia is a rare disease with an incidence of 1 in 10,000. For many years, ICA has been reported in many populations as a genetic condition inherited as an autosomal dominant condition with or without partial penetrance, syndromic autosomal recessive cases with hypogonadotrophic hypogonadism, or as an x-linked condition.3,12 -14
Genetic and clinical heterogeneity is well described in different olfactory dysfunction diseases such as KS and isolated hypogonadotrophic hypogonadism (IHH). Although more than 46 genes are known to be involved in syndromic and non-syndromic ICA, many patients still lack the known genetic variations.15,16 To date, only five genes have been associated with ICA (PROKR2, PROK2, CNGA2, SCN9A, and TENM1).3,8,17,18
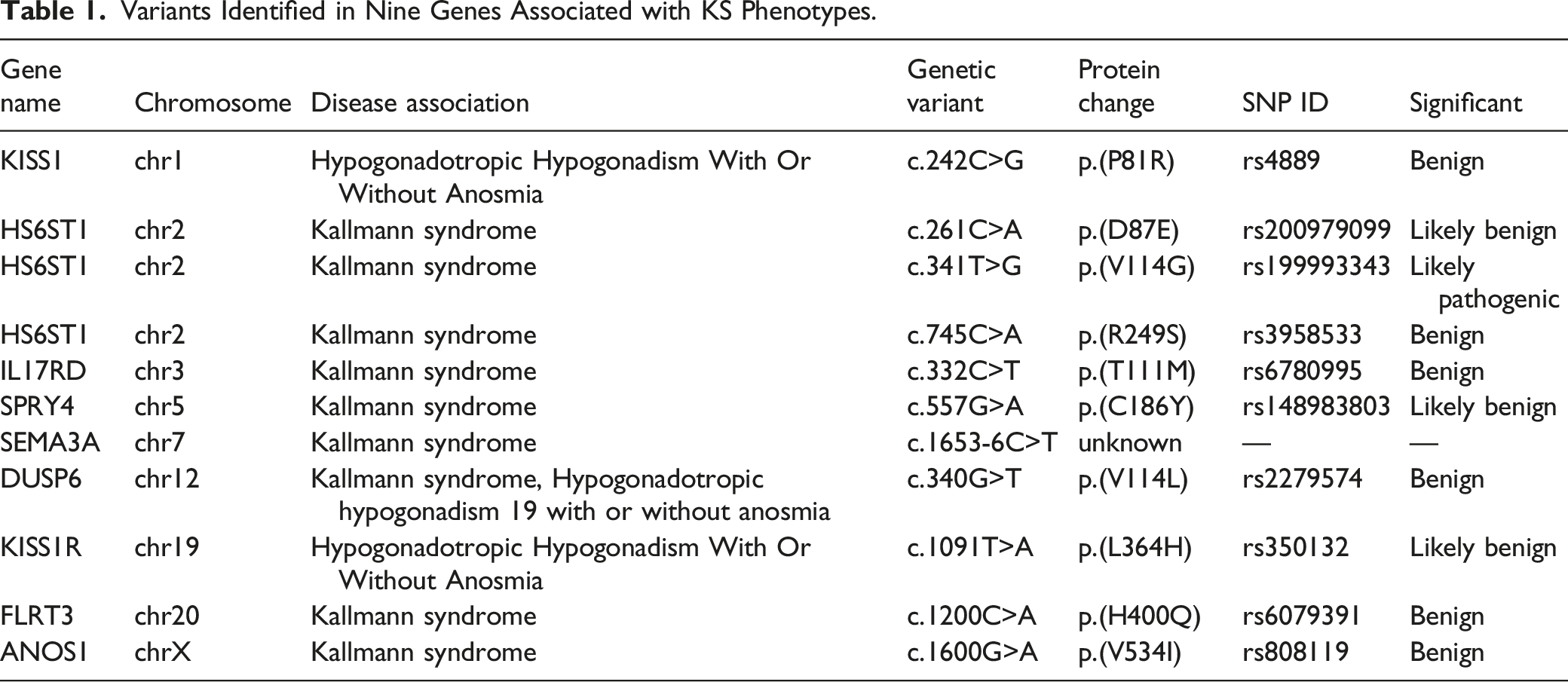
Variants Identified in Nine Genes Associated with KS Phenotypes.
Conclusions
Isolated congenital anosmia (ICA) is a rare entity worldwide with poorly understood genetic variation. The results of this patient’s genetic analysis add to the possible genetic culprits reported in ICA cases. Additional functional and expression analyses are required to validate mutations and understand the heterogeneity of the disease representation.
Footnotes
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.