
Abstract
Peripheral nerve sheath tumors encompass a spectrum of well-defined clinicopathologic entities, ranging from benign tumors, such as neurofibromas, to high grade malignant neoplasms termed malignant peripheral nerve sheath tumors. Morphologic variability of these tumors is wide, and they engender some of the most controversial, difficult differential diagnoses. Localized neurofibromas often involve a major nerve and result typically in fusiform expansion of the nerve trunk (intraneural subtype). We report a case of circumscribed solitary neurofibromas in a 14-year-old boy with NF1 who presented to our department with a left neck swelling. The neurofibromas lesion involved the anterior branch of the great auricular nerve. The sensory symptoms initially reported by the patient (paresthesia and hyperesthesia) in the lower preauricular region. Surgical treatment represents the therapeutic method of choice in the approach to neurofibromas, considering functional disorders and possible aesthetic deformities. The case described presented difficulties in surgical excision, based on risk of functional and aesthetic results.
Introduction
Neurofibromas are benign, slow-growing tumors arising from proliferating Schwann cells, perineural cells, and perineural fibroblasts of peripheral nerves. 1 They are classified in various subtypes depending on the clinicopathological aspects: localized cutaneous form or localized intraneural form, plexiform subtype, and diffuse and massive subtypes.1,2 Approximately 25% of all neurofibromas are found in the head and neck region and present as an isolated tumor or in a multiple form. 2 They generally affect patients aged 20–30 years without gender predilection.1–3 Localized neurofibromas often arise from cutaneous nerves, with occasional involvement of the deep nervous sheath, and there are no known causes in the 60–90% of cases. 3 In approximately 10% of the patients, there is an association with the neurofibromatosis type I (NF-1), an autosomal dominant disease, with complete penetrance and variable phenotypic expression that result from a spectrum of germline mutations in the NF-1 tumor-suppressor gene located in chromosome 17q11 coding for the protein neurofibromin. 4
Extracranial solitary neurofibromas in the head and neck areas are rare findings, frequently arising from the intratemporal portion of the facial nerve, and occasionally from the vagus nerve, glossopharyngeal and hypoglossal nerves, cervical sympathetic chain, and trigeminal nerve. 3
We report a case of a solitary neurofibroma of the left great auricular nerve (GAN) occurring in a 14-year-old boy with NF-1, presenting as a mass in the left retromandibular region. To our knowledge, this is the first report of an NF-1 patient presenting a single neurofibromatous lesion of this nerve without any other body localization.
Case report
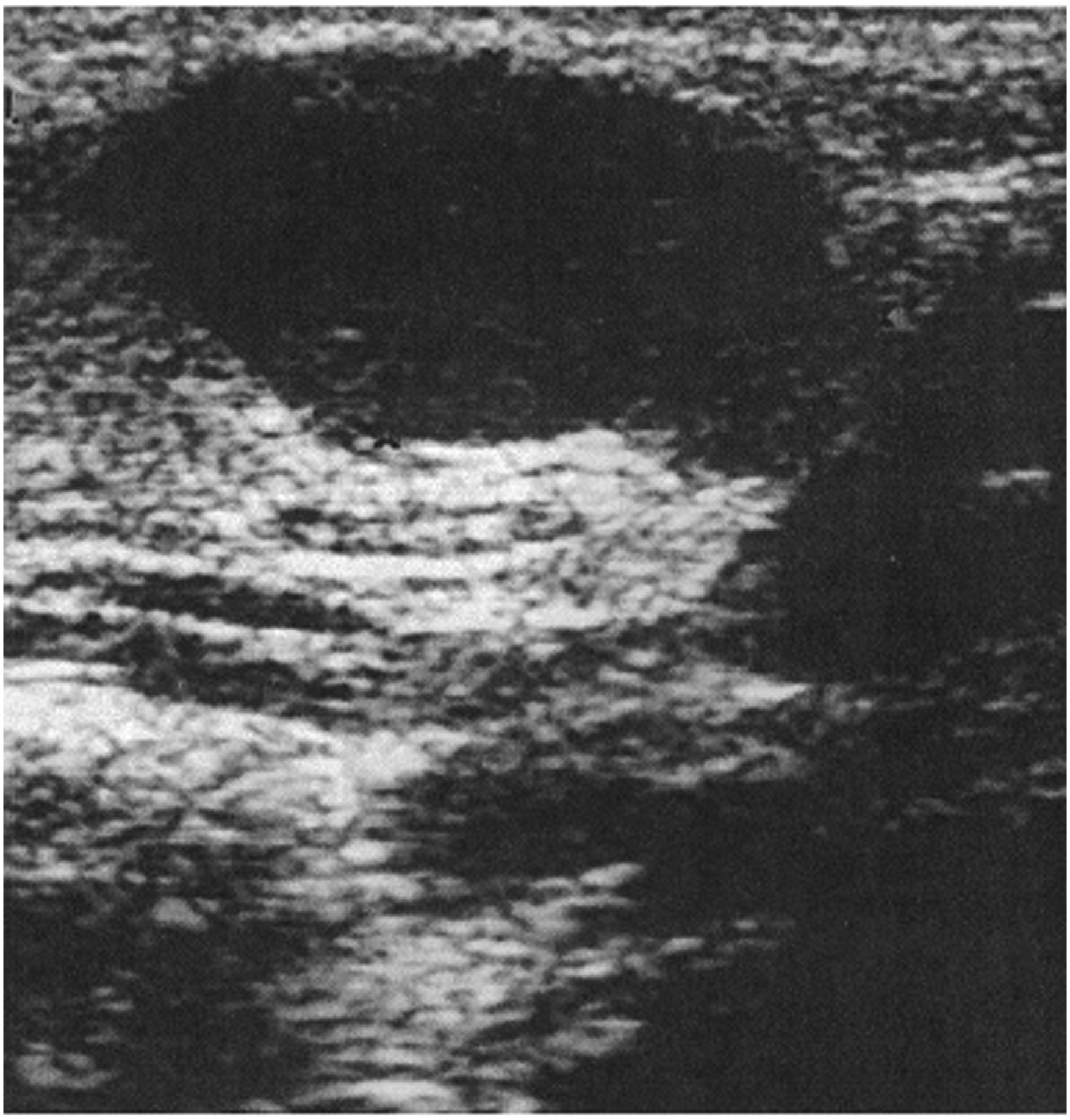
A 14-year-old child affected by NF-1 was referred to our Department from the Paediatric Unit of the University Federico II of Naples for proper evaluation and management of a symptomatic, slow growing, firm, not-tender neck mass with normal overlying skin, localized in the left retro-mandibular region (Figure 1). Diagnosis of NF-1 was made at the age of 20 months; the patient’s mother was affected by the same disease and died of intestinal hemorrhage at the age of 31. An overall clinical examination revealed more than seven randomly distributed cutaneous café-au-lait spots on the patient’s trunk and extremities; the largest spot was over 14 mm in diameter. Although these are typical signs of NF-1, no other indication of the syndrome such as Lisch nodules or cutaneous neurofibromas were evident; notably, skeletal abnormalities associated with scoliosis and pectus excavatum were present. The patient presented long-standing recurrent paresthesia and hyperesthesia in the lower pre-auricular region that was not associated with hearing impairment. Ultrasonography revealed an oval non-homogeneously hypoechoic mass in the left neck region measuring 24 mm × 13 mm × 19 mm in size (Figure 2). Neck mass localized in the left retro-mandibular region. An oval non-homogeneous hypo echoic mass.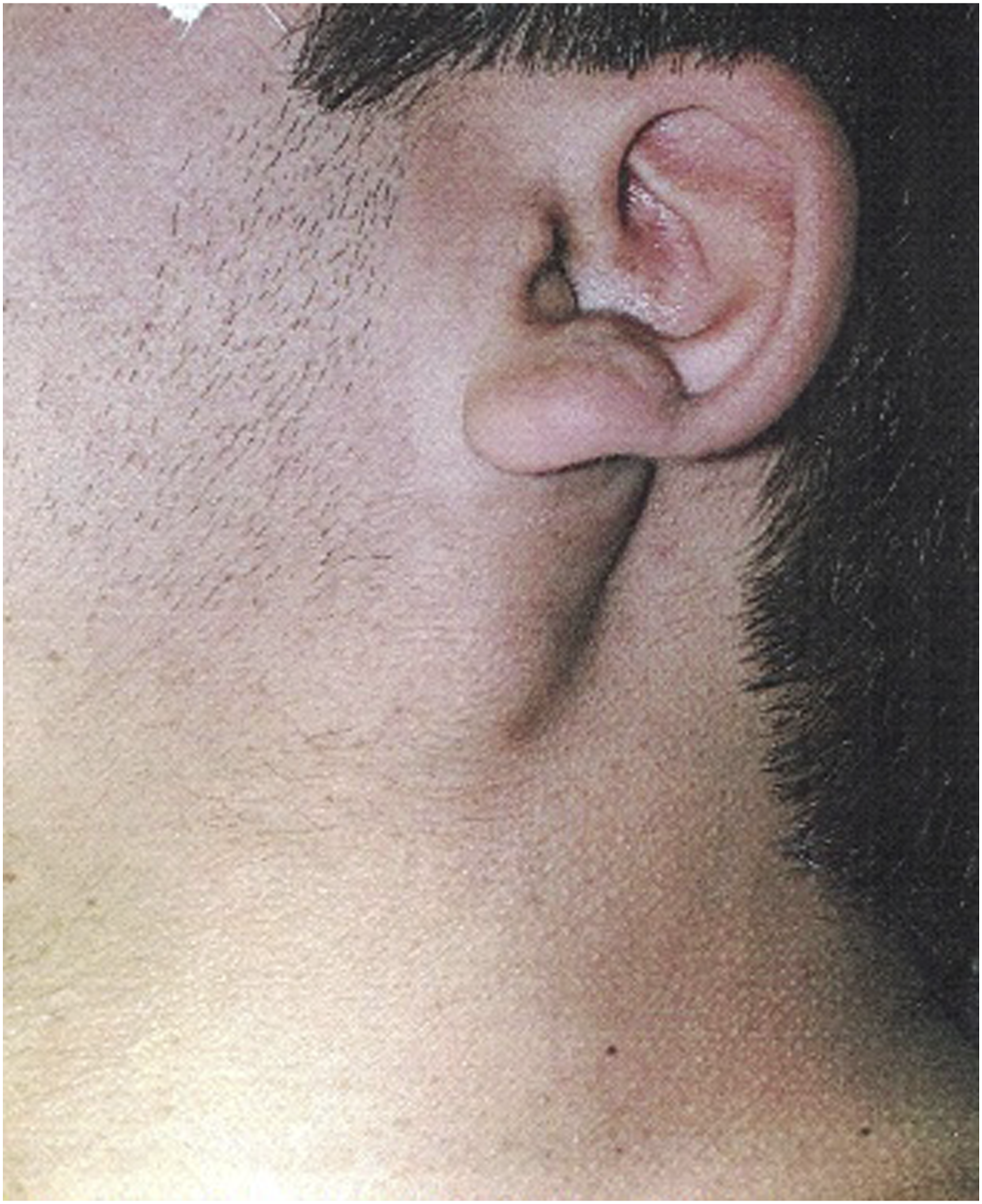
MR imaging of the head and neck revealed a solid cervical mass localized in the left GAN region. The lesions were moderately contrasted and enhanced on the post-contrast series with markedly hyper-intense signals on the T2-weighted series and were anteriorly dissociable from the retromandibular vein by the interposition of intact parotid parenchyma. Retro auricular skin profile was prominent because of a subcutaneous extension of the mass. The right parotid gland was normal, and an enlarged loco-regional lymphadenopathy was absent.
No pathological brain alterations were evident, except for a focal hyper-intensity in DP / T2 with a left pale area compatible with a hemartoma in NF-1 (Figure 3). MR imaging: a solid cervical mass localized in the area of the left great auricular nerve. The lesions were moderately contrast enhanced on the post-contrast series, with their signals markedly hyper intense on T2-weighted series.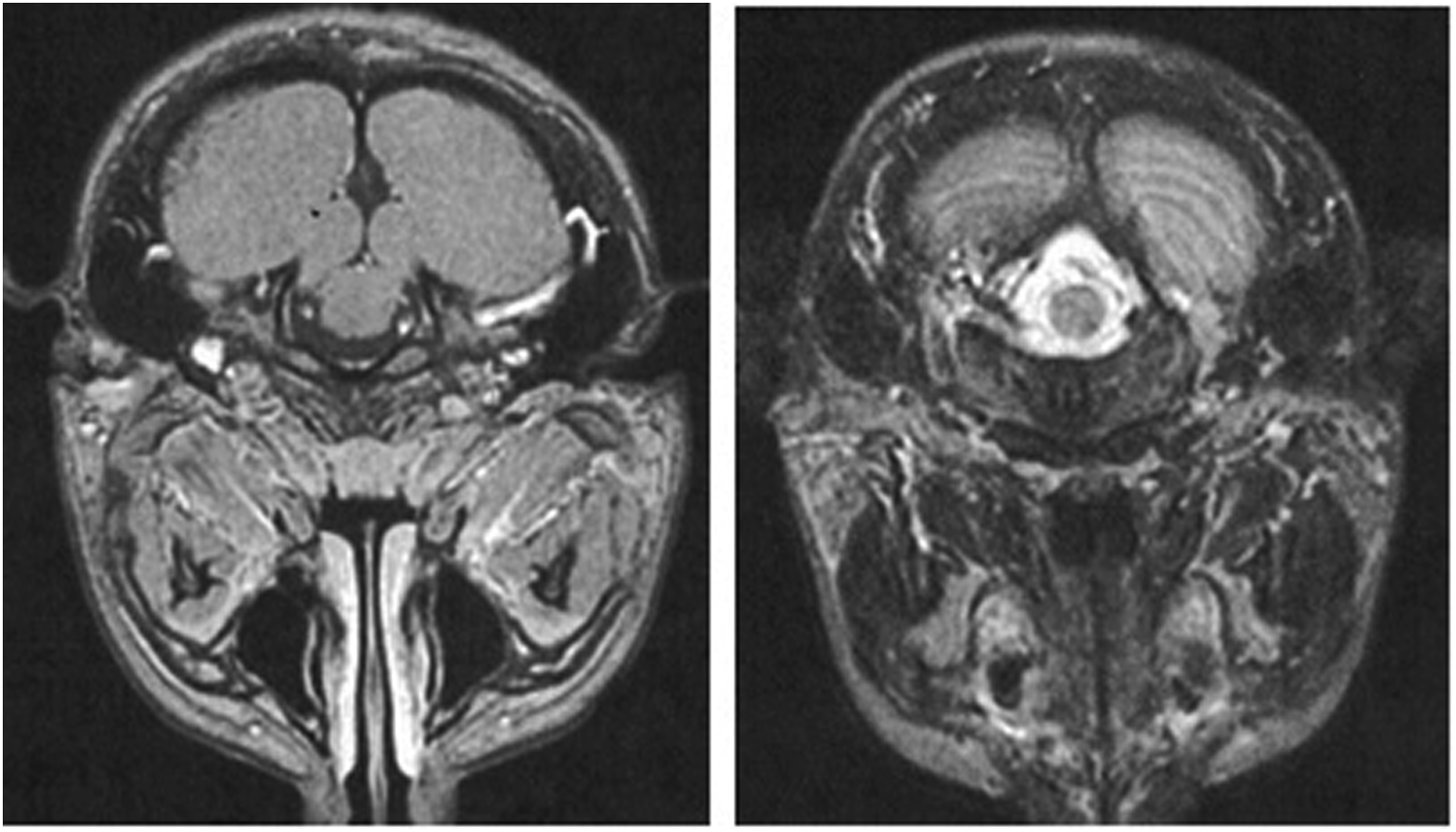
After informed consent was obtained from the father, surgical removal of the mass via postauricular approach under general anesthesia was performed. A skin incision of about 6 cm was made from the left mastoid tip to the mandibular angle, and the underlying submucosal and fascial layers were accurately dissected to reveal an oval, translucent whitish mass of firm consistency that was well-circumscribed, measuring 3 cm× 2 cm.
At both caudal and cranial extremities, the lesion adhered to the previously identified nerve structure, evaluating the anatomic area, as the anterior branch of the GAN. The mass was removed by sharp, accurate dissection under microscopic control to save the nerve’s integrity (Figure 4). All stitches were removed seven days after the surgery, and there were no complications such as wound dehiscence or infection. The patient complained of a temporary numbness of the left auricle and peripheral skin within the first 2 months, and no evidence of recurrence or neurological deficits was recorded at the five-year follow-up appointment. A) Surgical removal of the mass while great auricular nerve’s sparing. B) Left neck mass after surgical excision.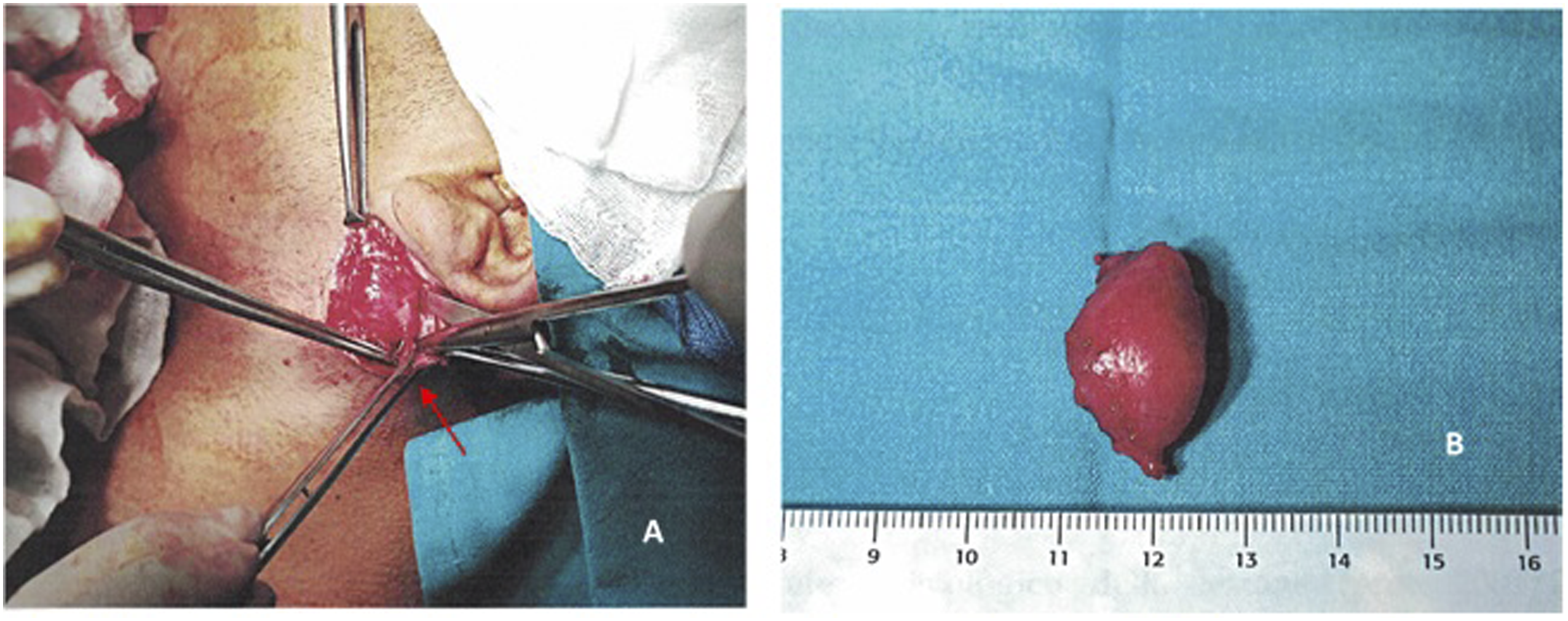
Macroscopically, the mass had a smooth and glistening surface. The histopathological study described a proliferation of Schwann cells with dark nuclei and a serpentine configuration mixed with mononuclear cells and occasional mast cells; a rich amount of collagen fibers arranged in bundles with a so-called “shredded carrots” appearance and a moderate amount of mucoid material separating the cells and collagen were observed; prominent vascularization was also present, and no areas of malignant transformation were observed. While immunohistochemistry identified a strong positivity for S-100 protein and vimentin, they were negative for smooth muscle actin, desmin, cytokeratin, and EMA (Figure 5); these findings strongly supported the final histopathologic diagnosis of intraneural neurofibroma, but not the plexiform subtype, which is generally reported in NF-1. A) A predominantly spindled cells lesion, with myxoid matrix and collagen bundles observed (hematoxylin–eosin, original magnification 40x). B) Collagen fibers arranged in bundles with a so-called “shredded carrots” appearance. Schwann cells of irregular shape and mononuclear cells are also visible (hematoxylin–eosin, original magnification 200x). C) Immunostaining for S-100 was positive in Schwann cells (immunoperoxidase staining, original magnification 100x). D) Immunostaining for S-100 negative in blood vessels provides an internal negative control (immunoperoxidase staining, original magnification 200x).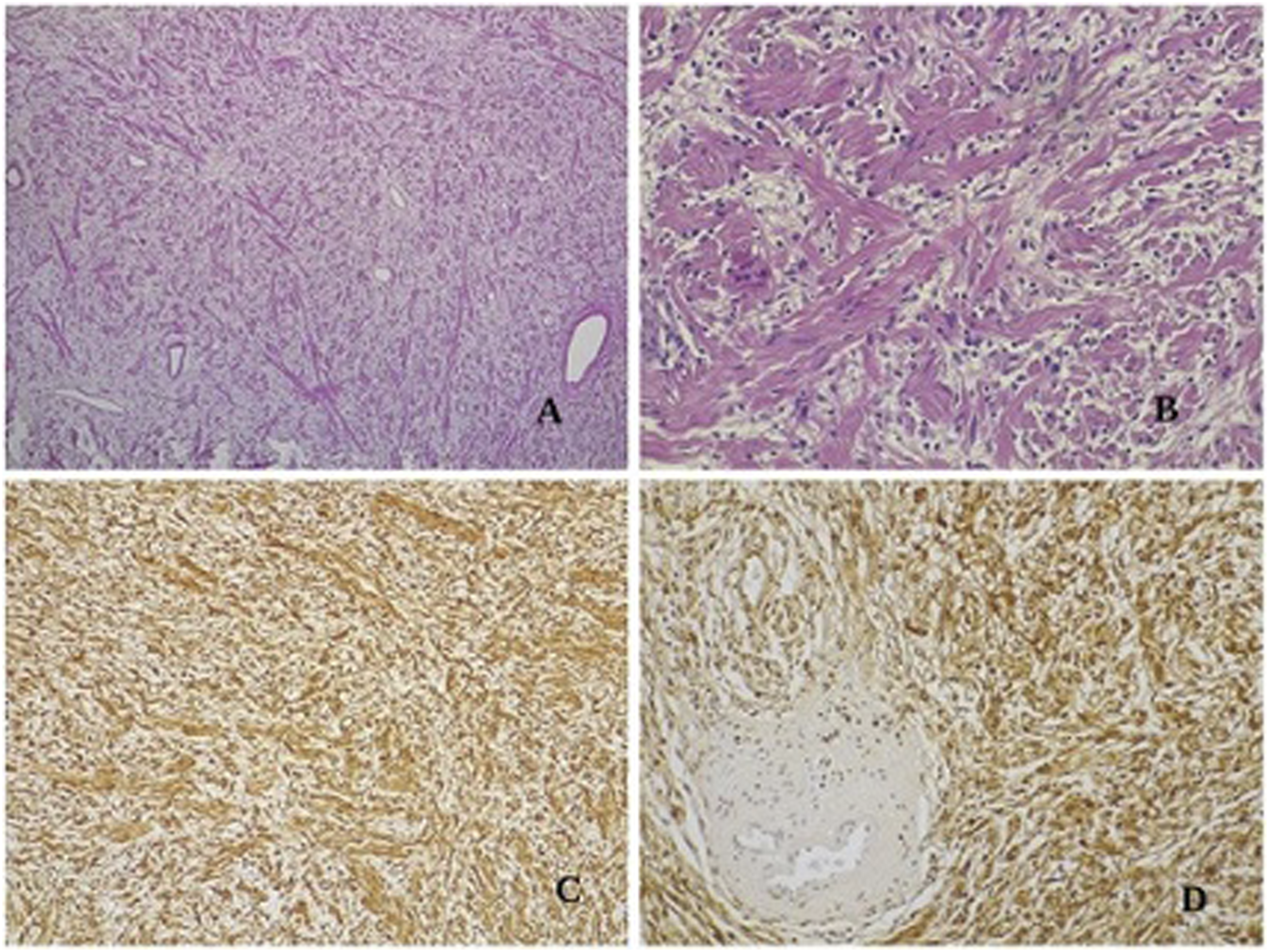
Discussion
Neurofibromas are well-differentiated, non-encapsulated nerve sheath tumors, primarily composed of Schwann cells, perineural-like cells, fibroblasts, and transitional cells and arising within the endoneurium. 1 Because the nerve fibers are incorporated within the tumor, complete excision often requires nerve sacrifice. There are two types of neurofibromas: dermal and plexiform. 2 Dermal neurofibroma is also called discrete or cutaneous neurofibroma, and it usually develops during adolescence and adulthood. These lesions tend to involve the terminal nerves and may be numerous, but the incidence of malignant transformation is rare.1–3 As the main problem to be faced is essentially cosmetic, removal is often required. Plexiform neurofibroma is usually congenital, developing in childhood and often extending deeply along the nerves with the involvement of skin, fascia, muscle, bone, and viscera. Malignant transformation is more common in plexiform tumors arising in the large nerve trunks of the neck or extremities, while superficial lesions of any size are rarely observed. 5
Neurofibromas may be sporadic or associated with NF-1 syndrome. The subtypes that strongly associated with NF-1 syndrome are multiple cutaneous subtype (i.e., NF-1 or N-2), massive subtype (i.e., NF-1), and plexiform subtype (i.e., NF-1). Systemic and hereditary factors are absent in the solitary type. 4
While approximately 25% of all neurofibromas are found in the head and neck area, solitary lesions are rare. 6 Neurofibromas of the facial nerve have been described, most of which are in the intratemporal portion and rarely involve the intraparotid portion of the nerve. Neurofibromas of the vagus nerves, 7 the sympathetic cervical chain, 8 and the trigeminal nerve 9 have occasionally been reported. When the nerve lesion affects the trigeminal dividing branches, various anatomical regions may be involved, such as the ear,10,11 nose,12,13 paranasal sinuses, 14 infratemporal fossa, 15 the para and retropharyngeal spaces, 16 and the mandibular region. 17 Solitary neurofibroma has also been reported in the larynx. 18
Neurofibromas in the auricular region were described by Cartellieri, 19 who recounted a case of a solitary plexiform neurofibroma of the auriculotemporal nerve without any other findings, which suggested NF-1. Shaida 20 reported a case of a neurofibroma of the pinna, and Ghosh 21 detailed a case of a neurofibroma of the external ear. Satar 22 documented two cases of neurofibroma in the auricular region that did not originate from the facial nerve: a patient affected by NF-1 with a plexiform neurofibromatous lesion of the pinna that likely originated in the pinna branches of the lesser occipital nerve or the GAN; and a neurofibroma that was localized in the external ear of a patient without café-au-lait spots on their body, the location of which implied that the lesion likely originated from the auriculotemporal nerve. Shi Nee 2 reported a solitary neurofibroma in the postauricular area of a patient with no family history of NF-1. Gur 4 described the first case of an NF-1 patient with bilateral symmetric localization in the GAN, which is associated with numerous neurofibromas at all spinal levels and along the vagus nerve.
Our case is the first report in the literature of a unilateral solitary non-plexiform neurofibroma involving the GAN in a 14-year-old boy who presented café-au-lait cutaneous spots without any other clinical findings pathognomonic for NF-1. The GAN is the largest of the cervical cutaneous nerves and is a purely sensory nerve that provides cutaneous sensory innervation of the parotid region, the external ear, and the posterior auricular region; this nerve arises from the C2 and C3 spinal roots and fuses to the main trunk before penetrating the middle part of the sternocleidomastoid muscle (SCM). The GAN is in its most superficial position at the SCM before bifurcating into anterior and posterior branches. The posterior branch, which is noses responsible for sensation on the posteroinferior surface of the auricle, travels along the SCM surface before arriving the mastoid area and ends at the post-auricular area; the anterior branch provides innervation to the skin of the parotid region and the lower preauricular region.23–25 In our case, the neurofibromatous lesion involved the anterior branch of the GAN; the sensory symptoms in the lower preauricular region initially reported by the patient (i.e., paresthesia and hyperesthesia) support the pathogenetic hypothesis.
Conclusions
Neurofibromas are commonly seen in patients with NF-1 and generally present as multiple plexiform lesions. In the rare case of a solitary neurofibroma, depending on its size, the site of the lesion, and whether the nerve trunk involved, complete excision is possible. Compared with similar benign tumors such as Schwannomas, preservation of the nerve integrity during surgery is difficult, because neurofibromas have no capsule and because the single cells tend to infiltrate the nerve fibers by invading the surrounding vital tissues and structures and are highly vascularized. As such, if potential damage to major motor nerves (i.e., the facial nerve or the vagus nerve) is expected, a “wait and see” attitude or subtotal resection could be advisable, even though the potential risk of malignant transformation, especially in patients with NF1, is possible; the occurrence of tumor regrowth is nearly inevitable in the case of partial excision. In our patient, a complete resection of the lesion was achieved with an accurate dissection under microscopic magnification, which allowed us to preserve the integrity of the nerve involved without any permanent functional deficits.
Footnotes
Declaration of conflicting interests
The author(s) declare no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.