
Abstract
Rosai-Dorfman disease (RDD) is a rare benign systemic histiocytic proliferation characterized by massive lymph node enlargement and sometimes associated with extranodal involvement. Even though it is considered to be benign, death can occur depending on the extent and location. Our case highlights a primary extranodal site of the right pinna with extension through the Eustachian tube to the subglottis. A previously healthy 15-year-old female presented with 1-year right pinna swelling, slowly enlarging and becoming more bothersome. An incisional biopsy was performed on the ear along with S100 staining yielding a diagnosis. After multidisciplinary case discussion, clofarabine monotherapy and systemic therapy for Langerhans cell histiocytosis has started. Rosai-Dorfman disease can be a general disorder, often affecting the lymph nodes. Unlike a nodal disease, extranodal disease could involve any site on the patient’s anatomy. Head and neck lesions are the most common extranodal lesions. Rosai-Dorfman disease is self-limited in more than 20% of the cases with spontaneous regression without intervention; 70% of the patients have noticeable symptoms and vital organ involvement requiring treatments such as surgery, steroids, radiation, and chemotherapy. In our case, the patient had wide involvement and presented without any serious breathing difficulties; we decided to start with monotherapy with chemotherapy and systematic glucocorticoid treatment.
Introduction
Rosai-Dorfman disease (RDD) is a rare benign systemic histiocytic proliferation in which there is an accumulation of activated histiocytes within the affected tissue. Rosai-Dorfman disease was first described by Destombes in 1965 and further by Rosai and Dorfman in 1969 as “sinus histiocytosis with massive lymphadenopathy” as a non-Langerhans cell histiocytosis (LCH). 1 There are 3 forms of this clinical entity, including nodal, extranodal, and cutaneous RDD. According to the revised histiocyte classification by the Histiocyte Society in 2016, the noncutaneous forms of RDD have been placed into the “R group” of histiocytoses, whereas the cutaneous form is considered a separate entity within the “C group.” 2,3 Rosai-Dorfman disease may occur as an association to an inheritable condition, malignancy, or autoimmune disease but may also occur spontaneously without associated comorbidities. 4
This is a relatively rare disease process with approximately 100 new cases per year in the United States and a prevalence of 1:200 000. 5 It tends to affect children and young adults with a mean age of 20.6 years. 4 Rosai-Dorfman disease classically presents with large bilateral cervical lymphadenopathy, but over 40% of cases present with extranodal disease. 4,6,7 Many extranodal sites within the head and neck can be involved. Most frequently described extranodal presentations in the head and neck are nasal cavity, paranasal sinuses, soft and hard palate, gingiva, oral mucosa, tongue, and tonsils. Less frequently involved are salivary glands, larynx, pharynx, and thyroid gland. 4,7 -10
Case reports have highlighted the presence of external ear lesions as cutaneous RDD. 11,12 In 2020, Flowers et al reported on the first known case of RDD involving the parapharyngeal space. 13 Involvement of the middle ear has been described relatively infrequently. Douleh et al documented the case of a 39-year-old woman with RDD of the entire tympanomastoid cavity and internal auditory canal with dural involvement. 14 In 2001, Ahsan et al described a patient initially diagnosed with hearing loss and asthma who was eventually found to have RDD involving the bilateral middle ear and external auditory canals as well as obstructive tracheobronchial lesions. 15 This report describes a particularly unusual presentation of RDD of the external pinna with extension along the skull base to the nasopharynx and parapharyngeal space with an associated lesion in the subglottis causing airway obstruction.
Case Report
A previously healthy 15-year-old female presented with 1 year of progressive right pinna swelling. She had seen various physicians with various workups during that time before admitting to our hospital for a definitive evaluation and diagnosis. The lesion first appeared approximately 1 year prior and was noted to be slowly enlarging, becoming more bothersome. It was associated with tinnitus, hearing loss, and painless drainage from the right external ear canal. She denied any previous trauma, previous or concurrent illnesses, fevers, chills, night sweats, or weight loss. Four months prior to presentation, and she endorsed shortness of breath that has progressively worsened, especially with physical activity. There were no voice changes, dysphagia, and aspiration.
Physical examination revealed a conglomeration of erythematous, firm nodules effacing the right external ear and obscuring the external auditory canal (Figure 1). She has no other cutaneous findings, nor did she have lymphadenopathy or hepatosplenomegaly. Flexible nasolaryngoscopy demonstrated submucosal right nasopharyngeal fullness, effacing the right Eustachian tube orifice, and a bulbous submucosal mass directly inferior to bilateral vocal folds. Vocal fold movement was normal bilaterally (Figure 2). There was mild inspiratory stridor detectable upon auscultation of the anterior neck but no respiratory distress.
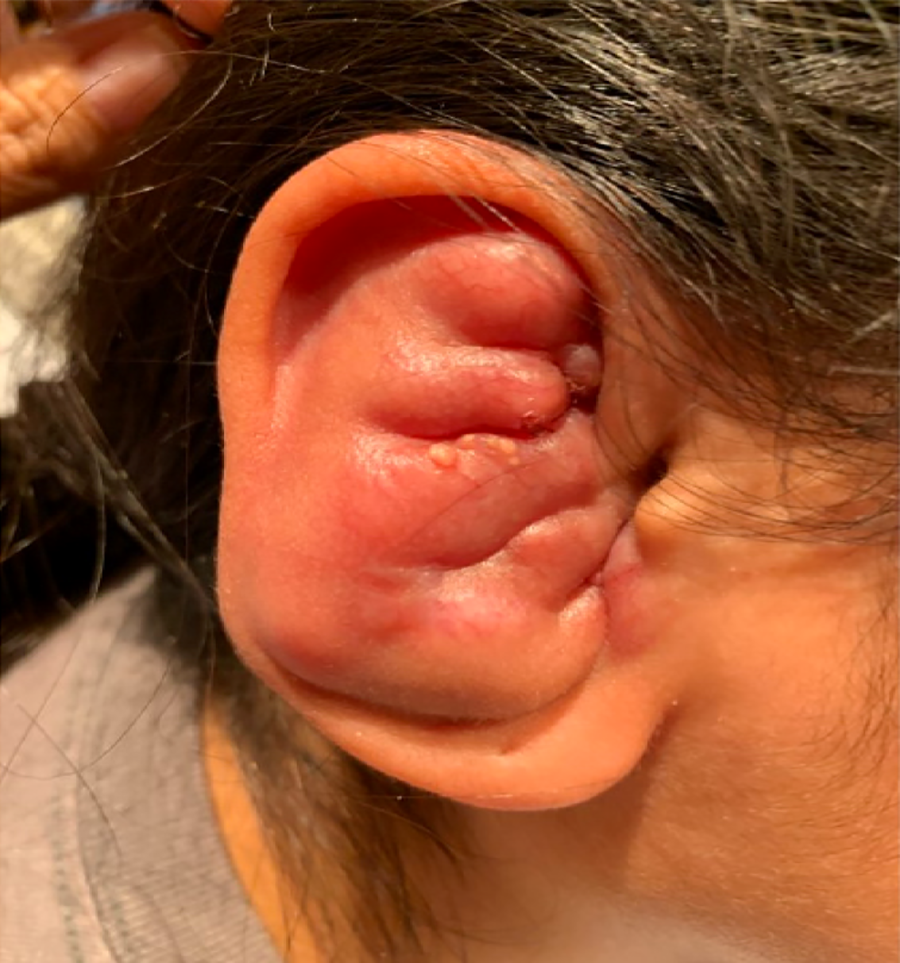
Multiple erythematous nodules of the right external ear.
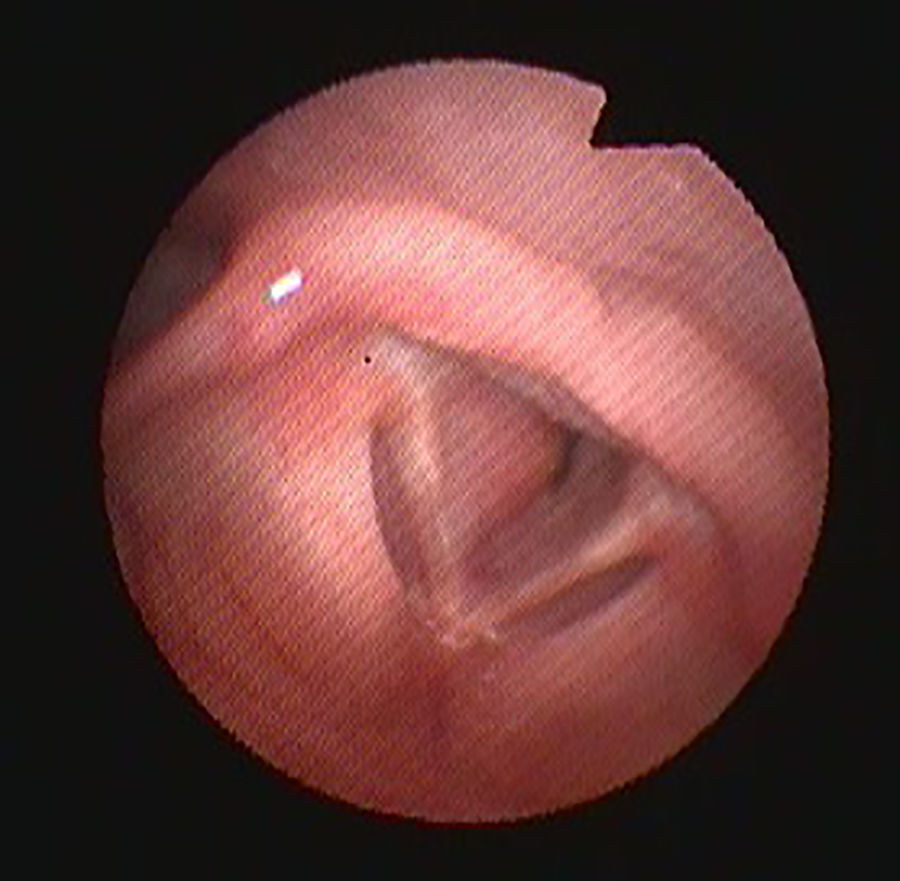
Subglottic involvement.
Imaging with computed tomography and magnetic resonance imaging (MRI) revealed an extensive, infiltrative, contrast-enhancing lesion that extended from the right external ear through the middle ear and Eustachian tube along with the skull base parapharyngeal space and nasopharynx (Figure 3). There was notable compression of the right internal jugular vein and focal area of skull base erosion adjacent to the precavernous internal carotid artery. Contrasted MRI also demonstrated focally enhancing T2-hyperintense lesions of the right subglottic region with mild airway narrowing (Figure 4). Positron emission tomography scan confirmed increased radiotracer uptake in the aforementioned regions without any evidence of distant lesions (Figure 5).
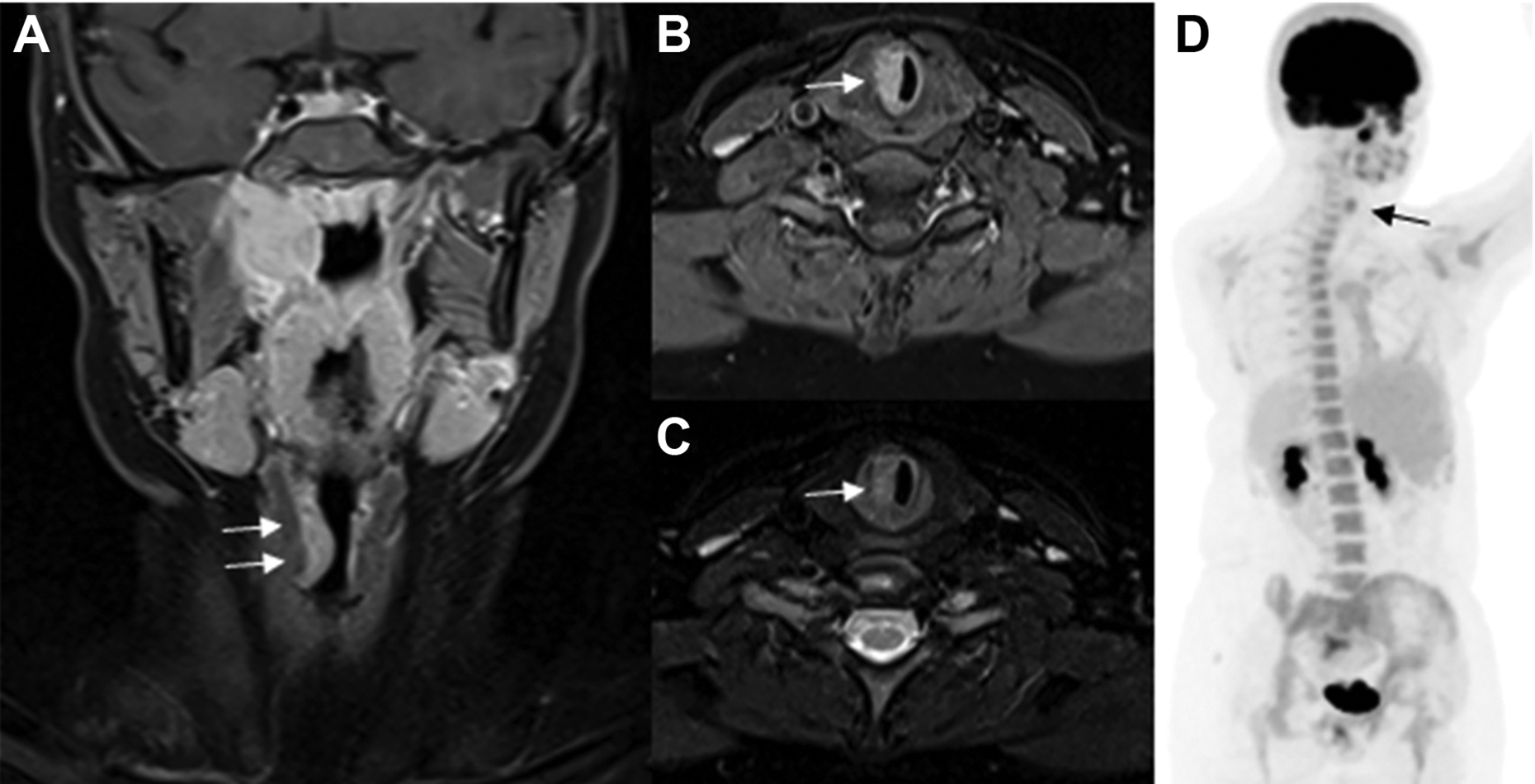
Coronal (A) and axial (B) contrast-enhanced T1-weighted (T1W) and axial T2-weighted (T2W) (C) Magnetic resonance (MR) images show a focal contrast enhancing, T2-hyperintense lesion involving the right subglottic region (arrows) with mild narrowing of the airway. (D) Nuclear medicine study shows a matching increased radiotracer uptake (black arrow) in addition to the right ear/parapharyngeal lesion.
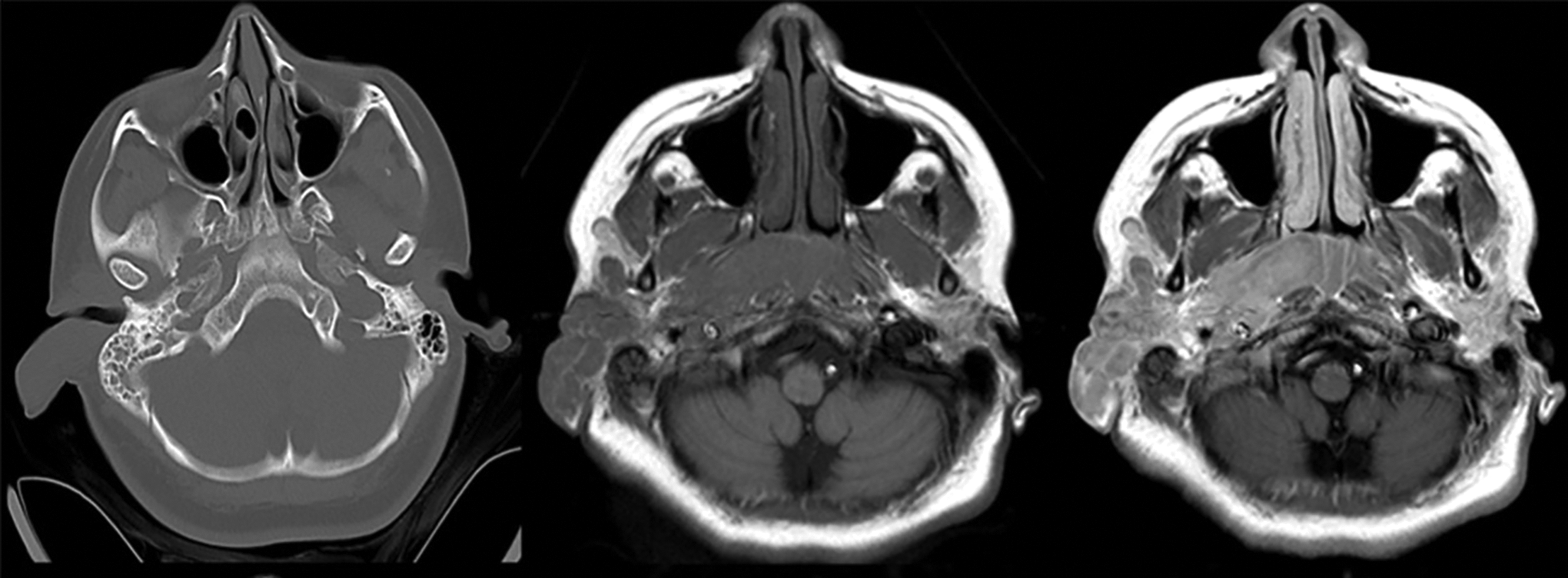
Axial computed tomography (CT), axial pre- and post-contrast-enhanced T1-weighted (T1W) magnetic resonance (MR) images reveal an extensive infiltrative, contrast-enhancing mass lesion extending from the right external ear along the skull base into the parapharyngeal space.
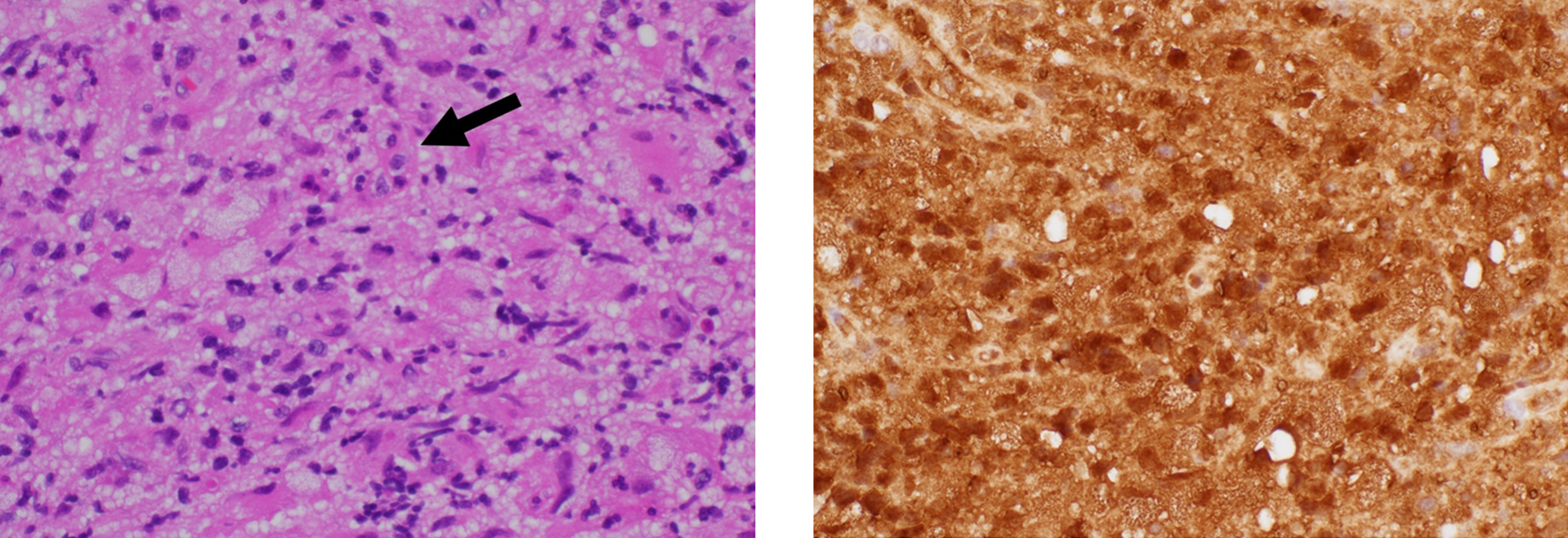
(Left) Many large histiocytes in a background of lymphoplasmacytic inflammation. Possible emperipolesis is noted focally (arrow). (H&E stain, 400x magnification) (Right) S100 immunohistochemistry shows strong, diffuse staining in both the nucleus and cytoplasm of the histiocytes (S100 immunohistochemical stain, 400x magnification) reject.
Incisional biopsy was performed under local anesthesia. The biopsy of the right ear mass shows an infiltrative histiocytic lesion involving the soft tissue. Many of the histiocytes are large, and focal areas of possible emperipolesis are also noted (Figure 5A). The histiocytes show strong, diffuse staining by S100 immunohistochemistry (Figure 5B). Immunohistochemistry for CD68 and CD163 is also positive. Immunohistochemical markers of LCH, CD1a, and langerin/CD207, are both negative. These findings are most consistent with the diagnosis of extranodal RDD. She was discussed at a multidisciplinary case conference and is currently undergoing systemic clofarabine and glucocorticoids. The patient is continuing to receive treatment for the last 6 months, and her hearing and breathing symptoms significantly improved.
Discussion
Rosai-Dorfman disease is a rare, benign systemic histiocytic proliferation, often characterized by massive lymph node involvement. The extranodal disease that may involve any anatomic site is not uncommon and occurs in over 40% of cases. 4,6,7 A wide variety of clinical manifestations for extranodal disease have been described in the literature, often as a diagnostic challenge. Slowly progressing lesions with subacute symptomatology should raise extranodal RDD in the differential diagnosis. This is highlighted in our case with a year-long slowly growing lesion of the external pinna with progressive shortness of breath. Diagnosis requires pathologic evaluation to support suspicions raised by clinical and radiographic evidence. 4 In workup and diagnosis, associated diseases such as inheritable diseases, autoimmune diseases, and malignancies must be ruled out so that treatment for an underlying condition is not neglected.
Histologically, RDD in nodal and extranodal sites includes large pale histiocytes, often with emperipolesis. Emperipolesis is not required for the diagnosis and may be seen in histiocytoses. Immunohistochemical staining demonstrates diffuse S100, CD68, and variable CD163/CD14 positivity. CD1a and CD207 are negative in contrast to LCH. 4
Rosai-Dorfman disease is often a self-limited disease and 20% to 50% of patients with the cutaneous or nodal disease having spontaneous regression without intervention. 4,16,17 Observation may be indicated in those patients without notable symptoms or vital organ involvement. Surgical intervention beyond biopsy is often limited. It can be curative in localized disease, and debulking procedures may be indicated for lesions causing airway compromise, spinal cord compression, and so on. Sinonasal involvement has been effective in symptomatic improvement. 8 Our patient presented with multiple head and neck involvement and no serious breathing difficulties, which led us to avoid surgical interventions like tracheostomy in favor of systemic treatment with chemotherapy and glucocorticoids.
Conclusion
Rosai-Dorfman disease is a rare lymphatic disease with a highly variable presentation that often presents a clinical and diagnostic dilemma frequently presents in the head and neck, and otolaryngologists should be aware of the diverse clinical manifestations of this disorder. Multidisciplinary evaluation and expertise are often required for the diagnosis and management of these patients.
Footnotes
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.