
Abstract
The diagnosis of desmoid fibromatosis or other spindle cell tumors in the sinonasal region is very rare in children and needs to be thoroughly confirmed with immunohistochemical and/or molecular tests. We report 2 patients with such rare tumors and describe the use of next-generation sequencing in their evaluation. A 3-year-old female had a 4.4-cm midline nasal cavity mass involving the bony septum and extending into the base of the skull bilaterally. The moderate cellular fibroblastic proliferation revealed areas of thick keloid-like collagen bands and other areas with myxoid edematous stroma. Deep targeted sequencing identified a novel G34V mutation in the CTNNB1 gene consistent with desmoid fibromatosis. An 11-month-old male infant presented with a right nasal mass that extended through the cribriform plate into the anterior cranial fossa and involved the right ethmoid sinus and adjacent right orbit. Histology revealed an infiltrative atypical fibrous proliferation with focal calcifications that was negative for CTNNB1 and GNAS mutations. A novel RET E511K variant was identified in the tumor and later was also found in the germline and hence rendered of unknown significance. Both cases highlight the utility of next-generation sequencing in the evaluation of pediatric sinonasal spindle cell tumors that may have overlapping pathologic features. Reporting of rare or novel variants in tumor-only sequencing should be cautiously evaluated in children and pairing with germline sequencing may be needed to avoid the pitfall of assigning uncommon variants.
Introduction
Fibroblastic or spindle cell proliferations in children are not uncommon but are rare in the head and neck, where they most commonly occur in the jaw and soft tissues of the neck. 1,2 They can be associated with bone formation or psammoma calcifications, and hence termed “fibro-osseous.” 3 Fibrous, fibro-osseous, and other spindle cell tumors are much rarer in the nasal region, paranasal sinuses, or skull base. 4,5 In these areas, scattered case reports reveal that fibrous dysplasia and juvenile ossifying fibroma are most frequent, while fibromatosis, myofibromatosis, solitary fibrous tumors, myxoma, and fibrosarcoma are exceptionally rare. 6 -8 Although most of these tumors are histologically benign, they are often infiltrative and may become locally destructive and difficult to manage.
Accurate diagnosis of sinonasal spindle cell tumors cannot be underemphasized as their biologic behavior can vary from being non-neoplastic to aggressive neoplasms such as fibrosarcoma. The majority of these tumors can be identified through their characteristic morphologic features aided with few ancillary studies. Fibroblastic lesions may exhibit variable immunohistochemical reactivity to muscle-specific or smooth muscle actin (SMA) and desmin. Although β-catenin nuclear labeling is fairly specific for desmoid fibromatosis, there is no diagnostic antibody test for most other fibro-osseous lesions. 9 Occasionally, fluorescent in situ hybridization (FISH), sequencing studies, and other diagnostic tests may be needed to assist with the diagnosis. 10 The recent improvements and expanded use of next-generation sequencing (NGS) technology in clinical practice have allowed the identification of characteristic tumor mutations and offered valuable information in the prognostic and therapeutic management of patients. 11
In this article, we report 2 rare pediatric cases of spindle cell proliferation involving the sinonasal tract and extending to the skull base, describe their unusual clinicopathologic features, and evaluate the clinical utility of NGS in identifying diagnostic variants.
Case Reports
Two cases were retrieved from the archived pathologic and clinical records of Children’s Mercy Hospital (CMH). Clinical presentation, radiologic data, pathologic reports, histologic slides, and results of all ancillary studies were reviewed including immunochemistry and genetic workup. Immunohistochemical stains for SMA (Cell Marque, Rocklin, California), myogenin (Dako, Santa Clara, California), myoD1 (Dako), desmin (Dako), β-catenin (Cell Marque), Alk1 (Dako), CD34 (Leica, Buffalo Grove, Illinois), epithelial membrane antigen (EMA; Dako), and S100 (Leica) were performed in a Leica Bond Max instrument (Buffalo Grove, Illinois) at CMH pathology laboratory using standard protocols that are validated for routine clinical use and which included antigen retrieval, a primary antibody, secondary antibody, polymer conjugate, and a coloring reagent. DNA from formalin-fixed paraffin-embedded tumor tissue was collected from the diagnostic specimens of the 2 cases for deep targeted sequencing which was done at an outside CLIA-certified/College of American Pathologists (CAP)-accredited laboratory. A capture-based NGS panel targeting 315 cancer-associated genes was performed. One variant identified by this method was subsequently confirmed in a blood sample from the same patient via targeted Sanger sequencing by our in-house CLIA-certified/CAP-accredited molecular genetics laboratory. Evaluation of stained slides and molecular results was performed by licensed and certified pathologists.
Case 1
A 3-year-old female presented to the Ear, Nose, Throat (ENT) clinic for evaluation of chronic mouth breathing, loud snoring, and sleep apnea. During a routine adenoidectomy, she was found to have a large midline nasal cavity mass involving the majority of the bony septum and extending into the base of the skull bilaterally. Magnetic resonance imaging (MRI) revealed a large T2 hyperintense, T1 iso- to hypointense mass involving the bilateral nasal cavities and anterior skull base. The lesion extended ventrally into the anterior ethmoid region and posteriorly with destruction of the sphenoid body immediately inferior to the sella. The mass measured 4.4 cm × 3.6 cm × 3.8 cm and was asymmetric to the right extending into the medial aspect of the right middle cranial fossa with resultant mass effect on the right uncal region (Figure 1A). The aggressive radiologic appearance provided the differential diagnosis of rhabdomyosarcoma, chondrosarcoma, lymphoma, and Ewing sarcoma.
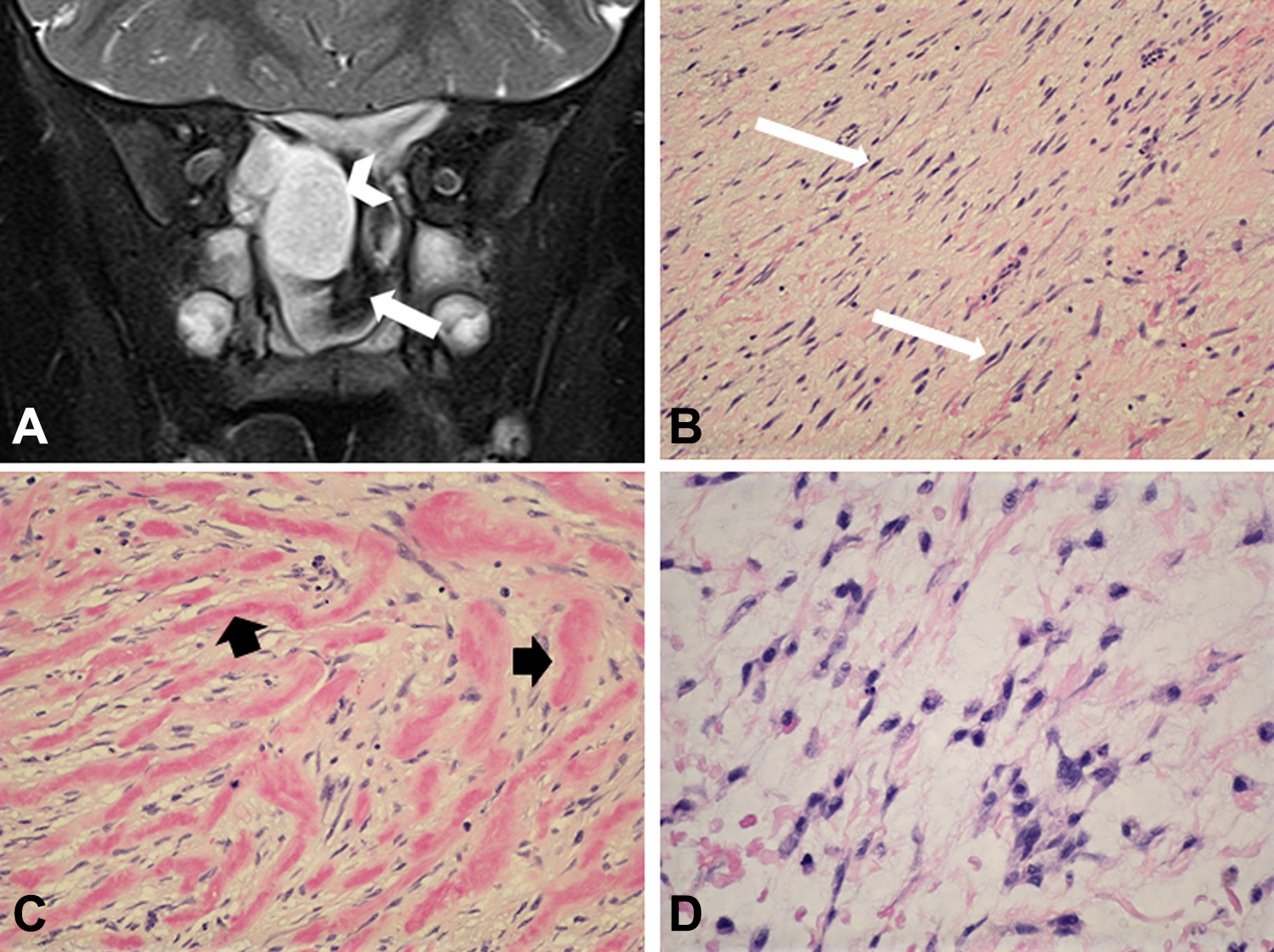
A, Magnetic resonance imaging (MRI) coronal T2 SPAIR demonstrating a sinonasal mass with involvement of the anterior skull base. The mass contains both T2 hyperintense (arrowhead) and hypointense (arrow) components. B, The tumor was composed of a moderately cellular proliferation of spindle cells (long arrows) with no significant pleomorphism or mitosis ( hematoloxylin-eosin, [H&E] × 100). C, In several areas, the tumor cells enclose broad collagen bands of dense keloid-like collagen, highlighted by short arrows (H&E × 200). D, Few areas reveal mucoid myxoid stroma in between the tumor cells (H&E × 200).
Biopsy of the mass revealed a moderately cellular proliferation of bland ovoid to spindle-shaped cells in a loose fibromyxoid to edematous stroma. The tumor cells had spindle tapering nuclei and were arranged haphazardly and in short fascicles (Figure 1B). In many areas, the tumor cells enclosed short thick keloid-like collagen bands while other areas revealed myxoid edematous stroma (Figure 1C and D). Mitotic figures were rare and there was no atypia, necrosis, pleomorphism, or other evidence of malignancy. Immunohistochemical staining showed strong diffuse nuclear staining with β-catenin and cytoplasmic positivity for SMA with no staining for myogenin, S100 protein, EMA, CD34, MyoD1, or desmin (Figure 1D). Because of β-catenin nuclear immunoreactivity, the differential diagnosis included desmoid fibromatosis, cranial fasciitis, and sinonasal sarcoma, all of which can have overlapping morphologic characteristics. Chromosome analysis revealed a normal 46, XX karyotype. Fluorescent in situ hybridization for FUS gene rearrangement was negative. Subsequent NGS sequencing revealed a G34V mutation in the CTNNB1 gene in a background of very few other alterations and a low tumor mutation burden overall (5 mutations per megabase). The morphology, immunophenotype, and molecular results were all consistent with the diagnosis of desmoid fibromatosis.
Case 2
An 11-month-old male infant presented to the ENT clinic with an incidentally discovered right nasal mass. The patient was noted to have congenital ptosis of the right eye as well as facial nerve palsy. A brain MRI revealed a large mass within the anterior skull base which extended into the anterior and inferior aspect of the cranium reaching the inferomedial aspects of the frontal lobes bilaterally. The lobular mass extended superiorly through the cribriform plate into the midline of the anterior cranial fossa and inferiorly into the posterior aspect of the right ethmoid sinus, posterior right nasal cavity, and the posterior/inferomedial aspect of the right orbit with associated body defects in these areas. It bulged anteriorly from the posterior/superior wall of the right maxillary sinus where it measured 3.7 cm along the craniocaudal axis (Figure 2A). The mass appeared heterogeneous in attenuation with multiple amorphous calcifications present involving its intracranial portion. No dilation of the brain ventricular system or intracranial hemorrhage was noted on computed tomography. The mass was completely resected via a multidisciplinary surgical team consisting of pediatric neurosurgery, ENT, and oculoplastics, but the tumor recurred in the right posterior ethmoid sinus after 9 months and was subsequently resected with no further recurrence.
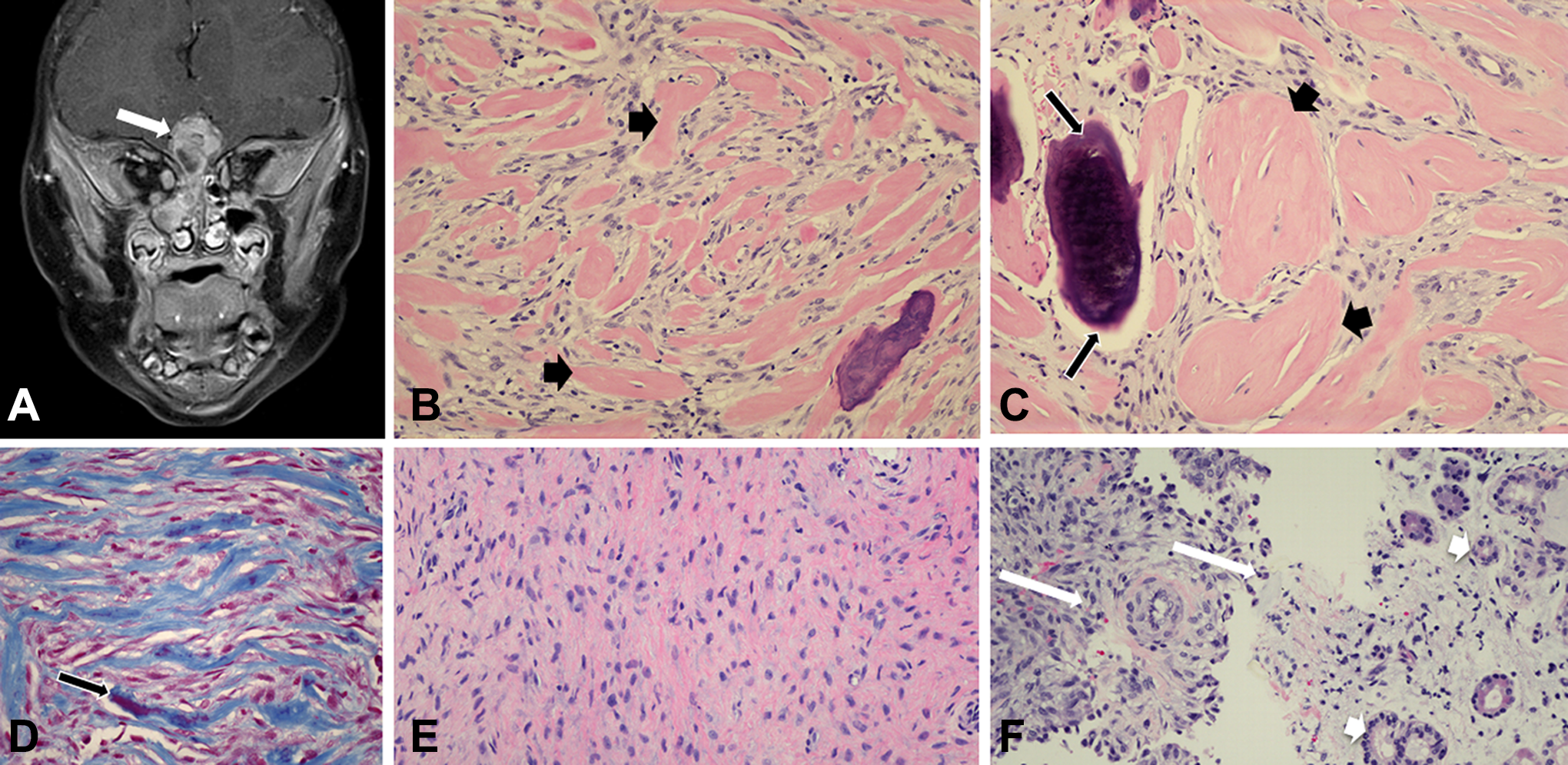
A, Magnetic resonance imaging (MRI) coronal T2 SPAIR demonstrating a sinonasal mass (arrows) with intracranial extension through the floor of the anterior cranial fossa. The mass contained mildly T2 hyperintense and hypointense components. B, The tumor was composed of spindle cells that enclose broad collagen bands, highlighted by short arrows (H&E × 200). C, The presence of dense collagen spherules and small calcifications (long arrows) simulated a fibro-osseous appearance (H&E × 400). D, Trichrome stain highlighting the collagenous bands. A small calcified area stained red (long arrow) (H&E × 200). E, In few areas, moderately cellular spindle cells were present with no collagen bands (H&E × 200). F, The tumor cells (long arrows) infiltrated the sinonasal mucosa (mucosal glands highlighted by short arrows) (H&E × 200).
A nasal endoscopic biopsy and subsequent excisions revealed a low to moderate cellular spindle cell proliferation. The tumor cells were spindle to ovoid with attenuated small nuclei. The poorly circumscribed proliferation was interspersed in most areas with dense keloid-like collagen bands, which focally became calcified (Figure 2B). Only focally were the tumor cells seen in a loose fibrous or fibromyxoid stroma (Figure 2C and D). Despite the aggressive radiologic appearance, the histology appeared bland with no significant atypia and only very rare mitoses. The tumor cells were seen invading the bone trabeculae of the nasal turbinates and the sinonasal mucosa (Figure 2E and F). By immunohistochemistry, the tumor cells were positive for SMA and negative for myogenin, myoD1, desmin, β-catenin nuclear labeling, Alk1, CD34, EMA, or S100.
Although the histologic features were suggestive of fibromatosis, the lack of β-catenin immunoreactivity was not consistent. Thus, the overall appearance was designated as “atypical fibrous or fibro-osseous proliferation” and could not be further specified despite expert consultation from multiple sources. Chromosome analysis showed a normal, 46, XY karyotype, and microarray analysis did not reveal any clonal abnormalities. FISH testing revealed ALK was not rearranged. Tumor targeted sequencing revealed the presence of an E511K mutation in the RET gene. No diagnostic variants in the CTNNB1 or GNAS genes were identified. Subsequent targeted sequencing of the DNA isolated from a sample of the patient’s blood identified the same RET variant.
Discussion
In the pediatric population, juvenile ossifying fibroma and fibrous dysplasia represent the most common spindle cell proliferations involving the sinonasal tract, 12 while desmoid fibromatosis is extremely rare with approximately 19 reported cases. 13 -18 When encountering such a rare entity, the diagnosis has to be firmly established with characteristic morphologic features and concordant results of immunohistochemical and molecular diagnostic tests. Desmoid fibromatosis exhibits characteristic CTNNB1 gene mutations in the majority of cases causing increased intranuclear accumulation of β-catenin. In the head and neck, genetic tests for CTNNB1 mutations can reliably differentiate desmoid fibromatosis from other fibrous or fibro-osseous lesions. 19,20 Although nuclear localization of β-catenin shows high correlation in identifying CTNNB1 mutations, it lacks sensitivity and specificity in the head and neck where cranial fasciitis, Gardner fibroma, and biphenotypic sinonasal sarcoma are all reported to be positive. 21
Compared to other molecular tests, NGS methods exhibit high sensitivity and specificity in detecting CTNNB1 mutations. 22 These mutations generally occur in exon 3 of CTNNB1, including the one observed in the present case, and are considered to be activating as they lead to increased stability of the β-catenin protein and activation of the wingless Int-1 (WNT) pathway. 23 Three different β-catenin missense mutations—T41A, S45F, and S45P—have been described in the vast majority of desmoid fibromatosis, and there is some evidence showing a poorer outcome in patients harboring the S45F mutation. 24 A pediatric case of sinonasal fibromatosis with a D32V mutation has also been reported. 18 Our patient in this series had a G34V mutation that has not been previously described in desmoid fibromatosis. 25 However, it is a known pathogenic mutation that has been confirmed to occur somatically and has been previously reported in a range of tumor types such as hepatoblastoma, gastric adenocarcinoma, medulloblastoma, adrenocortical carcinoma, melanoma, and squamous cell carcinoma of the head and neck. 26,27 Next-generation sequencing can also detect mutations that are characteristic of fibrous dysplasia or other tumors and help distinguish these from other spindle cell lesions. 28 Thus, in the diagnosis of pediatric sinonasal spindle cell proliferations, genetic molecular tests are often necessary in differentiating rare entities from more common ones. Next-generation sequencing methods allow for a smaller panel of immunohistochemical stains and supplant the need for FISH and other single molecular tests.
Desmoid fibromatosis arises from the musculoaponeurotic stromal elements in the head and neck and causes an aggressive proliferation of cytologically benign fibroblasts. The tumor often has infiltrative edges and poorly defined margins which make complete surgical excision difficult. 13,29 Although classified as an intermediate category lesion between benign fibrous lesions and fibrosarcoma, it often behaves like a low-grade sarcoma and tends to have high recurrence rates that may be life-threatening. 13 Head and neck fibromatosis is uncommon in general and is much rarer in children, where it may present as rapidly growing painless mass or cause secondary symptoms of pain, airway obstruction, and dysphagia depending on its exact location. 30 Most cases involve the neck, followed by the face, jaw region, oral cavity, and scalp. Involvement of the paranasal sinuses is rare, whereas the maxillary sinus was the most common site of involvement in a series of 25 adult cases. 13 The maxillary sinus was also the most commonly involved location in children. However, tumors generally show extension to more than one area, as seen in our 2 cases. Recurrence rate is higher in adults than children and increases with positive resection margin. 13,14 Most cases, including initial and recurrent ones, are treated by surgical resection.
The diagnosis of the spindle cell lesion in case 2 remained elusive after extensive immunohistochemical and molecular testing. Although the presence of focal calcifications suggested a fibro-osseous lesion such as fibrous dysplasia, that possibility was deemed unlikely in view of the absence of GNAS mutations. Due to its similar morphology to the tumor in case 1, at least focally, the possibility of fibromatosis could not be excluded. Rarely, desmoid fibromatosis may be negative for β-catenin nuclear immunoreactivity and sequencing to detect the characteristic CTNNB1 mutations may be necessary. 24 Interestingly, however, this tumor harbored an E511K variant in the RET gene, a feature that has not been previously reported with any known spindle cell proliferation in the head and neck. Generally, this variant occurs rarely in tumors, as it has only been reported in 2 cases in the COSMIC database: one, a carcinoma of the breast, and the other, a carcinoma of the rectum. 27 Furthermore, activating germline mutations in the RET gene are associated with multiple endocrine neoplasia type 2 (MEN2), medullary thyroid carcinoma (MTC), pheochromocytoma, and rarely in other carcinomas. 31,32
As only the tumor was profiled by NGS, and since the possibility existed that the RET variant was a germ line change and not a true somatic mutation, we pursued targeted Sanger sequencing for the RET variant, using DNA isolated from a sample of the patient’s blood. This variant, c.1531G>A (p.Glu511Lys), was detected in the blood at an allele frequency of ∼50%, confirming its germline status. Upon review, it was deemed by our laboratory to be a variant of unknown significance due to the following findings: Functional studies employing immortalized cell lines (mouse fibroblasts and human embryonic kidney cells) had shown this variant to increase RET kinase activity above that of wild type, but significantly less than other known pathogenic variants of the RET gene. Two published reports had identified 2 unrelated individuals carrying this variant as having presumably sporadic MTC. The first patient was referred at age 65 years. 31,32 She passed the variant to 3 of her 4 children (ages 49, 51, and 56), none of whom showed signs of MEN2, such as MTC, pheochromocytoma, or hyperparathyroidism. The second patient underwent thyroidectomy at age 66 and showed no features of MEN2 during 10 years of follow-up. 33 Furthermore, bioinformatic tools such as SIFT and PolyPhen provided conflicting evidence (tolerated and disease causing, respectively) regarding the deleterious nature of this variant, and the variant was not absent from the Broad Institute’s control population database gnomAD (highest frequency 0.03%, 37/114 812 Non-Finnish European alleles). Hence, this variant was classified as one of unknown significance, which was consistent with the interpretation made by a majority of other clinical genetics laboratories in the ClinVar database. 26 This case is a good reminder of how tumor-only NGS testing has the potential to identify both somatic and germ line variants. To minimize the cost and turnaround time, clinical tumor testing often does not involve analysis of a matched germline sample. Thus, it may not be fully apparent whether an identified variant is somatic or germline if only tumor DNA is sequenced. Thus, as exemplified by case 2, caution in interpretation of variants identified by tumor-only NGS testing and paired analysis with a matched germ line sample are recommended, particularly in children, to avoid misinterpretation of sequencing results.
Surgical excision is generally the standard, and also most effective, treatment for benign fibroblastic proliferations, including fibromatosis. However, resection in the head and neck region, especially in the sinuses and skull base, may be difficult because of the presence of vital structures. Due to spatial restrictions, en bloc resections are typically not possible without significant morbidity and, as such, a piecemeal approach to resection must be employed. In addition, negative margins are often not possible to obtain in this region given the proximity of the lesions to vital structures, which makes the risk of recurrence a significant concern. Since sinonasal fibromatosis is extremely rare, guidelines for the clinical management of residual and recurrent disease remain unclear. 30 Additionally, nonsurgical treatment such as radiotherapy, chemotherapy, hormonal therapy, and nonsteroidal anti-inflammatory drugs have all been explored with various benefits. 13 Identification of genetic mutations by NGS allows for targeted therapy or precision treatment of recurrent or incompletely resected tumors. Desmoid tumors with CTNNB1 mutations may be sensitive to mammalian target of rapamycin (mTOR) inhibitors, such as everolimus and temsirolimus, as well as other agents such as the COX-2 inhibitor celecoxib. In fact, a desmoid tumor with a T41A missense mutation in CTNNB1 in a 39-year-old man was successfully treated with celecoxib as evidenced by tumor regression. 34 Celecoxib has been shown to have an antitumor effect due to its suppression of the Wnt/β-catenin signaling pathway. Along the same lines, activating RET mutations, if present, could have been blocked by ponatinib, sunitinib, vandetanib, or other Food and Drug Administration–approved tyrosine kinase inhibitors. 35
In conclusion, sinonasal fibroblastic proliferations are rare in children and can be difficult to classify based on morphologic features alone. Because of the nonspecificity of immunohistochemistry, molecular testing may be required to differentiate aggressive fibromatosis from other entities. Next-generation sequencing can help identify diagnostic molecular alterations and its role in highlighting targets for possible precision therapy cannot be underestimated. Cautious interpretation of NGS data is needed to determine their specificity and applicability.
Footnotes
Acknowledgments
The authors would like to express gratitude to Drs Maryna Vazmitsel, Alexander Lazar, and Christopher Fletcher for expressing their diagnostic pathology opinions on these cases.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.